
De Novo Analysis of the Transcriptome of Meloidogyne enterolobii to Uncover Potential Target Genes for Biological Control

Abstract
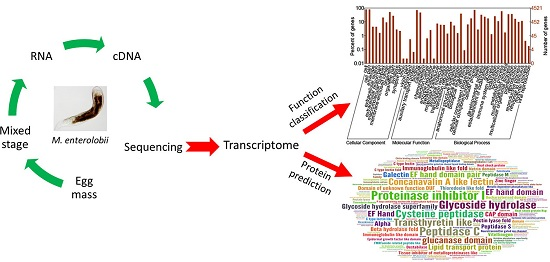
:
1. Introduction
2. Results
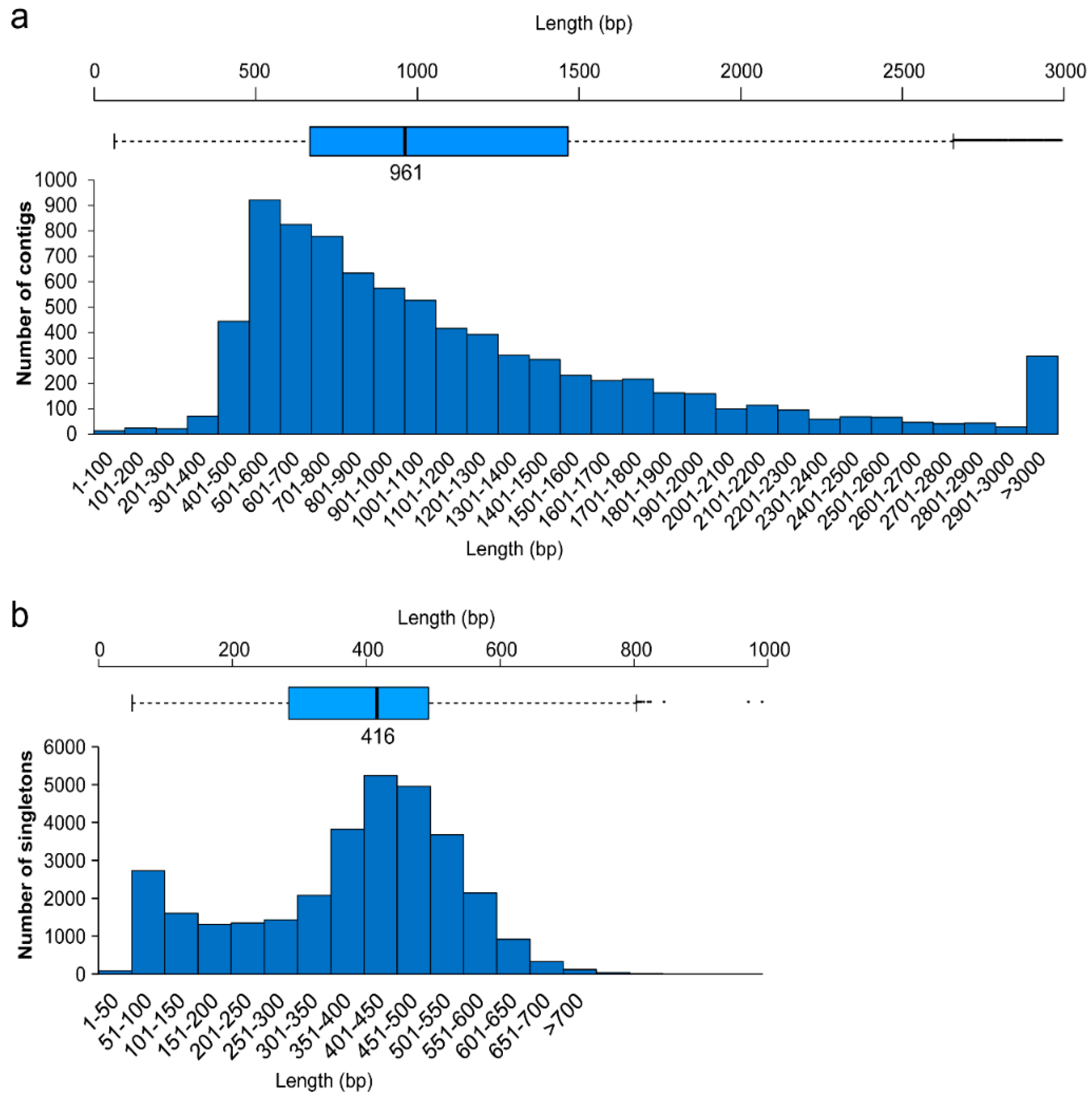
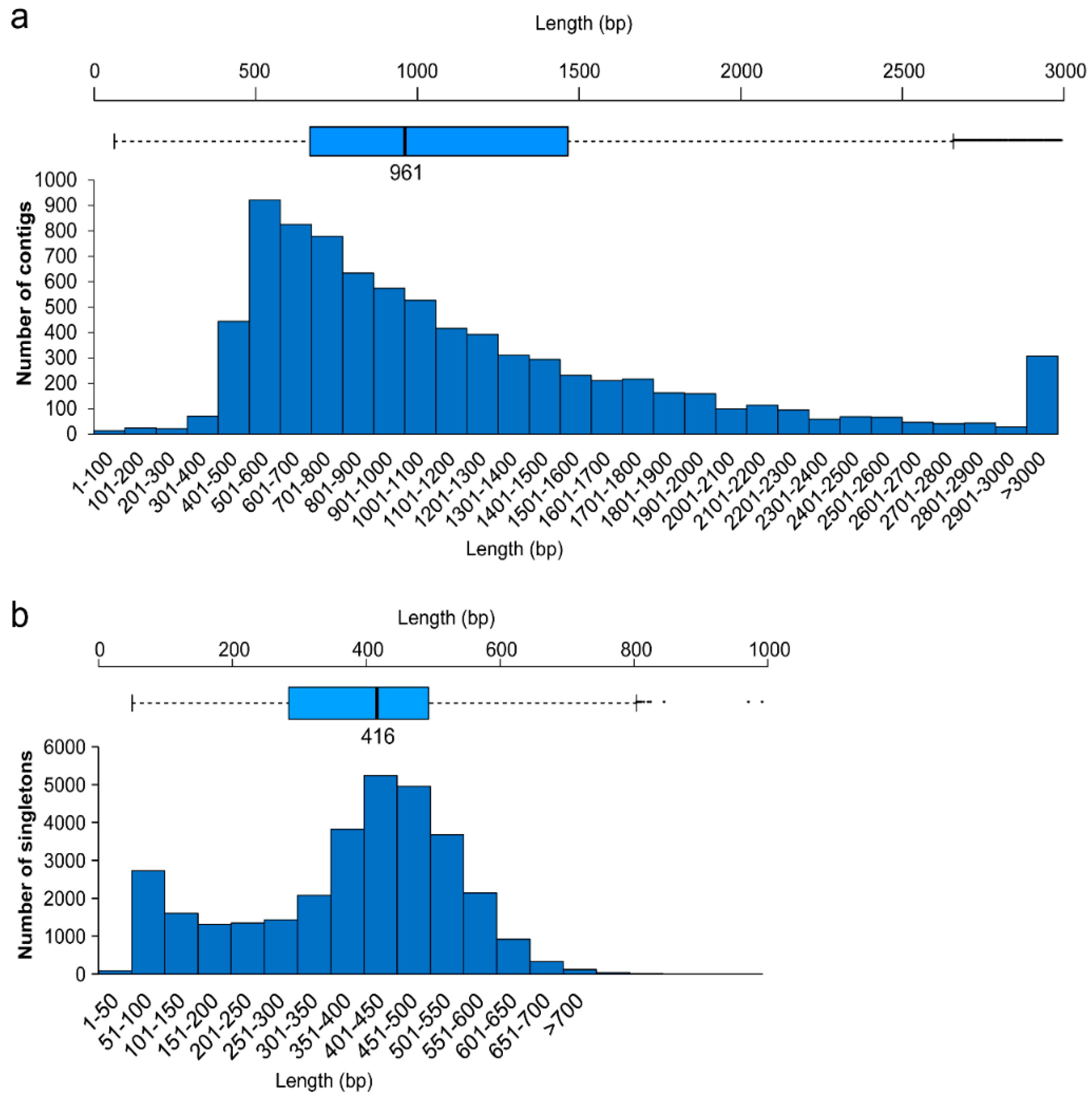
2.1. Assembly
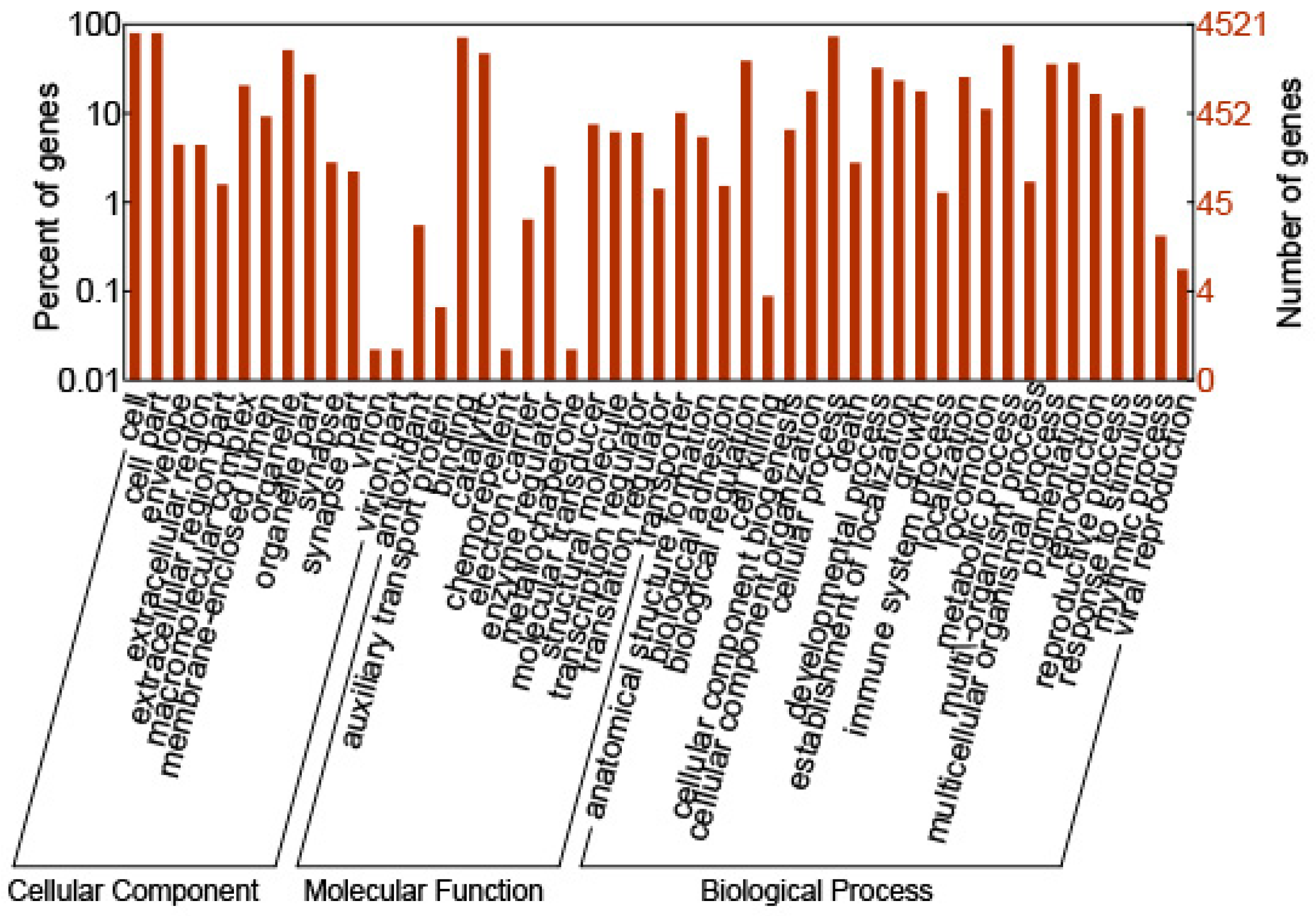
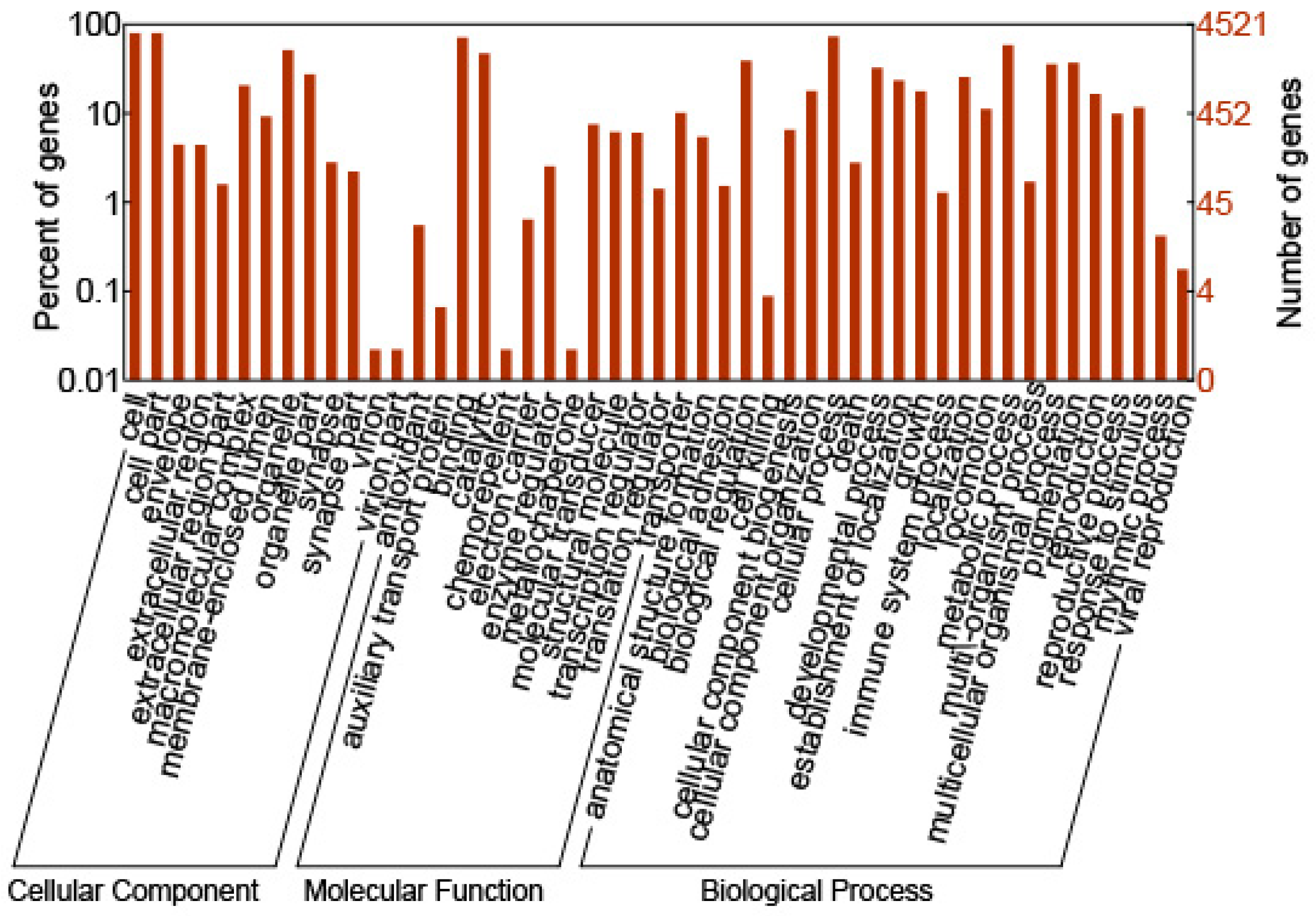
