
Nuclear Receptor Regulation of Aquaporin-2 in the Kidney

Abstract
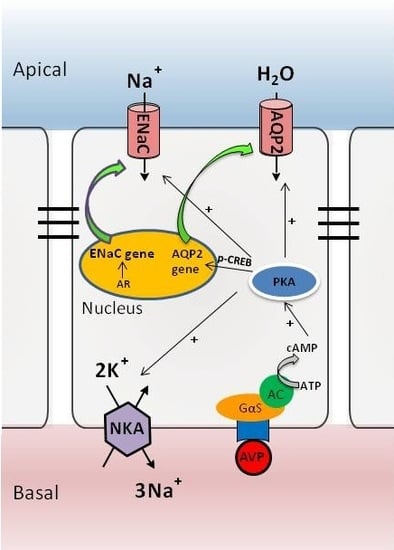
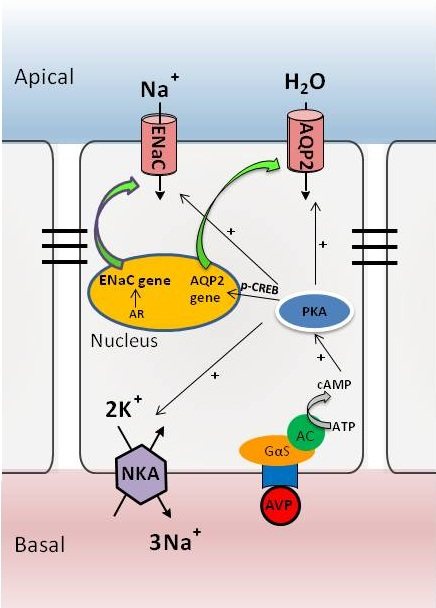
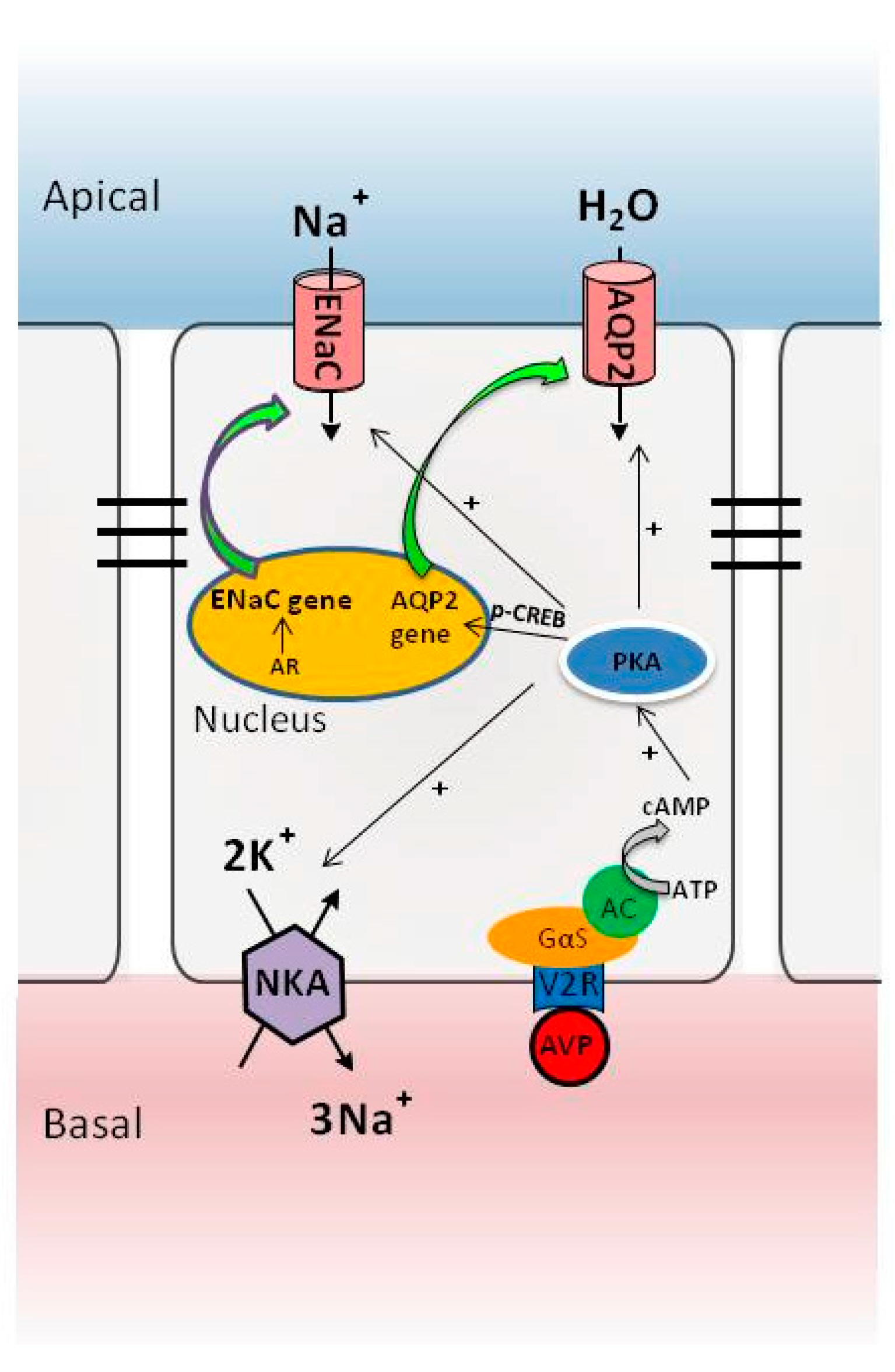
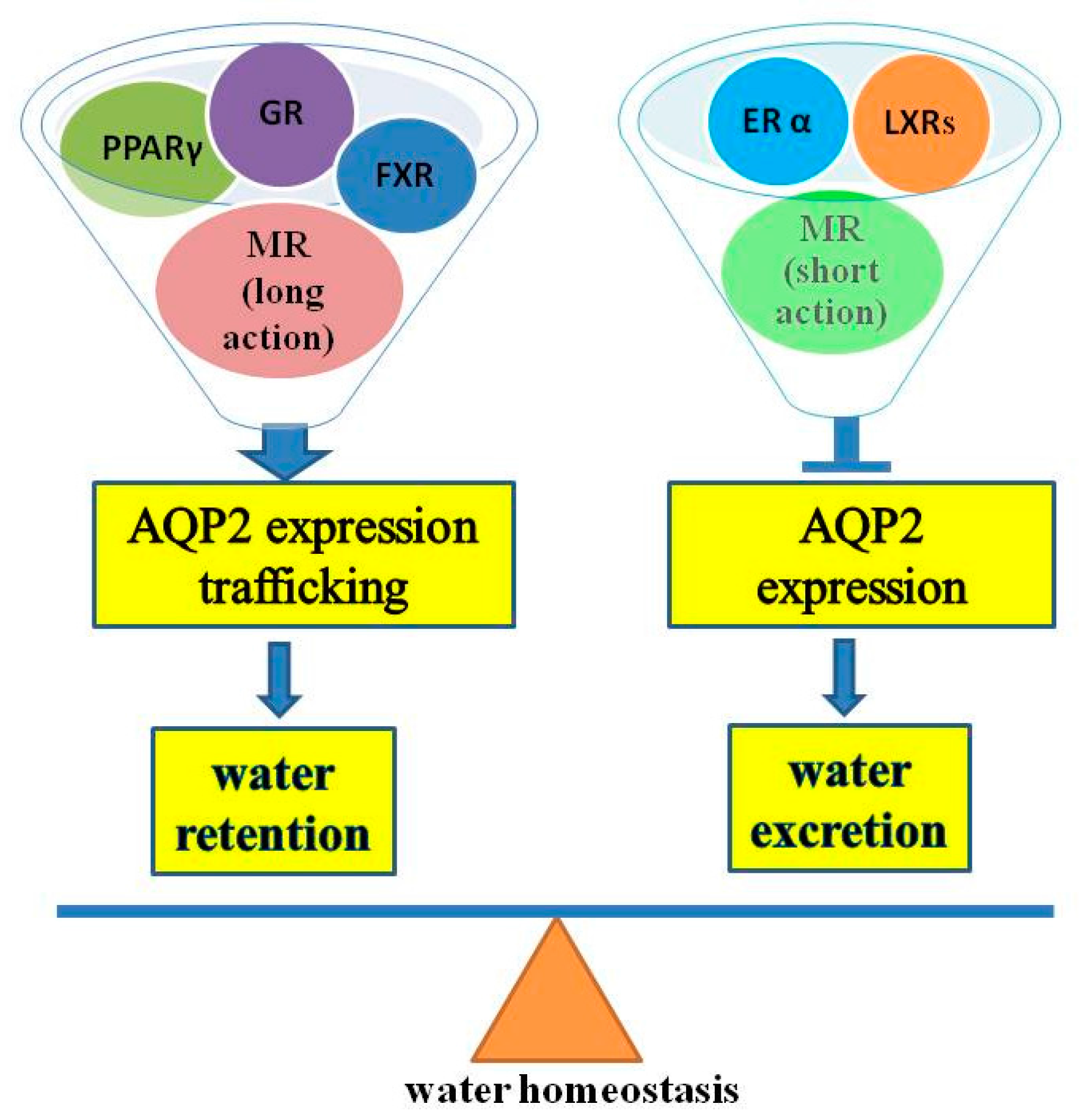
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. AQP Expression in the Kidney
3. The AVP-V2 Receptor System and Locally Produced Factors in Controlling AQP2 Expression
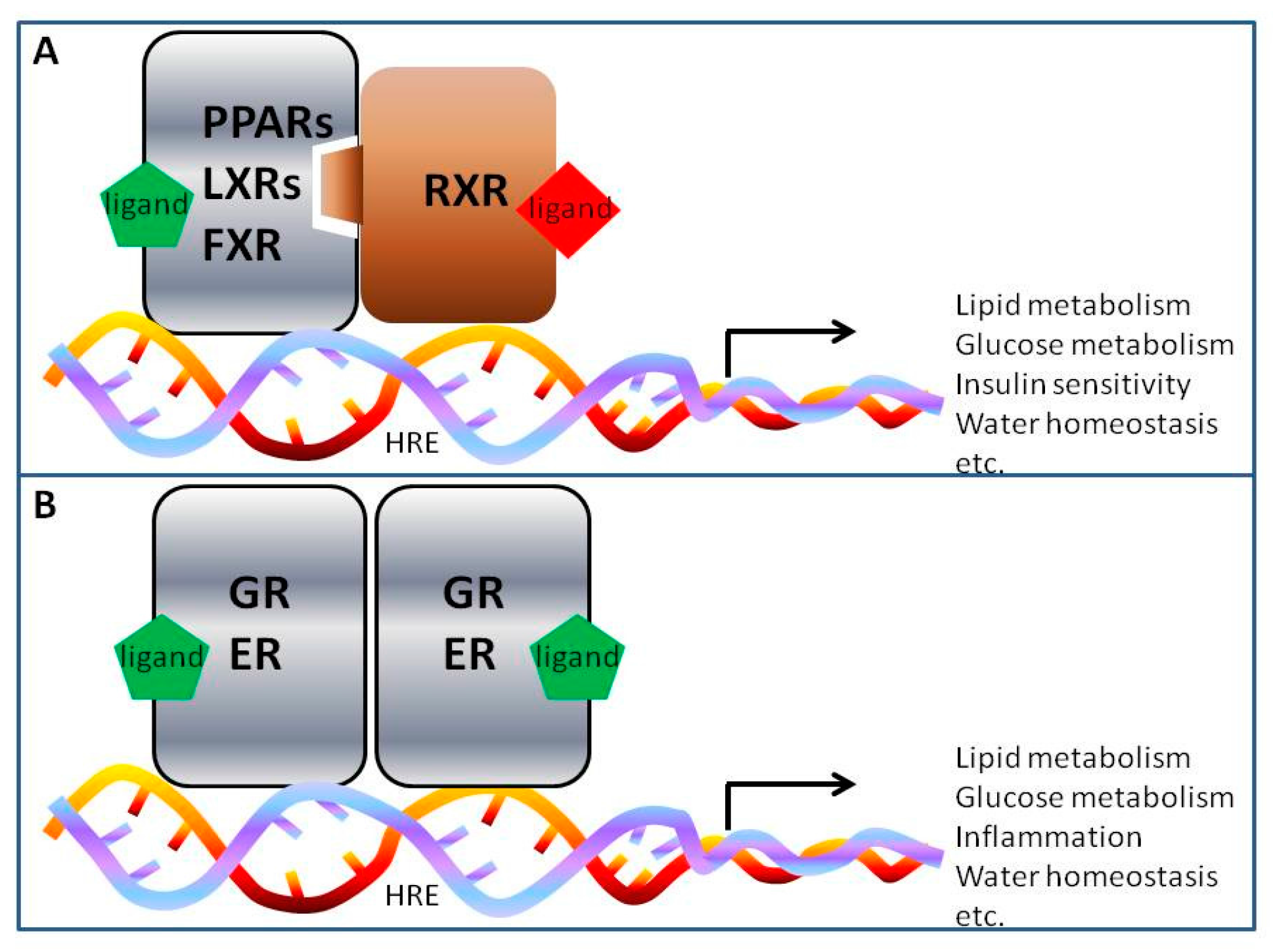
4. Nuclear Receptor Regulation of AQP2 in Collecting Ducts
4.1. Peroxisome Proliferator-Activated Receptor Gamma (PPARγ)
4.2. Glucocorticoid Receptor (GR)
4.3. Mineralocorticoid Receptor (MR)
4.4. Farnesoid X Receptor (FXR)
4.5. Liver X Receptors (LXRs)
4.6. Estrogen Receptor Alpha (ERα)
5. Perspective and Future Direction
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sands, J.M.; Layton, H.E. Advances in understanding the urine-concentrating mechanism. Annu. Rev. Physiol. 2014, 76, 387–409. [Google Scholar] [CrossRef] [PubMed]
- Esteva-Font, C.; Ballarin, J.; Fernandez-Llama, P. Molecular biology of water and salt regulation in the kidney. Cell. Mol. Life Sci. 2012, 69, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Fushimi, K.; Uchida, S.; Hara, Y.; Hirata, Y.; Marumo, F.; Sasaki, S. Cloning and expression of apical membrane water channel of rat kidney collecting tubule. Nature 1993, 361, 549–552. [Google Scholar] [CrossRef] [PubMed]
- Rojek, A.; Fuchtbauer, E.M.; Kwon, T.H.; Frokiaer, J.; Nielsen, S. Severe urinary concentrating defect in renal collecting duct-selective AQP2 conditional-knockout mice. Proc. Natl. Acad. Sci. USA 2006, 103, 6037–6042. [Google Scholar] [CrossRef] [PubMed]
- Deen, P.M.; Verdijk, M.A.; Knoers, N.V.; Wieringa, B.; Monnens, L.A.; van Os, C.H.; van Oost, B.A. Requirement of human renal water channel aquaporin-2 for vasopressin-dependent concentration of urine. Science 1994, 264, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, T.; Yaguchi, T.; Shimizu, K.; Kita, A.; Ishibashi, K.; Takata, K. The distribution and function of aquaporins in the kidney: Resolved and unresolved questions. Anat. Sci. Int. 2016. [Google Scholar] [CrossRef] [PubMed]
- Fujiyoshi, Y.; Mitsuoka, K.; de Groot, B.L.; Philippsen, A.; Grubmuller, H.; Agre, P.; Engel, A. Structure and function of water channels. Curr. Opin. Struct. Biol. 2002, 12, 509–515. [Google Scholar] [CrossRef]
