
Lipid Droplets: A Key Cellular Organelle Associated with Cancer Cell Survival under Normoxia and Hypoxia

Abstract
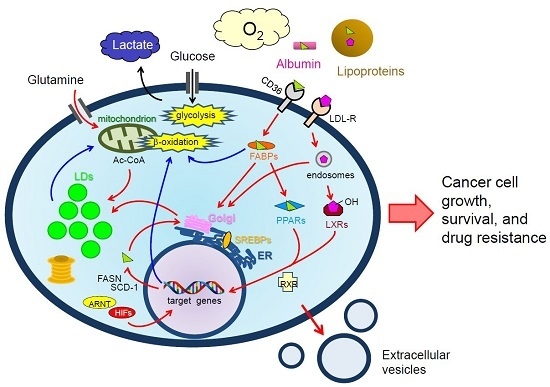
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Lipid Droplets: Origin and Components
3. Exogenous and Endogenous Sources of LCFAs and Cholesterol
4. Molecular Mechanisms Associated with the Biosynthesis of LDs in Cancer Cells
4.1. Decomposition of LDs through β-Oxidation
4.2. Production of LDs under Normoxia
4.2.1. Renal Clear Cell Carcinoma
4.2.2. HIF-Dependent Mechanisms of LD Synthesis in RCC Cells
4.2.3. HIF-Independent Mechanisms of LD Synthesis in RCC Cells
4.2.4. Prostate Cancer
4.2.5. Breast Cancer
4.2.6. Ovarian Cancer
4.2.7. Colon Cancer
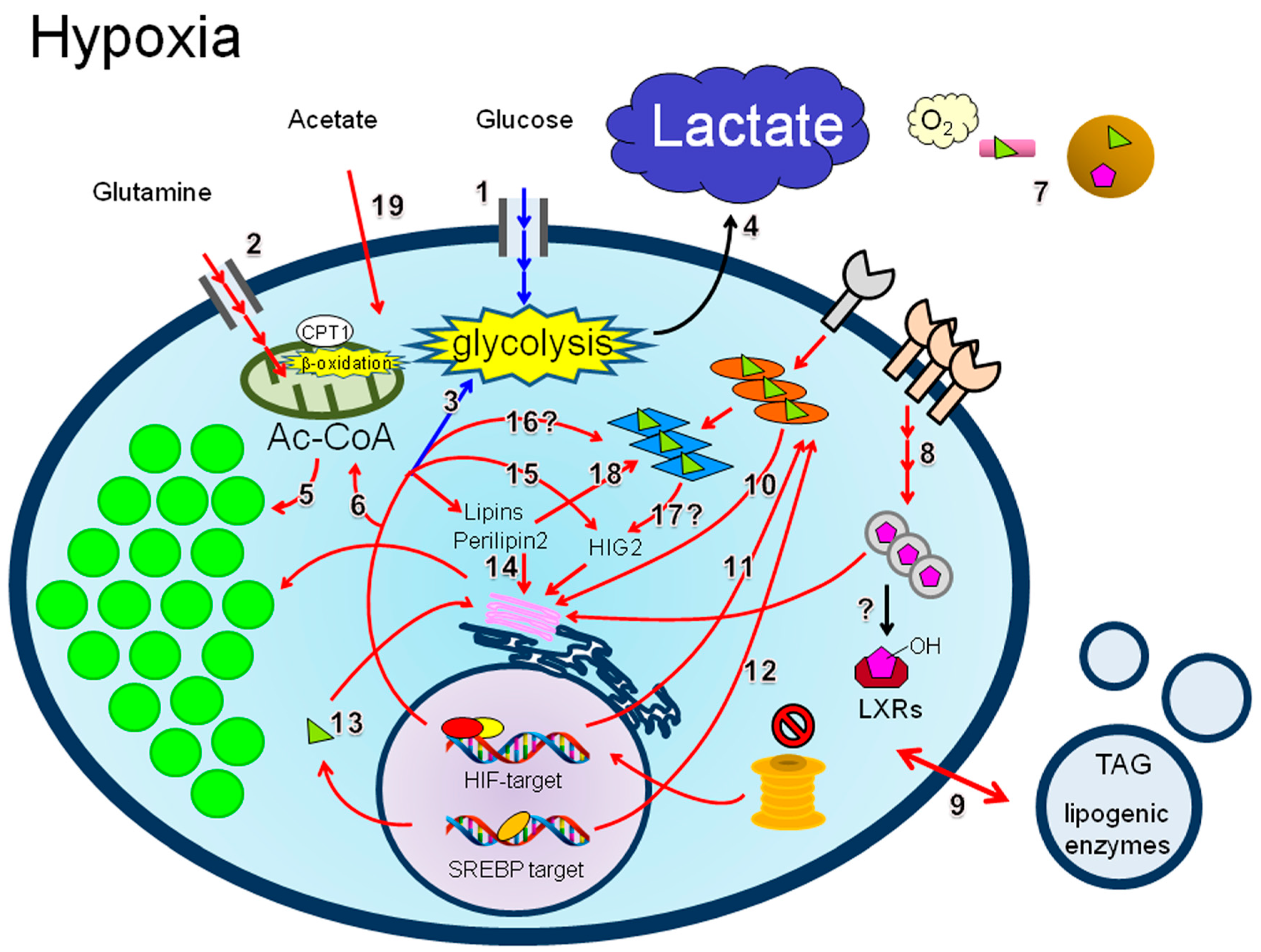
4.3. LD Synthesis under Hypoxia
4.3.1. Lipid Uptake
4.3.2. De Novo Lipogenesis
4.3.3. Lipins
4.3.4. Perilipin 2
4.3.5. HIG2
4.3.6. PPARs
4.3.7. Acetate
4.3.8. β-Oxidation and Hypoxia Reoxygenation
5. Function of LDs in Cancer Cells
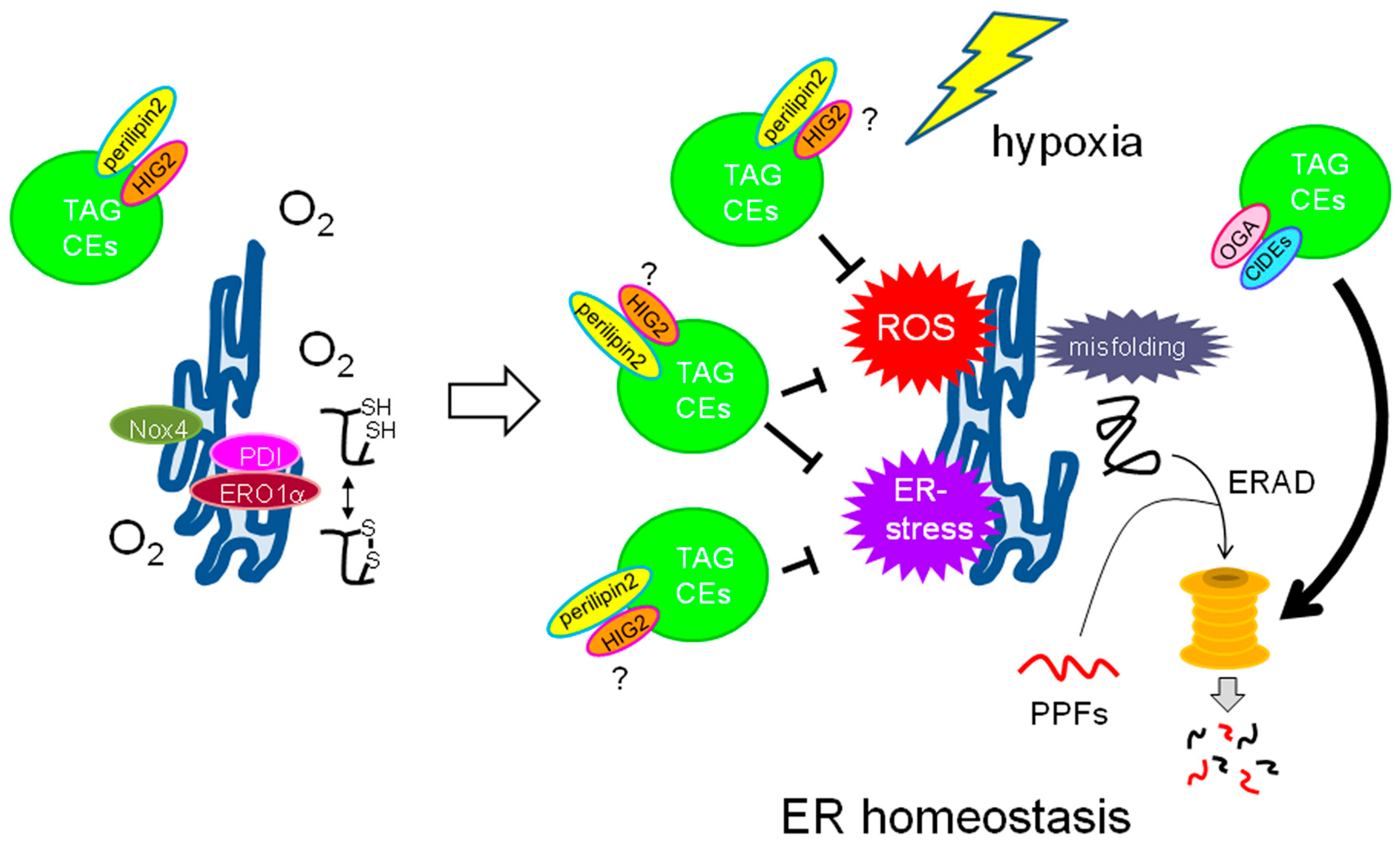
5.1. Roles of LDs in ER Homeostasis
5.2. Role of LDs as a ROS Scavenger
5.2.1. ROS Generation in Hypoxic Cancer Cells
5.2.2. LDs as Potential ROS Scavenging Organelles in Cancer Cells
5.3. Drug Resistance
6. Summary and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contribution to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumor growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Signaling in control of cell growth and metabolism. Cold Spring Harb. Perspect. Biol. 2012, 4, a006783. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr.; Thompson, C.B. Clues from cell metabolism. Nature 2010, 465, 562–564. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, M.W.; Berk, P.D. Cellular uptake of long chain free fatty acids: The structure and function of plasma membrane fatty acid binding protein. In Lipobiology; van der Vusse, G., Ed.; Elsevier: Amsterdam, The Netherlands, 2004; pp. 47–80. [Google Scholar]
- Wüstner, D. Intracellular cholesterol transport. In Cellular Lipid Metabolism; Ehnholm, C., Ed.; Springer-Verlag: Berlin & Heidelberg, Germany, 2009; pp. 157–190. [Google Scholar]
- Baenke, F.; Peck, B.; Miess, H.; Schulze, A. Hooked on fat; the role of lipid synthesis in cancer metabolism and tumor development. Dis. Model. Mech. 2013, 6, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, S.-V.; Boström, P.; Lagerstedt, J.; Anderson, L.; Adiels, M.; Permann, J. The lipid droplet: A dynamic organelle, not only involved in the storage and turnover of lipids. In Cellular Lipid Metabolism; Ehnholm, C., Ed.; Springer-Verlag: Berlin/Heidelberg, Germany, 2009; pp. 1–26. [Google Scholar]
- Farese, R.V.; Walther, T.C. Lipid droplets finally get a little R-E-S-P-E-C-T. Cell 2009, 25, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Beller, M.; Thiel, K.; Thul, P.J.; Jäckle, H. Lipid droplets: A dynamic organelle moves into focus. FEBS Lett. 2010, 584, 2176–2182. [Google Scholar] [CrossRef] [PubMed]
- Brasaemle, D.L.; Wolins, N.E. Packaging of fat: An evolving model of lipid droplet assembly and expansion. J. Biol. Chem. 2012, 287, 2273–2279. [Google Scholar] [CrossRef] [PubMed]
- Frayn, K.N.; Langin, D. Triacylglycerol metabolism in adipose tissue. In Lipobiology; van der Vusse, G., Ed.; Elsevier: Amsterdam, The Netherlands, 2004; pp. 337–356. [Google Scholar]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijon, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glode, L.M.; Eckel, R.H.; et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol. Cancer Ther. 2014, 13, 2361–2371. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Li, T.; Zhang, L.; Sun, L.; He, X.; Zhong, X.; Jia, D.; Song, L.; Semenza, G.L.; Gao, P.; et al. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 2014, 8, 1930–1942. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, C.C.; Pandolfi, P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Swierczynski, J.; Hebanowska, A.; Sledzinski, T. Role of abnormal metabolism in development, progression, diagnosis and therapy of pancreatic cancer. World J. Gastroenterol. 2014, 20, 2279–2303. [Google Scholar] [CrossRef] [PubMed]
- Peck, B.; Schulze, A. Lipid desaturation—The next step in targeting lipogenesis in cancer? FEBS J. 2016, 283, 2767–2778. [Google Scholar] [CrossRef] [PubMed]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.C.; Farese, R.V. Lipid droplets and cellular lipid metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. [Google Scholar] [CrossRef] [PubMed]