2.2. GO Annotation
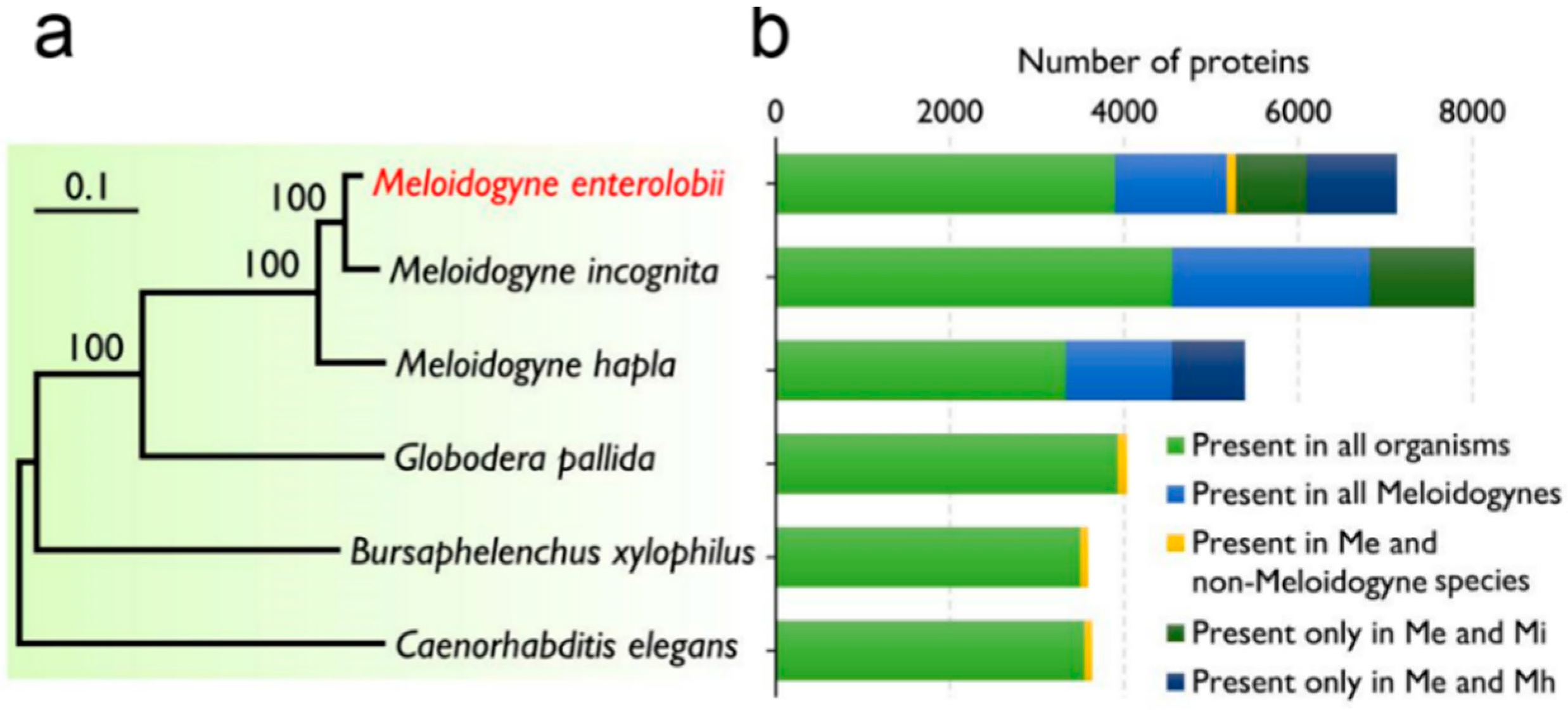
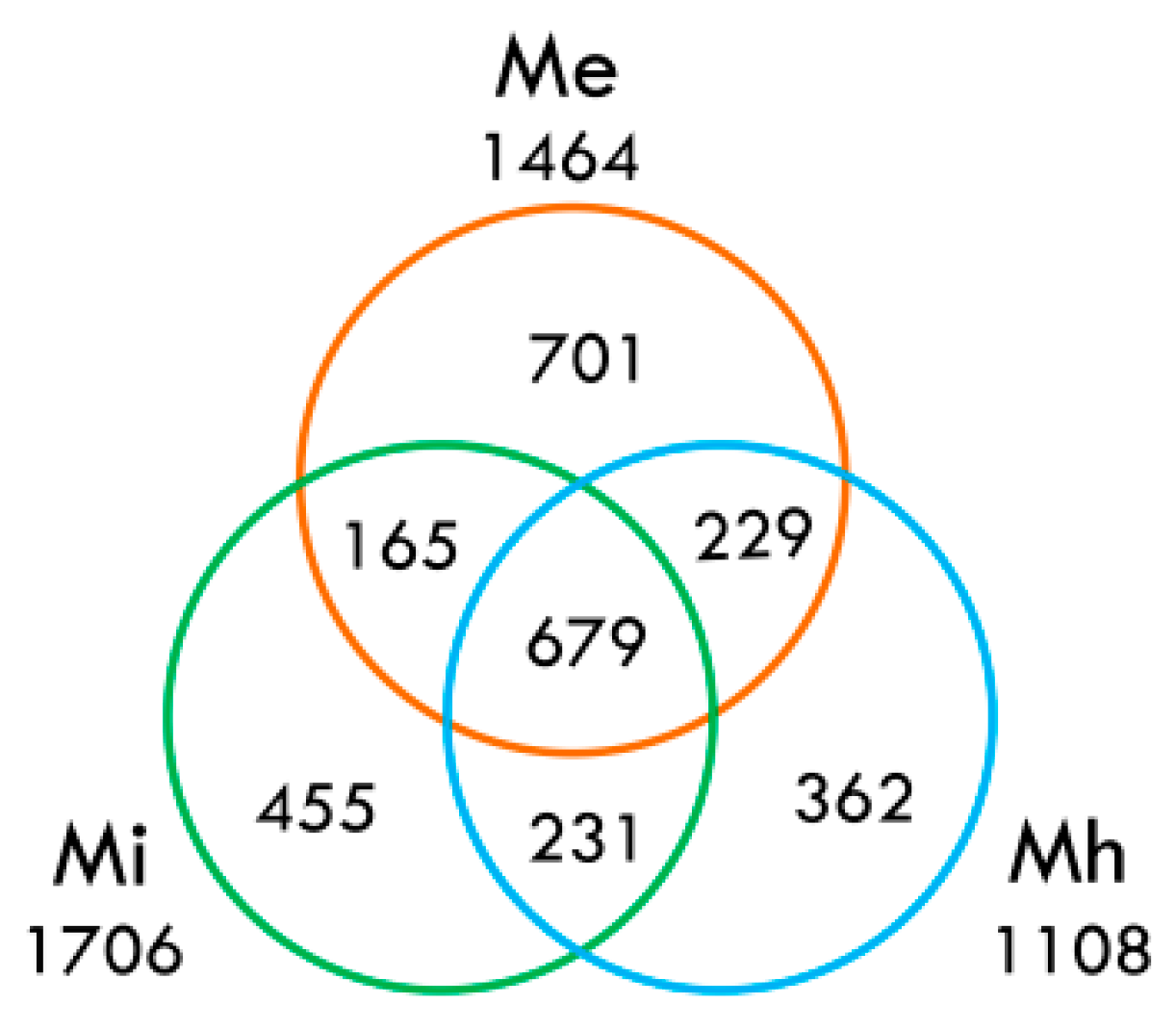
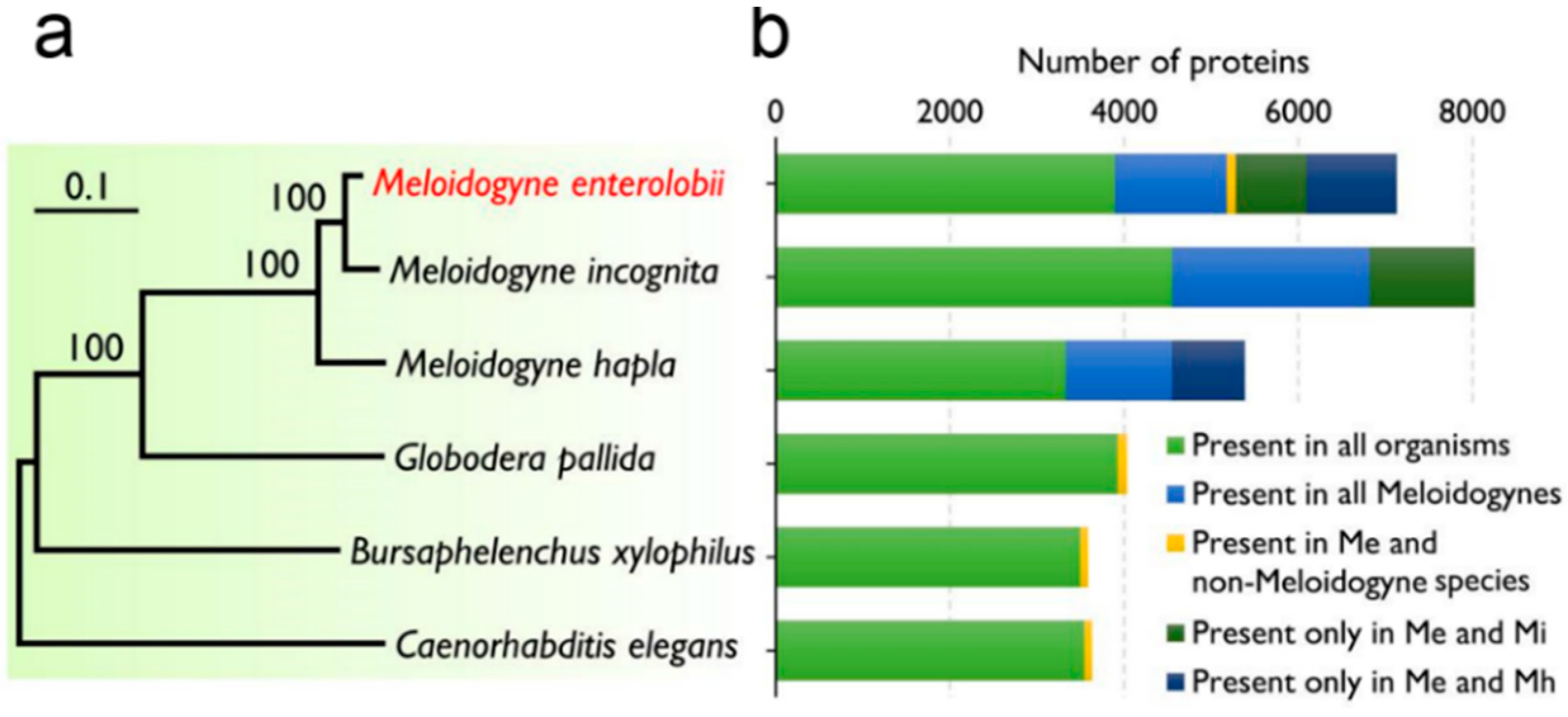
2.3. Comparative Analysis of the Transcriptome of M. enterolobii
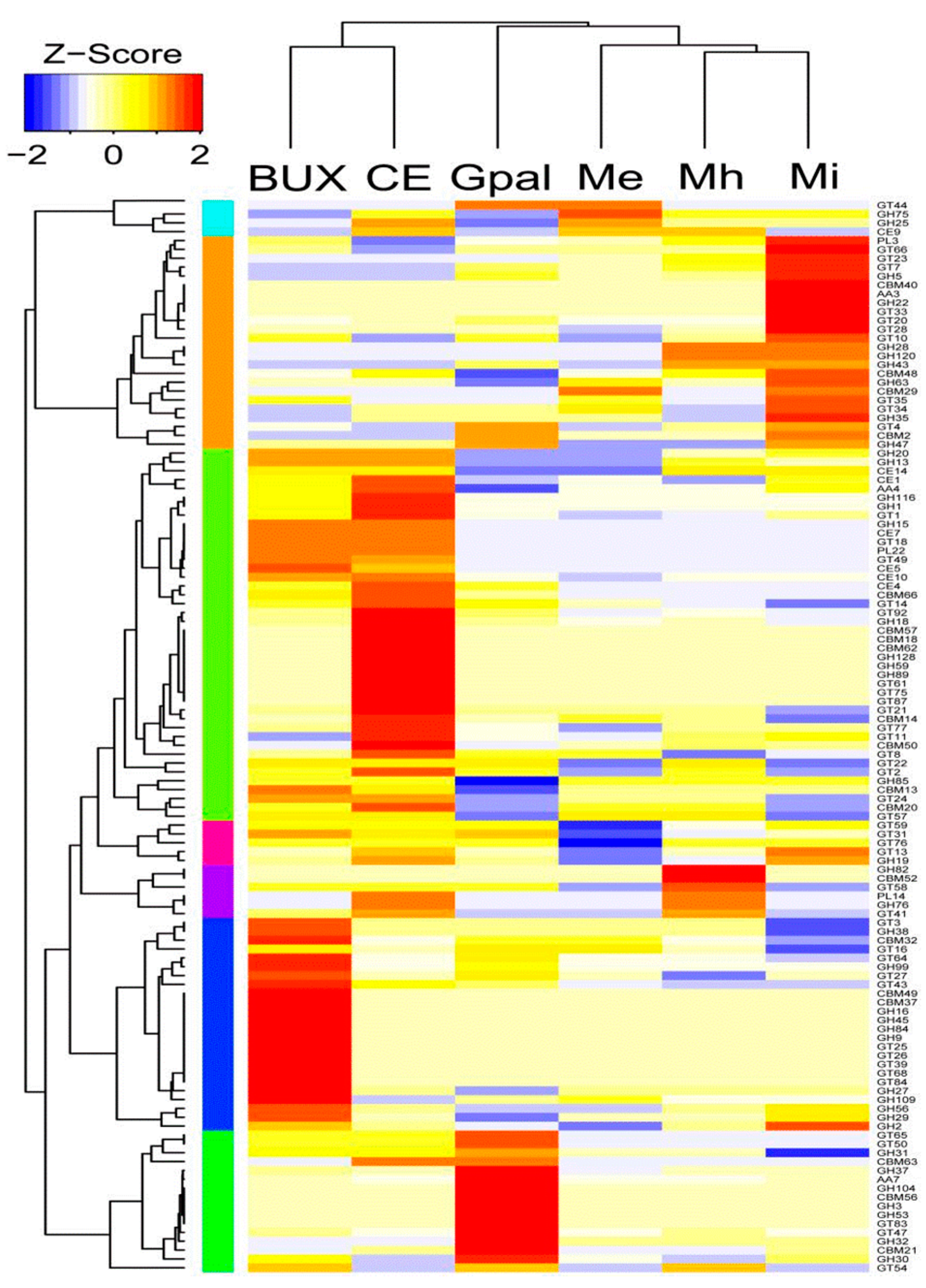
2.3.1. CAZymes
2.3.2. Kinases
2.3.3. Neuropeptides
2.3.4. RNAi Pathway
2.3.5. Immune Signaling