- Rojek, A.; Praetorius, J.; Frokiaer, J.; Nielsen, S.; Fenton, R.A. A current view of the mammalian aquaglyceroporins. Annu. Rev. Physiol. 2008, 70, 301–327. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S. Renal aquaporins: An overview. BJU Int. 2002, 90, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, K.; Hara, S.; Kondo, S. Aquaporin water channels in mammals. Clin. Exp. Nephrol. 2009, 13, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, R.A.; Marchissio, M.J. Mitochondrial aquaporin-8: A functional peroxiporin? Antioxid. Redox Signal. 2013, 19, 896. [Google Scholar] [CrossRef] [PubMed]
- Huber, V.J.; Tsujita, M.; Nakada, T. Aquaporins in drug discovery and pharmacotherapy. Mol. Asp. Med. 2012, 33, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Beitz, E.; Kozono, D.; Guggino, W.B.; Agre, P.; Yasui, M. Characterization of aquaporin-6 as a nitrate channel in mammalian cells. Requirement of pore-lining residue threonine 63. J. Biol. Chem. 2002, 277, 39873–39879. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, S.; Frokiaer, J.; Marples, D.; Kwon, T.H.; Agre, P.; Knepper, M.A. Aquaporins in the kidney: From molecules to medicine. Physiol. Rev. 2002, 82, 205–244. [Google Scholar] [CrossRef] [PubMed]
- Noda, Y.; Sohara, E.; Ohta, E.; Sasaki, S. Aquaporins in kidney pathophysiology. Nat. Rev. Nephrol. 2010, 6, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Zhai, X.Y.; Fenton, R.A.; Andreasen, A.; Thomsen, J.S.; Christensen, E.I. Aquaporin-1 is not expressed in descending thin limbs of short-loop nephrons. J. Am. Soc. Nephrol. 2007, 18, 2937–2944. [Google Scholar] [CrossRef] [PubMed]
- Kwon, T.H.; Frokiaer, J.; Nielsen, S. Regulation of aquaporin-2 in the kidney: A molecular mechanism of body-water homeostasis. Kidney Res. Clin. Pract. 2013, 32, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Loonen, A.J.; Knoers, N.V.; van Os, C.H.; Deen, P.M. Aquaporin 2 mutations in nephrogenic diabetes insipidus. Semin. Nephrol. 2008, 28, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Kortenoeven, M.L.; Pedersen, N.B.; Miller, R.L.; Rojek, A.; Fenton, R.A. Genetic ablation of aquaporin-2 in the mouse connecting tubules results in defective renal water handling. J. Physiol. 2013, 591, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Pearce, D.; Soundararajan, R.; Trimpert, C.; Kashlan, O.B.; Deen, P.M.; Kohan, D.E. Collecting duct principal cell transport processes and their regulation. Clin. J. Am. Soc. Nephrol. 