- Krahmer, N.; Farase, R.V.; Walther, T.C. Balancing the fat: Lipid droplets and human disease. EMBO Mol. Med. 2013, 5, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Wilfling, F.; Haas, J.T.; Walther, T.C.; Farase, R.V. Lipid biogenesis. Curr. Opin. Cell Biol. 2014, 29, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Welte, E.A. Expanding roles for lipid droplets. Curr. Biol. 2015, 25, 470–481. [Google Scholar] [CrossRef] [PubMed]
- Marcel, Y.L.; Ouimet, M.; Wang, M.-D. Cellular lipid traffic and lipid transporters: Regulation of efflux and HDL formation. In Cellular Lipid Metabolism; Ehnholm, C., Ed.; Springer-Verlag: Berlin & Heidelberg, Germany, 2009; pp. 73–106. [Google Scholar]
- Potocoava, M.C.; Futia, G.L.; Aughenbaugh, J.; Schlaepfer, I.R.; Gibson, E.A. Raman and coherent anti-Stokes Raman scattering microscopy studies of changes in lipid content and composition in hormone-treated breast and prostate cancer cells. J. Biomed. Opt. 2014, 19. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Wilson, M.; Mcconville, C.; Arvanitis, T.N.; Griffin, J.L.; Kauppinen, R.A.; Peet, A.C. Increased unsaturation of lipids in cytoplasmic lipid droplets in DAOY cancer cells in response to cisplatin treatment. Metabolomics 2013, 9, 722–729. [Google Scholar] [CrossRef] [PubMed]
- Yue, S.; Lee, S.-Y.; Lee, H.-J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaser, C.; Demmelmair, H.; Koletzko, B. High-throughput analysis of total plasma fatty acid composition with direct in situ transesterification. PLoS ONE 2010, 5, e12045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quehenberger, O.; Armando, A.M.; Brown, A.H.; Milne, S.B.; Myers, D.S.; Merrill, A.H.; Bandyopadhyay, S.; Jones, K.N.; Kelly, S.; Shaner, R.L. Lipidomics reveales a remarkable diversity of lipids in human plasma. J. Lipid Res. 2010, 51, 3299–3305. [Google Scholar] [CrossRef] [PubMed]
- Curry, S. Plasma albumin as a fatty acid carrier. In Lipobiology; van der Vusse, G., Ed.; Elsevier: Amsterdam, The Netherlands, 2004; pp. 29–46. [Google Scholar]
- Chang, T.-Y.; Chang, C.-Y.; Ohgami, N.; Yamaguchi, Y. Cholesterol sensing, trafficking, and esterification. Annu. Rev. Cell Biol. 2006, 22, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even Warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Keung, W.; Samokhvalov, V.; Wang, W.; Lopaschuk, G.D. Role of fatty acid uptake and fatty acid beta-oxidation in mediating insulin resistance in heart and skeletal muscle. Biochim. Biophys. Acta 2010, 1801, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Collins, L.B.; Wail, A.; Bigler, R.; Sun, W.; Bultman, S.J. The Warburg effect dictates the mechanism of butylate-mediated histone acetylation and cell proliferation. Mol. Cell 2012, 48, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Daniels, G.; Lee, P.; Monaco, M.E. Lipid metabolism in prostate cancer. Am. J. Clin. Exp. Urol. 2014, 2, 111–120. [Google Scholar] [PubMed]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Glatz, J.F.C.; Brinkman, J.F.F.; Bonen, A.; van der Vusse, G.J.; Luiken, J.J.F.P. Uptake of fatty acids by parenchymal cells: Role of FAT/CD36. In Lipobiology; van der Vusse, G., Ed.; Elsevier: Amsterdam, The Netherlands, 2004; pp. 89–98. [Google Scholar]
- Su, X.; Abumrad, N.A. Cellular fatty acid uptake: A pathway under construction. Trends Endoclinol. Metab. 2009, 20, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Haunerland, N.H.; Spener, F. Properties and physiological significance of fatty acid binding proteins. In Lipobiology; van der Vusse, G., Ed.; Elsevier: Amsterdam, The Netherlands, 2004; pp. 89–98. [Google Scholar]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.-W.; Cravatt, B.F. Monoacyl glycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Pyper, S.R.; Viswakarma, N.; Yu, S.; Reddy, J.K. PPARα: Energy combustion, hypolipidemia, inflammation and cancer. Nucl. Recept. Signal 2010, 8, e002. [Google Scholar] [CrossRef] [PubMed]