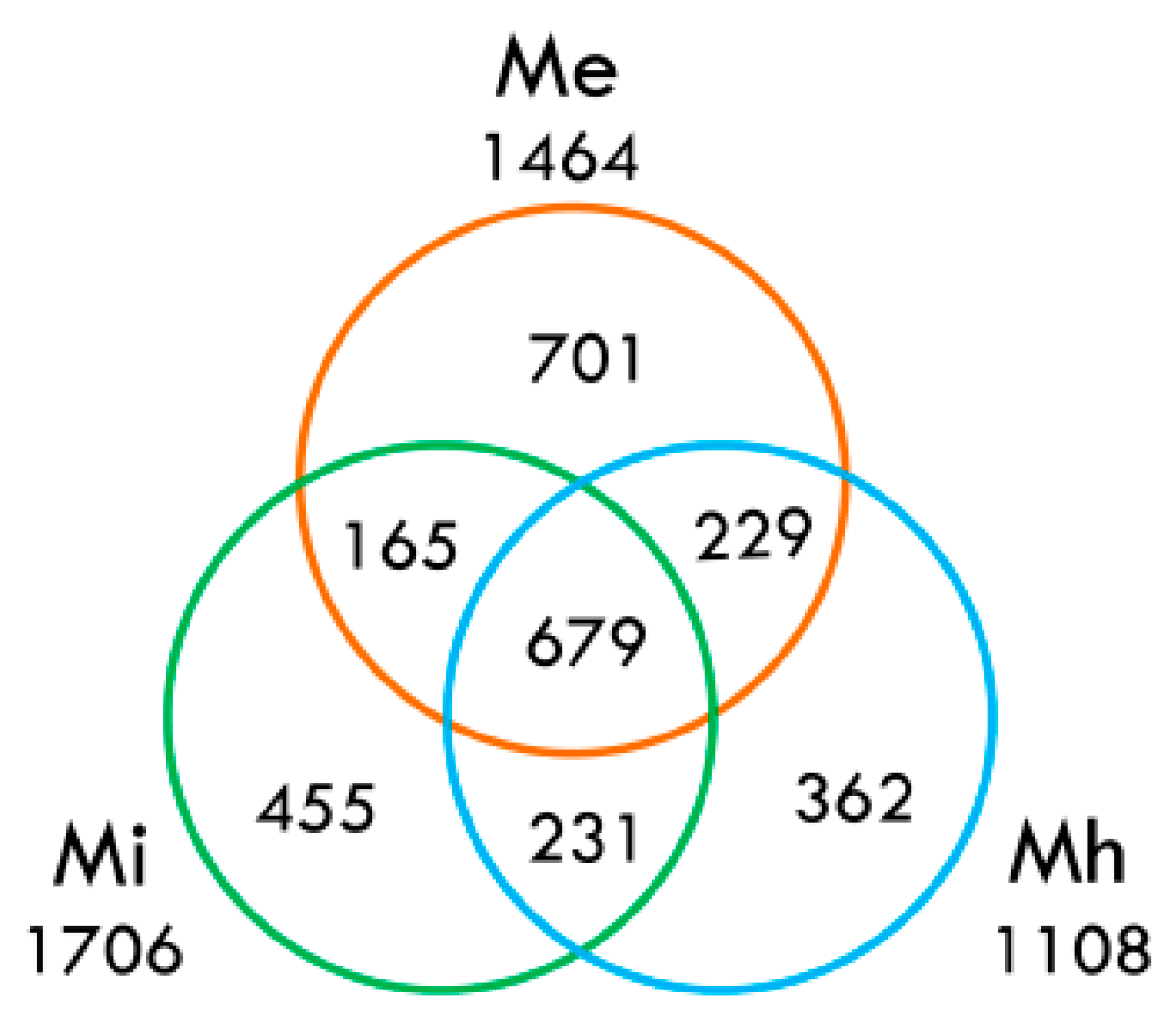
2.3.6. Secretory Proteins
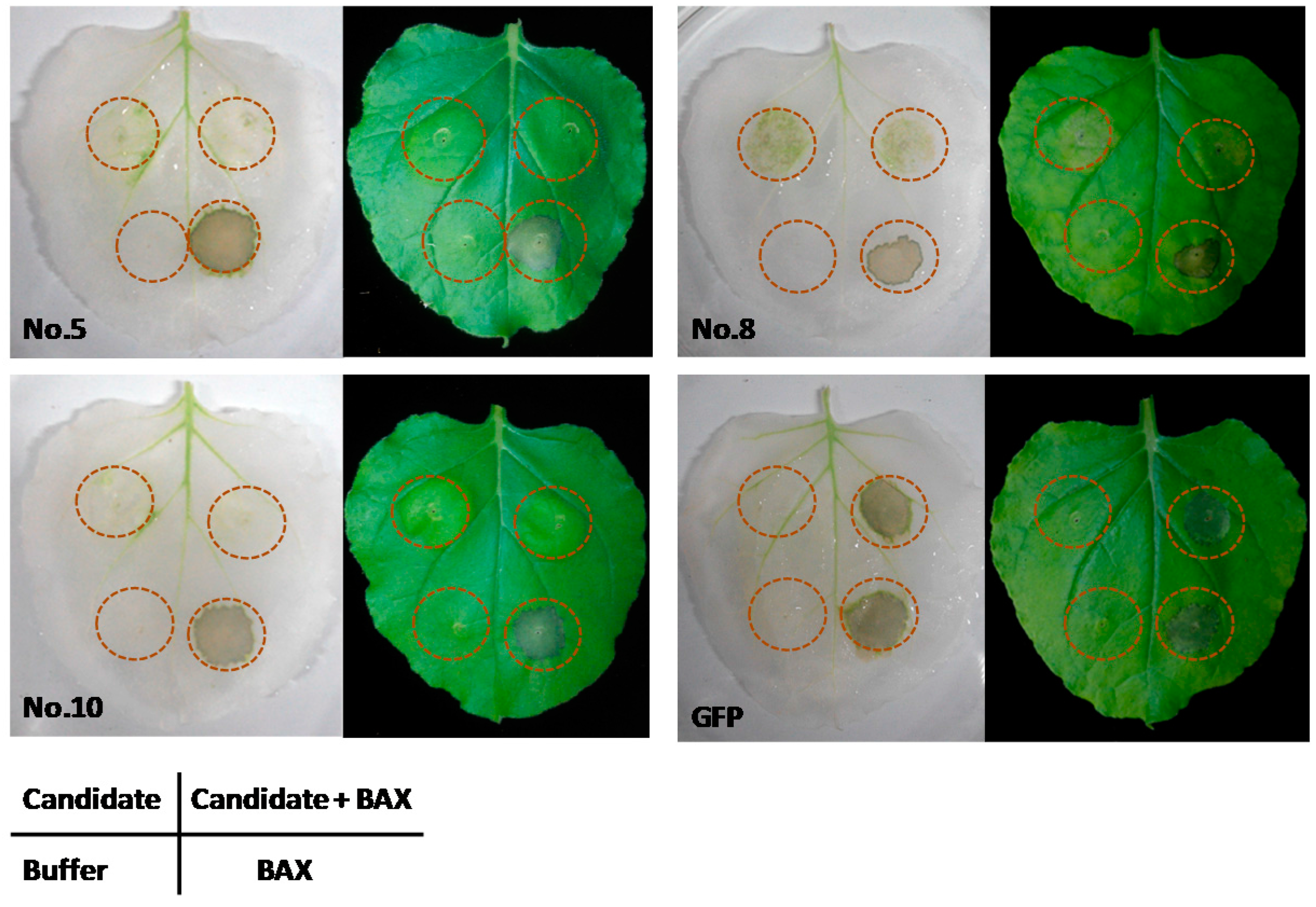
2.4. Expression of the Candidate Genes Suppressed BAX-Induced Cell Death in N. benthamiana
3. Discussion
4. Materials and Methods
4.1. Sample Treatment
4.2. RNA Extraction, cDNA Synthesis and Sequencing
4.3. Assembly and GO Annotation