2015, 10, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Cao, R.; Du, S.; Jia, X.; Zheng, S.; Huang, S.; Han, Q.; Liu, J.; Zhang, X.; Miao, Y.; et al. Disruption of prostaglandin E2 receptor EP4 impairs urinary concentration via decreasing aquaporin 2 in renal collecting ducts. Proc. Natl. Acad. Sci. USA 2015, 112, 8397–8402. [Google Scholar] [CrossRef] [PubMed]
- Vukicevic, T.; Schulz, M.; Faust, D.; Klussmann, E. The trafficking of the water channel aquaporin-2 in renal principal cells-A potential target for pharmacological intervention in cardiovascular diseases. Front. Pharmacol. 2016, 7, 23. [Google Scholar] [CrossRef]
- Knepper, M.A.; Kwon, T.H.; Nielsen, S. Molecular physiology of water balance. N. Engl. J. Med. 2015, 372, 1349–1358. [Google Scholar] [PubMed]
- Bustamante, M.; Hasler, U.; Kotova, O.; Chibalin, A.; Mordasini, D.; Rousselot, M.; Vandewalle, A.; Martin, P.Y.; Féraille, E. Insulin potentiates AVP-induced AQP2 expression in cultured renal collecting duct principal cells. Am. J. Physiol. Ren. Physiol. 2005, 288, F334–F344. [Google Scholar] [CrossRef] [PubMed]
- Hasler, U.; Mordasini, D.; Bianchi, M.; Vandewalle, A.; Feraille, E.; Martin, P.Y. Dual influence of aldosterone on AQP2 expression in cultured renal collecting duct principal cells. J. Biol. Chem. 2003, 278, 21639–21648. [Google Scholar] [CrossRef] [PubMed]
- Hasler, U.; Vinciguerra, M.; Vandewalle, A.; Martin, P.Y.; Feraille, E. Dual effects of hypertonicity on aquaporin-2 expression in cultured renal collecting duct principal cells. J. Am. Soc. Nephrol. 2005, 16, 1571–1582. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Song, I.K.; Jang, K.J.; Nielsen, J.; Frokiaer, J.; Nielsen, S.; Kwon, T.H. Increased AQP2 targeting in primary cultured IMCD cells in response to angiotensin II through AT1 receptor. Am. J. Physiol. Ren. Physiol. 2007, 292, F340–F350. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, W.; Rivard, C.J.; Lanaspa, M.A.; Summer, S.; Schrier, R.W. Molecular mechanisms of angiotensin II stimulation on aquaporin-2 expression and trafficking. Am. J. Physiol. Ren. Physiol. 2011, 300, F1255–F1261. [Google Scholar] [CrossRef] [PubMed]
- Olesen, E.T.; Rutzler, M.R.; Moeller, H.B.; Praetorius, H.A.; Fenton, R.A. Vasopressin-independent targeting of aquaporin-2 by selective E-prostanoid receptor agonists alleviates nephrogenic diabetes insipidus. Proc. Natl. Acad. Sci. USA 2011, 108, 12949–12954. [Google Scholar] [CrossRef] [PubMed]