- Hessvik, N.P.; Bakke, S.S.; Smith, R.; Ravna, A.W.; Sylte, I.; Rustan, A.C.; Thoresen, G.H.; Kase, E.T. The liver X receptor modulator 22(S)-hydroxycholesterol exerts cell-type specific effects on lipid and glucose metabolism. J. Steroid Biochem. Mol. Biol. 2012, 128, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Colgan, S.P.; Shelley, C.S. Hypoxia: The force that drives chronic kidney disease. Clin. Med. Res. 2016, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Brunet, J.-P.; Napoli, A.D.; Mertz, K.D.; Seeley, A.; Pires, M.M.; Linhart, D.; Worrell, R.A.; Moch, H.; Rubin, M.A.; et al. Patterns of gene expression and copy-number alterations in von-Hippel Lindau disease-associated and sporadic clear cell carcinoma of the kidney. Cancer Res. 2009, 69, 4674–4681. [Google Scholar] [CrossRef] [PubMed]
- Drabkin, H.A.; Gemmill, R.M. Obesity, cholesterol, and Clear-Cell Renal Cell Carcinoma (RCC). In Advances in Cancer Research; van der Vusse, G., Ed.; Elsevier: Amsterdam, The Netherlands, 2010; pp. 39–56. [Google Scholar]
- Sundelin, J.P.; Ståhlman, M.; Lundqvist, A.; Levin, M.; Parini, P.; Johansson, M.E.; Borén, J. Increased expression of the very low-density lipoprotein receptor mediates lipid accumulation in clear-cell renal cell carcinoma. PLoS ONE 2012, 7, e48694. [Google Scholar] [CrossRef] [PubMed]
- Togashi, A.; Katagiri, T.; Ashida, S.; Fujioka, T.; Maruyama, O.; Wakumoto, Y.; Sakamoto, Y.; Fujime, M.; Kawachi, Y.; Shuin, T.; et al. Hypoxia-inducible protein 2 (HIG2), a novel diagnostic marker for renal cell carcinoma and potential target for molecular therapy. Cancer Res. 2005, 65, 4817–4826. [Google Scholar] [CrossRef] [PubMed]
- Gimm, T.; Wiese, M.; Teschemacher, B.; Deggerich, A.; Schodel, J.; Knaup, K.X.; Hackenbeck, T.; Hellerbrand, C.; Amann, K.; Wiesener, M.S. Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1. FASEB J. 2010, 24, 4443–4458. [Google Scholar] [CrossRef] [PubMed]
- Qiu, B.; Ackerman, D.; Sanchez, D.J.; Li, B.; Ochocki, J.D.; Grazioli, A.; Bobrovnikova-Marjon, E.; Diehl, A.; Keith, B.; Simon, M.C. HIF2α-dependent lipid storage promotes endoplasmic reticulum homeostasis in clear-cell renal cell carcinoma. Cancer Discov. 2015, 5, 652–667. [Google Scholar] [CrossRef] [PubMed]
- Von Roemeling, C.A.; Marlow, L.A.; Wei, J.J.; Cooper, S.J.; Caulfield, T.R.; Wu, K.; Tan, W.W.; Tun, H.W.; Copland, J.A. Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell carcinoma. Clin. Cancer Res. 2013, 19, 2368–2380. [Google Scholar] [CrossRef] [PubMed]
- Gebhard, R.L.; Clayman, R.V.; Prigge, W.F.; Figenshau, R.; Staley, N.A.; Reesey, C.; Bear, A. Abnormal cholesterol metabolism in renal clear cell carcinoma. J. Lipid Res. 1987, 28, 1177–1184. [Google Scholar] [PubMed]
- Xu, G.; Jiang, Y.; Xiao, Y.; Liu, X.-D.; Yue, F.; Li, W.; Li, X.; He, Y.; Jiang, X.; Chen, Q.; et al. Fast clearlance of lipid droplets through MAP1S-activated autophagy suppresses clear renal cell carcinomas and promotes patient survival. Oncotarget 2016, 7, 6255–6265. [Google Scholar] [PubMed]
- Huang, W.-C.; Li, X.; Liu, J.; Lin, J.; Chung, L.W.K. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Mol. Cancer Res. 2011, 10, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Gang, X.; Yang, Y.; Zhong, J.; Pan, Y.; Karnes, R.J.; Zhang, J.; Xu, W.; Wang, G.; Huang, H. p300 acetyltransferase regulates fatty acid synthase expression, lipid metabolism and prostate cancer growth. Oncotarget 2016, 7, 15135–15149. [Google Scholar] [PubMed]
- Kaini, R.R.; Hu, C.-A.A. Synergistic killing effect of chloroquine and androgen deprivation in LNCaP cells. Biochem. Biophys. Res. Commun. 2012, 425, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Kaini, R.R.; Sillerud, L.O.; Zhaorigetsu, S.; Hu, C.-A.A. Autophagy regulates lipolysis and cell survival through lipid droplet degradation in androgen-sensitive prostate cancer cells. Prostate 2012, 72, 1412–1422. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Hitz, C.A.; Gijon, M.A.; Bergman, B.C.; Eckel, R.H.; Jacobsen, B.M. Progestin modulates the lipid profile and sensitivity of breast cancer cells to docetaxel. Mol. Cell. Endocrinol. 2012, 363, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Oba, T.; Ono, M.; Iesato, A.; Hanamura, T.; Watanabe, T.; Ito, T.; Kanai, T.; Maeno, K.; Ito, K.; Tateishi, A. Lipid-rich carcinoma of the breast that is strongly positive for estrogen receptor: A case report and literature review. OncoTarget Ther. 2016, 9, 1641–1646. [Google Scholar]