4.4. Phylogenetic and Comparative Analysis of Orthologous Families
4.5. Comparative Analysis of the Transcriptome
4.6. Functional Verification of Three Putative Effectors
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Khan, S. Pathogenic Effects of Pratylenchus-Zeae on Sugarcane, Phytopathology, 1959; The American Phytopathological Society: St. Paul, MN, USA, 1959; p. 543. [Google Scholar]
- Ramírez-Suárez, A.; Rosas-Hernández, L.; Alcasio-Rangel, S.; Powers, T. First report of the root-knot nematode Meloidogyne enterolobii parasitizing watermelon from Veracruz, Mexico. Plant Dis. 2014. [Google Scholar] [CrossRef]
- Yang, B.; Eisenback, J. Meloidogyne enterolobii n. sp. (Meloidogynidae), a root-knot nematode parasitizing pacara earpod tree in China. J. Nematol. 1983, 15, 381–391. [Google Scholar] [PubMed]
- Brito, J.; Powers, T.O.; Mullin, P.; Inserra, R.; Dickson, D. Morphological and molecular characterization of Meloidogyne mayaguensis isolates from Florida. J. Nematol. 2004, 36, 232–240. [Google Scholar] [PubMed]
- Long, H.; Liu, H.; Xu, J. Development of a pcr diagnostic for the root-knot nematode Meloidogyne enterolobii. Acta Phytopathol. Sin. 2006, 36, 109–115. [Google Scholar]
- Civerolo, E. Alternatives to Methyl Bromide: Assessment of Research Needs and Priorities: Proceedings from the Usda Workshop on Alternatives to Methyl Bromide; US Department of Agriculture; Agriculture Research Service: Arlington, VA, USA, 1993.