- Tamma, G.; Wiesner, B.; Furkert, J.; Hahm, D.; Oksche, A.; Schaefer, M.; Valenti, G.; Rosenthal, W.; Klussmann, E. The prostaglandin E2 analogue sulprostone antagonizes vasopressin-induced antidiuresis through activation of Rho. J. Cell Sci. 2003, 116, 3285–3294. [Google Scholar] [CrossRef] [PubMed]
- Zelenina, M.; Christensen, B.M.; Palmer, J.; Nairn, A.C.; Nielsen, S.; Aperia, A. Prostaglandin E2 interaction with AVP: Effects on AQP2 phosphorylation and distribution. Am. J. Physiol. Ren. Physiol. 2000, 278, F388–F394. [Google Scholar]
- Jang, K.J.; Cho, H.S.; Kang, D.H.; Bae, W.G.; Kwon, T.H.; Suh, K.Y. Fluid-shear-stress-induced translocation of aquaporin-2 and reorganization of actin cytoskeleton in renal tubular epithelial cells. Integr. Biol. 2011, 3, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Jung, H.J.; Kwon, T.H. Extracellular pH affects phosphorylation and intracellular trafficking of AQP2 in inner medullary collecting duct cells. Am. J. Physiol. Ren. Physiol. 2015, 308, F737–F748. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Jung, H.J.; Lee, Y.J.; Kwon, T.H. Vasopressin-regulated miRNAs and AQP2-targeting miRNAs in kidney collecting duct cells. Am. J. Physiol. Ren. Physiol. 2015, 308, F749–F764. [Google Scholar] [CrossRef] [PubMed]
- Burris, T.P.; Solt, L.A.; Wang, Y.; Crumbley, C.; Banerjee, S.; Griffett, K.; Lundasen, T.; Hughes, T.; Kojetin, D.J. Nuclear receptors and their selective pharmacologic modulators. Pharmacol. Rev. 2013, 65, 710–778. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y. Peroxisome proliferator-activated receptor family and its relationship to renal complications of the metabolic syndrome. J. Am. Soc. Nephrol. 2004, 15, 2801–2815. [Google Scholar] [CrossRef] [PubMed]
- Moeller, H.B.; Rittig, S.; Fenton, R.A. Nephrogenic diabetes insipidus: Essential insights into the molecular background and potential therapies for treatment. Endocr. Rev. 2013, 34, 278–301. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Gong, L.; Fang, Y.; Zhan, Q.; Liu, H.X.; Lu, Y.; Guo, G.L.; Lehman-McKeeman, L.; Fang, J.; Wan, Y.J. The role of retinoic acid in hepatic lipid homeostasis defined by genomic binding and transcriptome profiling. BMC Genom. 2013, 14, 575. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Hao, C.; Cha, D.R.; Rao, R.; Lu, W.; Kohan, D.E.; Magnuson, M.A.; Redha, R.; Zhang, Y.; Breyer, M.D. Thiazolidinediones expand body fluid volume through PPARgamma stimulation of ENaC-mediated renal salt absorption. Nat. Med. 2005, 11, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Gabbi, C.; Kong, X.; Suzuki, H.; Kim, H.J.; Gao, M.; Jia, X.; Ohnishi, H.; Ueta, Y.; Warner, M.; Guan, Y.; et al. Central diabetes insipidus associated with impaired renal aquaporin-1 expression in mice lacking liver X receptor β. Proc. Natl. Acad. Sci. USA 2012, 109, 3030–3034. [Google Scholar] [CrossRef] [PubMed]
- Cheema, M.U.; Irsik, D.L.; Wang, Y.; Miller-Little, W.; Hyndman, K.A.; Marks, E.S.; Frokiaer, J.; Boesen, E.I.; Norregaard, R. Estradiol regulates AQP2 expression in the collecting duct: A novel inhibitory role for estrogen receptor α. Am. J. Physiol. Ren. Physiol. 2015, 309, F305–F317. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, Y.; Ogawa, D.; Wada, J.; Yamamoto, N.; Shikata, K.; Sato, C.; Tachibana, H.; Toyota, N.; Makino, H. Activation of peroxisome proliferator-activated receptor delta inhibits streptozotocin-induced diabetic nephropathy through anti-inflammatory mechanisms in mice. Diabetes 2011, 60, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y. Targeting peroxisome proliferator-activated receptors (PPARs) in kidney and urologic disease. Minerva Urol. Nefrol. 2002, 54, 65–79. [Google Scholar] [PubMed]
- Zhou, L.; Panasiuk, A.; Downton, M.; Zhao, D.; Yang, B.; Jia, Z.; Yang, T. Systemic PPARgamma deletion causes severe disturbance in fluid homeostasis in mice. Physiol. Genom. 2015, 47, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Knepper, M.A.; Hu, X.; Verbalis, J.G.; Ecelbarger, C.A. Rosiglitazone activates renal sodium- and water-reabsorptive pathways and lowers blood pressure in normal rats. J. Pharmacol. Exp. Ther. 2004, 308, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Zhang, Y.; Davis, L.; Breyer, M.D. Expression of peroxisome proliferator-activated receptors in urinary tract of rabbits and humans. Am. J. Physiol. 1997, 273, F1013–F1022. [Google Scholar] [PubMed]