- Antalis, C.J.; Arnold, T.; Rasool, T.; Lee, B.; Buhman, K.K.; Siddiqui, R.A. High ACAT1 expression in estrogen receptor negative basal-like breast cancer cells is associated with LDL-induced proliferation. Breast Cancer Res. Treat. 2010, 122, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Antalis, C.J.; Uchida, A.; Buhman, K.K.; Siddiqui, R.A. Migration of MDA-MB-231 breast cancer cells depends on the availability of exogenous lipids and cholesterol esterification. Clin. Exp. Metastasis 2011, 28, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, X.; Lee, J.-S.; Wang, X.; Yang, Z.-Q.; Zhang, K. Endoplasmic reticulum factor ERLIN2 regulates cytosolic lipid content in cancer cells. Biochem. J. 2012, 446, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Pucer, A.; Brglez, V.; Payre, C.; Pungercar, J.; Lambeau, G.; Petan, T. Group X secreted phospholipase A2 induces lipid droplet formation and prolongs breast cancer cell survival. Mol. Cancer 2013, 12, 111. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Vela, A.; Aguilar-Gallardo, C.; Martinez-Arroyo, A.M.; Soriano-Navarro, M.; Ruiz, V.; Simon, C. Specific unsaturated fatty acids enforce the transdifferentiation of human cancer cells toward adipocyte-like cells. Stem Cell Rev. Rep. 2011, 7, 898–909. [Google Scholar] [CrossRef] [PubMed]
- Burgermeister, E.; Tencer, L.; Liscovitch, M. Peroxisome proliferator-activated receptor-γ upregulates caveolin-1 and caveolin-2 expression in human carcinoma cells. Oncogene 2003, 22, 3888–3900. [Google Scholar] [CrossRef] [PubMed]
- Selvendrian, K.; Ahmed, S.; Dayton, A.; Ravi, Y.; Kuppusamy, M.L.; Bratasz, A.; Rivera, B.K.; Kálai, T.; Hideg, K.; Kuppusamy, P. HO-3867, a synthetic compound, inhibits the migration and invasion of ovarian carcinoma cells through downregulation of fatty acid synthase and focal adhesion kinase. Mol. Cancer Res. 2010, 8, 1188–1197. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Wu, J.; Barbour, S.; Fang, X. Lysophosphatidic acid activates lipogenic pathways and de novo lipid synthesis in ovarian cancer cells. J. Biol. Chem. 2012, 287, 24990–25000. [Google Scholar] [CrossRef] [PubMed]
- Pyragius, C.E.; Fuller, M.; Ricciardelli, C.; Oehler, M.K. Aberrant lipid metabolism: An emerging diagnostic and therapeutic target in ovarian cancer. Int. J. Mol. Sci. 2013, 14, 7742–7756. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.K.; Xiong, X.; Mustafi, S.B.; Saha, S.; Dhanasekaran, D.; Mandal, N.A.; McMeekin, S.; Bhattacharya, R.; Mukherjee, P. Role of cystathionine beta synthase in lipid metabolism in ovarian cancer. Oncotarget 2015, 6, 37367–37384. [Google Scholar] [PubMed]
- Roy, D.; Mondal, S.; Wang, C.; He, X.; Khurana, A.; Giri, S.; Hoffmann, R.; Jung, D.-B.; Kim, S.H.; Chini, E.N.; et al. Loss of HSulf-1 promotes altered lipid metabolism in ovarian cancer. Cancer Metab. 2014, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Tsuda, H.; Ito, K.; Takano, M.; Terai, Y.; Jobo, T.; Kigawa, J.; Sugiyama, T.; Yaegashi, N.; Aoki, D. Differential expression of hypoxia-inducible protein 2 among different histological types of epitherial ovarian cancer and in clear cell adenocarcinomas. Int. J. Gynecol. Cancer 2010, 20, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Accioly, M.T.; Pacheco, P.; Maya-Monteiro, C.M.; Carrossini, N.; Robbs, B.K.; Oliveira, S.S.; Kaufmann, C.; Morgado-Diaz, J.A.; Bozza, P.T.; Viola, J.P.B. Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res. 2008, 68, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Fitchev, P.S.; Cornwell, M.L.; Greenberg, J.; Cabe, M.; Weber, C.R.; Roy, H.K.; Crawford, S.E.; Savkovic, S.D. FOXO3 growth inhibition of colonic cells is dependent on intraepitherial lipid droplet density. J. Biol. Chem. 2013, 288, 16274–16281. [Google Scholar] [CrossRef] [PubMed]
- Penrose, H.; Heller, S.; Cable, C.; Makboul, R.; Chadalawada, G.; Chen, Y.; Crawford, S.E.; Savkovic, S.D. Epidermal growth factor receptor mediated proliferation depends on increased lipid droplet density regulated via a negative regulatory loop with FOXO3/Sirtuin6. Biochem. Biophys. Res. Commun. 2016, 469, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Yosef, H.K.; Mavanari, M.; Maghnouj, A.; Hahn, S.; El-Mashtoly, S.F.; Gerwert, K. In vitro prediction of the efficacy of molecularly targeted cancer therapy by Raman spectral imaging. Anal. Bioanal. Chem. 2015, 407, 8321–8331. [Google Scholar] [CrossRef] [PubMed]
- Tirinato, L.; Liberale, C.; Franco, S.D.; Candeloro, P.; Benfante, A.; la Rocca, R.; Potze, L.; Marotta, R.; Ruffilli, R.; Rajamanickam, V.P.; et al. Lipid droplets: A new player in colorectal cancer stem cells unveiled by spectroscopic imaging. Stem Cells 2015, 33, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappa, G.; Mercapide, J.; Anzanello, F.; Le, T.T.; Johlfs, M.G.; Fiscus, R.R.; Wilsch-Bräuninger, M.; Corbeil, D.; Lorico, A. Wnt interaction and extracellular release of promin-1/CD133 in human malignant melanoma cells. Exp. Cell Res. 2013, 319, 810–819. [Google Scholar] [CrossRef] [PubMed]