- Panchapakesan, U.; Pollock, C.A.; Chen, X.M. The effect of high glucose and PPAR-γ agonists on PPAR-γ expression and function in HK-2 cells. Am. J. Physiol. Ren. Physiol. 2004, 287, F528–F534. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, G.; Jia, Z.; Yang, K.T.; Sun, Y.; Kakizoe, Y.; Liu, M.; Zhou, S.; Chen, R.; Yang, B.; Yang, T. Increased susceptibility of db/db mice to rosiglitazone-induced plasma volume expansion: Role of dysregulation of renal water transporters. Am. J. Physiol. Ren. Physiol. 2013, 305, F1491–F1497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, A.; Kohan, D.E.; Nelson, R.D.; Gonzalez, F.J.; Yang, T. Collecting duct-specific deletion of peroxisome proliferator-activated receptor γ blocks thiazolidinedione-induced fluid retention. Proc. Natl. Acad. Sci. USA 2005, 102, 9406–9411. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yang, B.; McNulty, J.A.; Clifton, L.G.; Binz, J.G.; Grimes, A.M.; Strum, J.C.; Harrington, W.W.; Chen, Z.; Balon, T.W.; et al. GI262570, a peroxisome proliferator-activated receptor γ agonist, changes electrolytes and water reabsorption from the distal nephron in rats. J. Pharmacol. Exp. Ther. 2005, 312, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Blasi, E.R.; Heyen, J.R.; McHarg, A.D.; Ecelbarger, C. Time course of AQP-2 and ENaC regulation in the kidney in response to PPAR agonists associated with marked EDEMA in rats. Pharmacol. Res. 2008, 57, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Procino, G.; Gerbino, A.; Milano, S.; Nicoletti, M.C.; Mastrofrancesco, L.; Carmosino, M.; Svelto, M. Rosiglitazone promotes AQP2 plasma membrane expression in renal cells via a Ca-dependent/cAMP-independent mechanism. Cell Physiol. Biochem. 2015, 35, 1070–1085. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.B.; George, B.C.; Gonzalez-Auvert, C.; Dingman, J.F. Increased plasma arginine vasopressin in clinical adrenocortical insufficeincy and its inhibition by glucosteroids. J. Clin. Investig. 1967, 46, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Linas, S.L.; Berl, T.; Robertson, G.L.; Aisenbrey, G.A.; Schrier, R.W.; Anderson, R.J. Role of vasopressin in the impaired water excretion of glucocorticoid deficiency. Kidney Int. 1980, 18, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, S.; Schrier, R.W. Effect of arginine vasopressin antagonist on renal water excretion in glucocorticoid and mineralocorticoid deficient rats. Kidney Int. 1982, 22, 587–593. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Ishikawa, S.E.; Ando, F.; Higashiyama, M.; Nagasaka, S.; Sasaki, S. Vasopressin-dependent upregulation of aquaporin-2 gene expression in glucocorticoid-deficient rats. Am. J. Physiol. Ren. Physiol. 2000, 279, F502–F508. [Google Scholar]