- Rappa, G.; Mercapide, J.; Anzanello, F.; Pope, R.M.; Lorico, A. Biochemical and biological characterization of exosomes containing promin-1/CD133. Mol. Cancer 2013, 12. [Google Scholar] [CrossRef] [PubMed]
- Bozza, P.T.; Viola, J.P.B. Lipid droplets in inflammation and cancer. Prostaglandins Leukot. Essent. Fatty Acids 2010, 82, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Zaytseva, Y.Y.; Harris, J.W.; Mitov, M.I.; Kim, J.T.; Butterfield, D.A.; Lee, E.Y.; Weiss, H.L.; Gao, T.; Evers, B.M. Increased expression of fatty acid synthase provides a survival advantage to colorectal cancer cells via upregulation of cellular respiration. Oncotarget 2015, 6, 18891–18904. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V.; Le, A.; Gao, P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. 2009, 15, 6479–6483. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Vincent, E.E.; Griss, T.; Samborska, B.; Izreig, S.; Svenson, R.U.; Mamer, O.A.; Avizonis, D.; Shackelford, D.B.; Shaw, R.J. Loss of tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1α. Proc. Natl. Acad. Sci. USA 2014, 111, 2554–2559. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Ward, P.S.; Shay, J.E.S.; Cross, J.R.; Gruber, J.J.; Sachdeva, U.M.; Platt, J.M.; DeMatteo, R.G.; Simon, M.C.; Thompson, C.B. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of a-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 2011, 108, 19611–19616. [Google Scholar] [CrossRef] [PubMed]
- Mettallo, C.M.; Gameiro, P.A.; Bell, E.L.; Mattaini, K.R.; Yang, J.; Hiller, K.; Jewell, C.M.; Johnson, Z.R.; Irvine, D.J.; Guarente, L.; et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2012, 48, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.C.; Denko, N.C. Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. 2014, 19, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Gouv, A.M.; Toal, G.G.; Felsher, D.W. Metabolic vulnerabilities of MYC-induced cancer. Oncotarget 2016, 7, 29879–29880. [Google Scholar]
- Furuta, E.; Pai, S.K.; Zhang, R.; Bandyopadhyay, S.; Watabe, M.; Mo, Y.-Y.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; et al. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 2008, 68, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, H.; Zhang, J.; Lv, J.; Huang, Y. Positive feedback loop and synergistic effects between hypoxia-inducible factor-2α and stearoyl‒CoA desaturase-1 promote tumorigenesis in clear cell renal cell carcinoma. Cancer Sci. 2013, 104, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Valli, A.; Rodriguez, M.; Moutsianas, L.; Fischer, R.; Fedele, V.; Huang, H.-L.; Stiphout, R.V.; Jones, D.; Mccarthy, M.; Vinaxia, M.; et al. Hypoxia induces a lipogenic cancer cell phenotype via HIF1α-dependent and -independent pathways. Oncotarget 2014, 6, 1920–1941. [Google Scholar] [CrossRef] [PubMed]
- Montel, V.; Gaultier, A.; Lester, R.D.; Campana, W.M.; Gonias, S.L. The low-density lipoprotein receptor-related protein regulates cancer cell survival and metastasis development. Cancer Res. 2007, 67, 9817–9824. [Google Scholar] [CrossRef] [PubMed]
- Kamphorst, J.J.; Cross, J.R.; Fan, J.; de Stanchina, E.; Mathew, R.; White, E.P.; Thompson, C.B.; Rabinowitz, J.D. Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. USA 2013, 110, 8882–8887. [Google Scholar] [CrossRef] [PubMed]
- Schlaepfer, I.R.; Nambia, D.K.; Ramteke, A.; Kumar, R.; Dhar, D.; Agarwal, C.; Bergman, B.; Graner, M.; Maroni, P.; Singh, R.P. Hypoxia induces triglycerides accumulation in prostate cancer cells and extracellular vesicles supporting growth and invasiveness following reoxygenation. Oncotarget 2015, 6, 22836–22856. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Durden, D.L.; van Meir, E.G.; Brat, D.J. Pseudopalisading necrosis in glioblastoma: A familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J. Neuropathol. Exp. Neurol. 2006, 65, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Galeffi, F.; Turner, D.A. Exploiting metabolic differences in glioma therapy. Curr. Drug Discov. Technol. 2012, 9, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Zoula, S.; Rijken, P.F.J.W.; Peters, J.P.W.; Farion, R.; van der Sanden, B.P.J.; van der Kogel, A.J.; Décorps, M.; Rémy, C. Pimonidazole binding in C6 rat brain glioma: Relation with lipid droplet detection. Br. J. Cancer 2003, 88, 1439–1444. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.-L.; et al. Fatty acid uptake and lipid strage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.A.; Brault, C.; Peck, B.; Bensaad, K.; Griffiths, B.; Mitter, R.; Chakravarty, P.; East, P.; Dankworth, B.; Alibhai, D.; et al. SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 2015, 34, 5128–5140. [Google Scholar] [CrossRef] [PubMed]
- Sano, S.; Izumi, Y.; Yamaguchi, T.; Yamazaki, T.; Tanaka, M.; Shiota, M.; Osada-Oka, M.; Nakamura, Y.; Wei, M.; Wanibuchi, H.; et al. Lipid synthesis is promoted by hypoxic adipocyte-derived exosomes in 3T3-L1 cells. Biochem. Biophys. Res. Commun. 2014, 445, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Sun, J.; Dai, Y.; Cao, P.; Zhang, L.; Peng, S.; Zhou, Y.; Li, G.; Tang, J.; Xiang, J. HIF-1A and C/EBPs transcriptionally regulate adipogenic differentiation of bone marrow-derived MSCs in hypoxia. Stem Cell Res. Ther. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Elzschig, H.K.; Carmeliet, P. Hypoxia and inflammation. N. Engl. J. Med. 2011, 364, 656–665. [Google Scholar]
- Crucet, M.; Wust, S.J.A.; Spielmann, P.; Luscher, T.F.; Wegner, R.H.; Matter, C.M. Hypoxia enhances lipid uptake in macrophages: Roles of the scavenger receptors Lox1, SRA, and CD36. Atherosclerosis 2013, 229, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Paton, C.M.; Ntambi, M. Biochemical and physiological function of stearoyl-CoA desaturase. Am. J. Physiol. Endocrinol. Metab. 2009, 297, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Sembongi, H.; Befani, C.; Liakos, P.; Siniossoglou, S.; Simos, G. Hypoxia causes triglyceride accumulation by HIF-1-mediated stimulation of lipin 1 expression. J. Cell Sci. 2012, 125, 3485–3493. [Google Scholar] [CrossRef] [PubMed]
- Kourti, M.; Ikonomou, G.; Giakoumakis, N.-N.; Rapsomaniki, M.A.; Landegren, U.; Siniossoglou, S.; Lygerou, Z.; Simos, G.; Mylonis, I. CK1δγ restrains lipin-1 induction, lipid droplet formation and cell proliferation under hypoxia by reducing HIF-1α/ARNT complex formation. Cell Signal. 2015, 27, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Shah, T.; Krishnamachary, B.; Wildes, F.; Mironchik, Y.; Kakkad, S.M.; Jacob, D.; Artemov, D.; Bhujwalla, Z.M. HIF isoforms have divergent effects on invasion, metastasis, metabolism and formation of lipid droplets. Oncotarget 2015, 6, 28104–28119. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, J.; Suter, M.; Windak, R.; Krebs, T.; Felley, A.; Montessuit, C.; Tokarska-Schlattner, M.; Aasum, E.; Bogdanova, A.; Perriard, E.; et al. Activation of a HIF1α-PPARγ axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009, 9, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Laurenti, G.; Benedetti, E.; D’Angelo, B.; Cristiano, L.; Cinque, B.; Raysi, S.; Alecci, M.; Ceru, M.P.; Cifone, M.G.; Galzio, R.; et al. Hypoxia induces peroxisome proliferator-activated receptor α (PPARα) and lipid metabolism peroxisomal enzymes in human glioblastoma cells. J. Cell. Biochem. 2011, 112, 3891–3901. [Google Scholar] [CrossRef] [PubMed]
- Galzio, R.; Cristiano, L.; Fidoamore, A.; Cifone, M.G.; Benedetti, E.; Cinque, B.; Menghini, P.; Dehcordi, S.R.; Ippoliti, R.; Giordano, A.; et al. Hypoxia modulation of peroxisome proliferator-activated receptors (PPARs) in human glioblastoma stem cells. Implications for therapy. J. Cell. Biochem. 2012, 113, 3342–3352. [Google Scholar] [CrossRef] [PubMed]
- Mattijssen, F.; Georgiadi, A.; Andasarie, T.; Szalowska, E.; Zota, A.; Krones-Herzig, A.; Heier, C.; Ratman, D.; de Bosscher, K.; Qi, L.; et al. Hypoxia-inducible lipid droplet-associated (HILPDA) is a novel peroxisome proliferator-activated receptor (PPAR) target involved in hepatic triglyceride secretion. J. Biol. Chem. 2014, 289, 19279–19293. [Google Scholar] [CrossRef] [PubMed]
- Donkor, J.; Zhang, P.; Wong, S.; O’Loughlin, L.; Dewald, J.; Kok, B.P.C.; Brindley, D.N.; Reue, K. A conserved serine residue is required for the phosphatidate phosphatase activity but not the transcriptional coactivator functions of lipin-1 and lipin-2. J. Biol. Chem. 2009, 284, 29968–29978. [Google Scholar] [CrossRef] [PubMed]