- Wang, W.; Li, C.; Summer, S.N.; Falk, S.; Cadnapaphornchai, M.A.; Chen, Y.C.; Schrier, R.W. Molecular analysis of impaired urinary diluting capacity in glucocorticoid deficiency. Am. J. Physiol. Ren. Physiol. 2006, 290, F1135–F1142. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Cadnapaphornchai, M.A.; Summer, S.N.; Falk, S.; Li, C.; Wang, W.; Schrier, R.W. Molecular mechanisms of impaired urinary concentrating ability in glucocorticoid-deficient rats. J. Am. Soc. Nephrol. 2005, 16, 2864–2871. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Cai, H.; Klein, J.D.; Laur, O.; Chen, G. Dexamethasone increases aquaporin-2 protein expression in ex vivo inner medullary collecting duct suspensions. Front. Physiol. 2015, 6, 310. [Google Scholar] [CrossRef] [PubMed]
- Jaisser, F.; Farman, N. Emerging roles of the mineralocorticoid receptor in pathology: Toward new paradigms in clinical pharmacology. Pharmacol. Rev. 2016, 68, 49–75. [Google Scholar] [CrossRef] [PubMed]
- Jonassen, T.E.; Promeneur, D.; Christensen, S.; Petersen, J.S.; Nielsen, S. Decreased vasopressin-mediated renal water reabsorption in rats with chronic aldosterone-receptor blockade. Am. J. Physiol. Ren. Physiol. 2000, 278, F246–F256. [Google Scholar]
- Nielsen, J.; Kwon, T.H.; Praetorius, J.; Frokiaer, J.; Knepper, M.A.; Nielsen, S. Aldosterone increases urine production and decreases apical AQP2 expression in rats with diabetes insipidus. Am. J. Physiol. Ren. Physiol. 2006, 290, F438–F449. [Google Scholar]
- Ohara, M.; Cadnapaphornchai, M.A.; Summer, S.N.; Falk, S.; Yang, J.; Togawa, T.; Schrier, R.W. Effect of mineralocorticoid deficiency on ion and urea transporters and aquaporin water channels in the rat. Biochem. Biophys. Res. Commun. 2002, 299, 285–290. [Google Scholar] [CrossRef]
- Kwon, T.H.; Nielsen, J.; Masilamani, S.; Hager, H.; Knepper, M.A.; Frokiaer, J.; Nielsenm, S. Regulation of collecting duct AQP3 expression: Response to mineralocorticoid. Am. J. Physiol. Ren. Physiol. 2002, 283, F1403–F1421. [Google Scholar] [CrossRef] [PubMed]