- Schug, Z.T.; Peck, B.; Jones, D.T.; Zhang, Q.; Grosskurth, S.; Alam, I.S.; Goodwin, L.M.; Smethurst, E.; Mason, S.; Blyth, K.; et al. Acetyl-CoA synthase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 2015, 27, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewhirst, M.W.; Cao, Y.; Moeller, B. Cyling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat. Rev. Cancer 2008, 8, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.-R.; Chae, H.-J. Endoplasmic reticulum stress and associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [PubMed]
- Boelens, J.; Lust, S.; Offner, F.; Bracke, M.E.; Vanhoecke, B.W. The endoplasmic reticulum: A target for new anticancer drugs. In Vivo 2007, 21, 215–226. [Google Scholar] [PubMed]
- Ohsaki, Y.; Cheng, J.; Fujita, A.; Tokumoto, T.; Fujimoto, T. Cytoplasmic lipid droplets are sites of convergence of proteasomal and autophagic degradation of apolipoprotein. Mol. Biol. Cell 2006, 17, 2674–2683. [Google Scholar] [CrossRef] [PubMed]
- Keembiyehetty, C.N.; Krzeslak, A.; love, D.C.; Hanover, J.A. A lipid-droplet-targeted O-GlcNAcase isoform is a key regulator of the proteasome. J. Cell Sci. 2011, 24, 2851–2860. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Zhou, S.; Kim, J.Y.; Tillison, K.; Majors, D.; Rearick, D.; Lee, J.H.; Fernandez-Boyanapalli, R.F.; Barricklow, K.; Houston, M.S. Functional analysis of FSP27 protein regions for lipid droplet localization, caspase-dependent apoptosis, and dimerization with CIDEA. Am. J. Physiol. Endocrinol. Metab. 2009, 297, 1395–1413. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wang, H.; Zhao, J.; Yuan, Y.; Wang, C.; Li, J.; Zhang, L.; Zhang, L.; Li, Q.; Ye, J. Expression of CIDE proteins in clear cell renal carcinoma and their prognostic significance. Mol. Cell. Biochem. 2013, 378, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Gu, Y.; Zhang, F.; Li, J.; Liu, F.; Zhang, Z.; Ye, J.; Li, Q. Cidec promotes the differentiation of human adipocytes by degradation of AMPKα through ubiquitin-proteasome pathway. Biochim. Biophys. Acta 2015, 1850, 2552–2562. [Google Scholar] [CrossRef] [PubMed]
- Young, R.M.; Ackerman, D.; Quinn, Z.L.; Mancuso, A.; Gruber, M.; Liu, L.; Giannoukos, D.N.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Keith, B.; et al. Dysregulated mTORC1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. Genes Dev. 2013, 27, 1115–1131. [Google Scholar] [CrossRef] [PubMed]
- Ackerman, D.; Simon, M.C. Hypoxia, lipids, and cancer: Surviving the harsh tumor microenvironment. Trends Cell Biol. 2014, 24, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Piatkov, K.I.; Brower, C.S.; Varshavsky, A. The N-end rule pathway counteracts cell death by destroying proapoptotic protein fragments. Proc. Natl. Acad. Sci. USA 2012, 109, 1839–1847. [Google Scholar] [CrossRef] [PubMed]
- Eldeeb, M.A.; Fahlman, R.P. The anti-apoptotic form of tyrosine kinase Lyn that is generated by proteolysis is degraded by the N-end rule pathway. Oncotarget 2014, 5, 2714–2722. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, C.; Rowicka, M.; Kudlicki, A.; Nowis, D.; McConnell, E.; Kujawa, M.; DeMartino, G.N. Valosin-containing protein (p97) is a regulator of endoplasmic reticulum stress and of the degradation of N-end rule and ubiquitin-fusion degradation pathway substrates in mammalian cells. Mol. Biol. Cell. 2006, 17, 4606–4618. [Google Scholar] [CrossRef] [PubMed]
- Eldeeb, M.; Fahlman, R. The N-end rule: The beginning determines the end. Protein Pept. Lett. 2016, 23, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Koizume, S.; Ito, S.; Nakamura, Y.; Yoshihara, M.; Furuya, M.; Yamada, R.; Miyagi, E.; Hirahara, F.; Takano, Y.; Miyagi, Y. Lipid starvation and hypoxia synergistically activates ICAM1 and multiple genes in an Sp1-dependent manner to promote the growth of ovarian cancer. Mol. Cancer 2015, 14. [Google Scholar] [CrossRef] [PubMed]
- Koritzinsky, M.; Wouters, B.G. The roles of reactive oxygen species and autophagy in mediating the tolerance of tumor cells to cycling hypoxia. Semin. Radiat. Oncol. 2013, 23, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Puskas, L.G.; Feher, L.Z.; Vizler, C.; Ayaydin, F.; Raso, E.; Molnar, E.; Magyary, I.; Kanizsai, I.; Gyuris, M.; Madacsi, R.; et al. Polyunsaturated fatty acids synergize with lipid droplet binding thalidomide analogus to induce oxidative stress in cancer cells. Lipids Health Dis. 2010, 9. [Google Scholar] [CrossRef] [PubMed]