- Stanton, B.; Janzen, A.; Klein-Robbenhaar, G.; DeFronzo, R.; Giebisch, G.; Wade, J. Ultrastructure of rat initial collecting tubule. Effect of adrenal corticosteroid treatment. J. Clin. Investig. 1985, 75, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Funder, J.W.; Pearce, P.T.; Smith, R.; Smith, A.I. Mineralocorticoid action: Target tissue specificity is enzyme, not receptor, mediated. Science 1988, 242, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Brooks, H.L.; Ageloff, S.; Kwon, T.H.; Brandt, W.; Terris, J.M.; Seth, A.; Michea, L.; Nielsen, S.; Fenton, R.; Knepper, M.A. cDNA array identification of genes regulated in rat renal medulla in response to vasopressin infusion. Am. J. Physiol. Ren. Physiol. 2003, 284, F218–F228. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.C.; Livingstone, D.E.; Kenyon, C.J.; Jansen, M.A.; Dear, J.W.; Mullins, J.J.; Bailey, M.A. A urine-concentrating defect in 11β-hydroxysteroid dehydrogenase type 2 null mice. Am. J. Physiol. Ren. Physiol. 2014, 303, F494–F502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, S.; Gao, M.; Liu, J.; Jia, X.; Han, Q.; Zheng, S.; Miao, Y.; Li, S.; Weng, H.; et al. Farnesoid X receptor (FXR) gene deficiency impairs urine concentration in mice. Proc. Natl. Acad. Sci. USA 2014, 111, 2277–2282. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Wang, F.; Xu, C.; Soodvilai, S.; Peng, K.; Su, J.; Zhao, L.; Yang, K.T.; Feng, Y.; Zhou, S.F.; et al. Soluble (pro)renin receptor via β-catenin enhances urine concentration capability as a target of liver X receptor. Proc. Natl. Acad. Sci. USA 2016, 113, E1898–E1906. [Google Scholar] [CrossRef] [PubMed]
- Ohara, M.; Martin, P.Y.; Xu, D.L.; St John, J.; Pattison, T.A.; Kim, J.K.; Schrier, R.W. Upregulation of aquaporin 2 water channel expression in pregnant rats. J. Clin. Investig. 1998, 101, 1076–1083. [Google Scholar] [CrossRef] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.-Y.; Wang, B.; Guan, Y.-F. Nuclear Receptor Regulation of Aquaporin-2 in the Kidney. Int. J. Mol. Sci. 2016, 17, 1105. https://doi.org/10.3390/ijms17071105
Zhang X-Y, Wang B, Guan Y-F. Nuclear Receptor Regulation of Aquaporin-2 in the Kidney. International Journal of Molecular Sciences. 2016; 17(7):1105. https://doi.org/10.3390/ijms17071105
Chicago/Turabian StyleZhang, Xiao-Yan, Bing Wang, and You-Fei Guan. 2016. "Nuclear Receptor Regulation of Aquaporin-2 in the Kidney" International Journal of Molecular Sciences 17, no. 7: 1105. https://doi.org/10.3390/ijms17071105
APA StyleZhang, X.-Y., Wang, B., & Guan, Y.-F. (2016). Nuclear Receptor Regulation of Aquaporin-2 in the Kidney. International Journal of Molecular Sciences, 17(7), 1105. https://doi.org/10.3390/ijms17071105