- Baily, A.P.; Koster, G.; Guillermier, C.; Hirst, E.M.A.; Macrae, J.I.; Lechene, C.P.; Postle, A.D.; Gould, A.P. Antioxidant role for lipid droplets in a stem cell niche of Drosophila. Cell 2015, 163, 340–353. [Google Scholar] [CrossRef] [PubMed]
- Rysman, E.; Brusselmans, K.; Scheys, K.; Timmermans, L.; Derua, R.; Munck, S.; van Veldhoven, P.P.; Waltregny, D.; Daniels, V.W.; Machiels, J.; et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010, 70, 8117–8126. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, H.; Li, Z.; Zhao, Z.; Yip-Schneider, M.; Fan, Q.; Schmidt, C.M.; Chiorean, E.G.; Xie, J.; Cheng, L.; et al. Role of fatty acid synthase in gemcitabine and radiation resistance of pancreatic cancers. Int. J. Biochem. Mol. Biol. 2011, 2, 89–98. [Google Scholar] [PubMed]
- Greening, D.W.; Lee, S.T.; Ji, H.; Simpson, R.J.; Rigopoulos, A.; Murone, C.; Fang, C.; Gong, S.; O’Keefe, G.; Scott, A.M. Molecular profiling of cetuximab and bevacizumab treatment of colorectal tumours reveals perturbations in metabolic and hypoxic response pathways. Oncotarget 2015, 6, 38166–38180. [Google Scholar] [PubMed]
- Zietkowski, D.; Payne, G.S.; Nagy, E.; Mobberley, M.A.; Ryder, T.A.; deSouza, N.M. Comparison of NMR lipid profiles in mitotic arrest and apoptosis as indicators of paclitaxel resistance in cervical cell lines. Magn. Reson. Med. 2012, 68, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Rak, S.; DeZan, T.; Stefuji, J.; Kosovic, M.; Gamulin, O.; Osmak, M. FTIR spectroscopy reveales lipid droplets in drug resistant laryngeal carcinoma cells through detection of increased ester vibrational bands intensity. Analyst 2014, 139, 3407–3415. [Google Scholar] [CrossRef] [PubMed]
- Montopoli, M.; Bellanda, M.; Lonardoni, F.; Ragazzi, E.; Dorigo, P.; Froldi, G.; Mammi, S.; Caparrotta, L. Metabolic reprogramming in ovarian cancer cells resistant to cisplatin. Curr. Cancer Drug Targets 2011, 11, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Rappa, G.; Fargeas, C.A.; Le, T.T.; Corbeil, D.; Lorico, A. Letter to the editor: An intriguing relationship between lipid droplets, cholesterol-binding protein CD133 and Wnt/β-catenin signaling pathway in carcinogenesis. Stem Cells 2015, 33, 1366–1370. [Google Scholar] [CrossRef] [PubMed]
- Verbrugge, S.E.; Al, M.; Assaraf, Y.G.; Kammerer, S.; Chandrupatla, D.M.S.H.; Honeywell, R.; Musters, R.P.J.; Giovannetti, E.; O’Toole, T.; Scheffer, G.L.; et al. Multifactorial resistance to aminopeptidase inhibitor prodrug CHR2863 in myeloid leukemia cells: Down-regulation of carboxylesterase 1 drug sequestration in lipid droplets and prosurvival activation ERK/Akt/mTOR. Oncotarget 2015, 7, 5240–5257. [Google Scholar]
- Gaida, N.M.; Mayer, C.; Dapunt, U.; Stegmaier, S.; Schirmacher, P.; Wabnitz, G.H.; Hansch, M. Expression of the bitter receptor T2R38 in pancreatic cancer: Localization in lipid droplets and activation by a bacteria-derived quorum-sencing molecule. Oncotarget 2016, 7, 12623–12632. [Google Scholar] [PubMed]
- Morjani, H.; Aouali, N.; Behoussine, R.; Veldan, R.J.; Levade, T.; Manfait, M. Elevation of glucosylceramide in multidrug-resistant cancer cells and accumulation in cytoplasmic droplets. Int. J. Cancer 2001, 94, 157–165. [Google Scholar] [CrossRef] [PubMed]
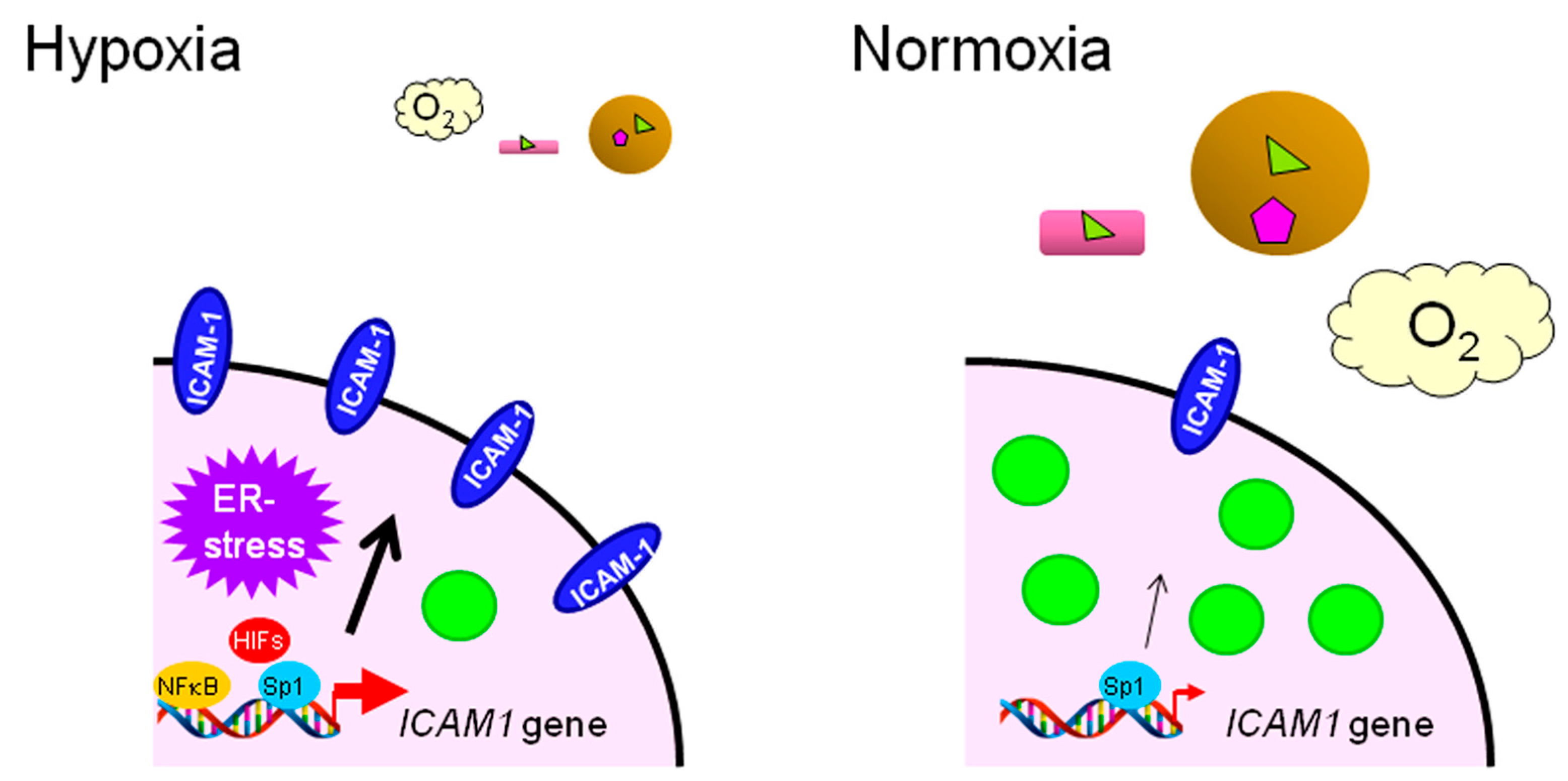
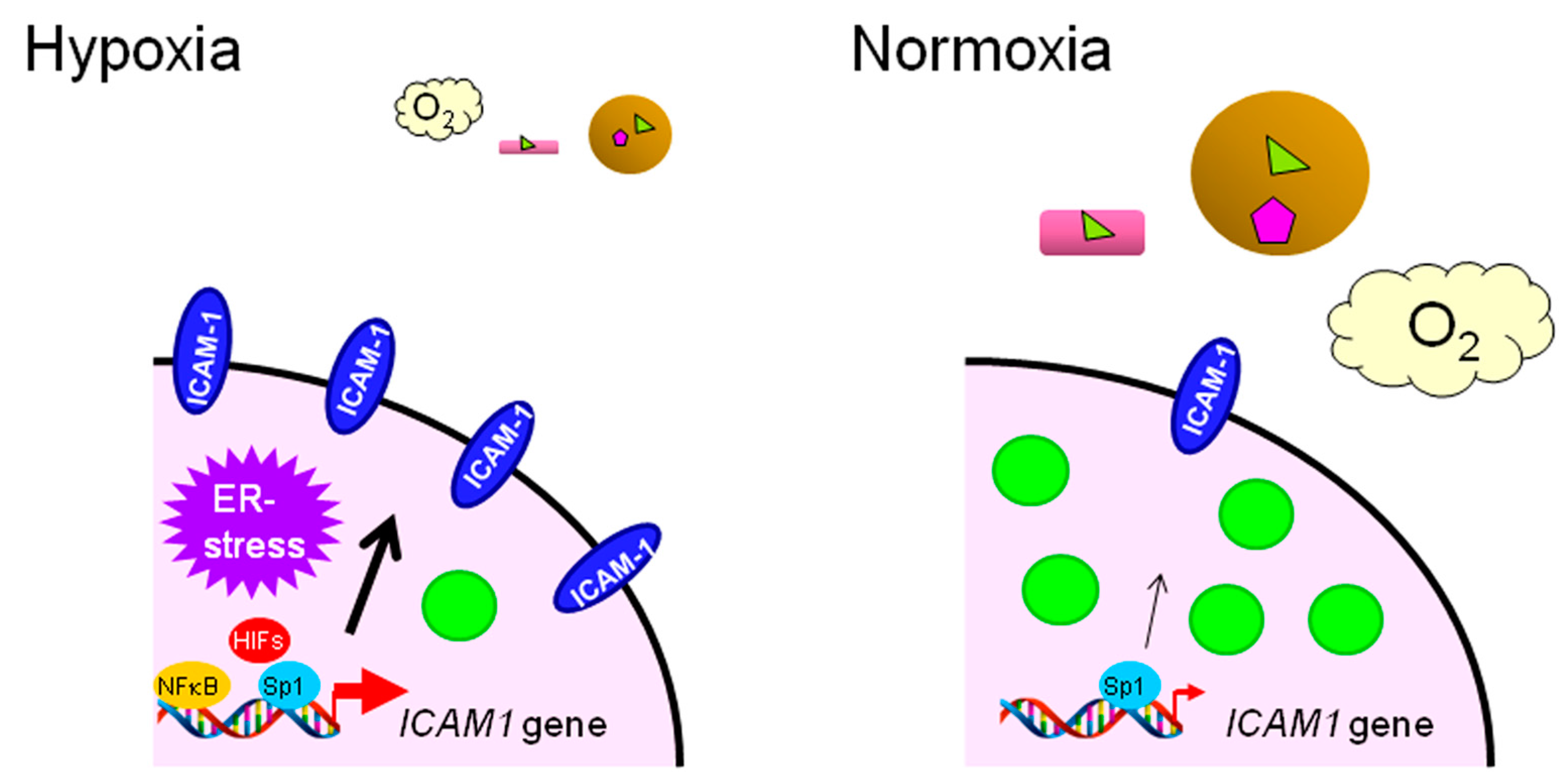
 : LCFAs,
: LCFAs,  : cholesterol,
: cholesterol,  : transporter,
: transporter,  : receptor.
: LCFAs, : cholesterol, : transporter, : receptor.
: receptor.
: LCFAs, : cholesterol, : transporter, : receptor.


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koizume, S.; Miyagi, Y. Lipid Droplets: A Key Cellular Organelle Associated with Cancer Cell Survival under Normoxia and Hypoxia. Int. J. Mol. Sci. 2016, 17, 1430. https://doi.org/10.3390/ijms17091430
Koizume S, Miyagi Y. Lipid Droplets: A Key Cellular Organelle Associated with Cancer Cell Survival under Normoxia and Hypoxia. International Journal of Molecular Sciences. 2016; 17(9):1430. https://doi.org/10.3390/ijms17091430
Chicago/Turabian StyleKoizume, Shiro, and Yohei Miyagi. 2016. "Lipid Droplets: A Key Cellular Organelle Associated with Cancer Cell Survival under Normoxia and Hypoxia" International Journal of Molecular Sciences 17, no. 9: 1430. https://doi.org/10.3390/ijms17091430
APA StyleKoizume, S., & Miyagi, Y. (2016). Lipid Droplets: A Key Cellular Organelle Associated with Cancer Cell Survival under Normoxia and Hypoxia. International Journal of Molecular Sciences, 17(9), 1430. https://doi.org/10.3390/ijms17091430