- Chitwood, D.J. Phytochemical based strategies for nematode control 1. Annu. Rev. Phytopathol. 2002, 40, 221–249. [Google Scholar] [CrossRef] [PubMed]
- Williamson, V.M.; Gleason, C.A. Plant–nematode interactions. Curr. Opin. Plant Biol. 2003, 6, 327–333. [Google Scholar] [CrossRef]
- Mitchum, M.G.; Hussey, R.S.; Baum, T.J.; Wang, X.; Elling, A.A.; Wubben, M.; Davis, E.L. Nematode effector proteins: An emerging paradigm of parasitism. New Phytol. 2013, 199, 879–894. [Google Scholar] [CrossRef] [PubMed]
- Haegeman, A.; Joseph, S.; Gheysen, G. Analysis of the transcriptome of the root lesion nematode Pratylenchus coffeae generated by 454 sequencing technology. Mol. Biochem. Parasitol. 2011, 178, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Haegeman, A.; Bauters, L.; Kyndt, T.; Rahman, M.M.; Gheysen, G. Identification of candidate effector genes in the transcriptome of the rice root knot nematode Meloidogyne graminicola. Mol. Plant Pathol. 2013, 14, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Nicol, P.; Gill, R.; Fosu-Nyarko, J.; Jones, M.G. De novo analysis and functional classification of the transcriptome of the root lesion nematode, Pratylenchus thornei, after 454 GS FLX sequencing. Int. J. Parasitol. 2012, 42, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Gantasala, N.P.; Roychowdhury, T.; Thakur, P.K.; Banakar, P.; Shukla, R.N.; Jones, M.G.; Rao, U. De novo transcriptome sequencing and analysis of the cereal cyst nematode, Heterodera avenae. PLoS ONE 2014, 9, e96311. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, D.; Wang, Z.; Dong, A.; Liu, L.; Wang, B.; Chen, Q.; Liu, X. Transcriptomic analysis of the rice white tip nematode, Aphelenchoides besseyi (nematoda: Aphelenchoididae). PLoS ONE 2014, 9, e91591. [Google Scholar] [CrossRef] [PubMed]
- Palomares-Rius, J.E.; Hirooka, Y.; Tsai, I.J.; Masuya, H.; Hino, A.; Kanzaki, N.; Jones, J.T.; Kikuchi, T. Distribution and evolution of glycoside hydrolase family 45 cellulases in nematodes and fungi. BMC Evol. Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Morton, D.B. Invertebrates yield a plethora of atypical guanylyl cyclases. Mol. Neurobiol. 2004, 29, 97–116. [Google Scholar] [CrossRef]
- Li, C.; Nelson, L.S.; Kim, K.; Nathoo, A.; Hart, A.C. Neuropeptide gene families in the nematode Caenorhabditis elegans. Ann. N. Y. Acad. Sci. 1999, 897, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Nelson, L.S.; Rosoff, M.L.; Li, C. Disruption of a neuropeptide gene, flp-1, causes multiple behavioral defects in Caenorhabditis elegans. Science 1998, 281, 1686–1690. [Google Scholar] [CrossRef] [PubMed]
- Waggoner, L.E.; Hardaker, L.A.; Golik, S.; Schafer, W.R. Effect of a neuropeptide gene on behavioral states in Caenorhabditis elegans egg-laying. Genetics 2000, 154, 1181–1192. [Google Scholar] [PubMed]
- Fargette, M.; Phillips, M.; Blok, V.C.; Waugh, R.; Trudgill, D. An RFLP study of relationships between species, populations and resistance-breaking lines of tropical species of Meloidogyne. Fundam. Appl. Nematol. 1996, 19, 193–200. [Google Scholar]
- Brito, J.; Stanley, J.; Cetintas, R.; Powers, T.; Inserra, R.; McAvoy, G.; Mendes, M.; Crow, B.; Dickson, D. Identification and host preference of Meloidogyne mayaguensis and other root-knot nematodes from Florida, and their susceptibility to Pasteuria Penetrans. J. Nematol. 2004, 36, 308–309. [Google Scholar]
- Brito, J.A.; Stanley, J.; Mendes, M.; Cetintas, R.; Dickson, D. Host status of selected cultivated plants to Meloidogyne mayaguensis in Florida. Nematropica 2007, 37, 65–72. [Google Scholar]
- Alegado, R.A.; Campbell, M.C.; Chen, W.C.; Slutz, S.S.; Tan, M.W. Characterization of mediators of microbial virulence and innate immunity using the Caenorhabditis elegans host-pathogen model. Cell. Microbiol. 2003, 5, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Kurz, C.L.; Tan, M.W. Regulation of aging and innate immunity in C. elegans. Aging Cell 2004, 3, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Savage-Dunn, C.; Tokarz, R.; Wang, H.; Cohen, S.; Giannikas, C.; Padgett, R.W. SMA-3 smad has specific and critical functions in DBL-1/SMA-6 TGFβ-related signaling. Dev. Biol. 2000, 223, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Davis, E.L.; Hussey, R.S.; Baum, T.J. Getting to the roots of parasitism by nematodes. Trends Parasitol. 2004, 20, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Torto-Alalibo, T.; Collmer, C.W.; Lindeberg, M.; Bird, D.; Collmer, A.; Tyler, B.M. Common and contrasting themes in host cell-targeted effectors from bacterial, fungal, oomycete and nematode plant symbionts described using the gene ontology. BMC Microbiol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Alkharouf, N.W.; Klink, V.P.; Matthews, B.F. Identification of heterodera glycines (soybean cyst nematode [SCN]) cDNA sequences with high identity to those of Caenorhabditis elegans having lethal mutant or RNAi phenotypes. Exp. Parasitol. 2007, 115, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Castagnone-Sereno, P.; Deleury, E.; Danchin, E.G.; Perfus-Barbeoch, L.; Abad, P. Data-mining of the Meloidogyne incognita degradome and comparative analysis of proteases in nematodes. Genomics 2011, 97, 29–36. [Google Scholar] [CrossRef] [PubMed]
- De Boer, J.; Yan, Y.; Smant, G.; Davis, E.; Baum, T. In-situ hybridization to messenger RNA in heterodera glycines. J. Nematol. 1998, 30, 309–312. [Google Scholar] [PubMed]
- Gao, B.; Allen, R.; Maier, T.; Davis, E.L.; Baum, T.J.; Hussey, R.S. The parasitome of the phytonematode heterodera glycines. Mol. Plant. Microbe Interact. 2003, 16, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Haegeman, A.; Jacob, J.; Vanholme, B.; Kyndt, T.; Mitreva, M.; Gheysen, G. Expressed sequence tags of the peanut pod nematode ditylenchus africanus: The first transcriptome analysis of an anguinid nematode. Mol. Biochem. Parasitol. 2009, 167, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.; Mitreva, M.; Vanholme, B.; Gheysen, G. Exploring the transcriptome of the burrowing nematode Radopholus similis. Mol. Genet. Genom. 2008, 280, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Aikawa, T.; Kosaka, H.; Pritchard, L.; Ogura, N.; Jones, J.T. Expressed sequence tag (EST) analysis of the pine wood nematode Bursaphelenchus xylophilus and B. mucronatus. Mol. Biochem. Parasitol. 2007, 155, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Gao, B.-L.; Kong, L.-A.; Yu, Q.; Huang, W.-K.; He, X.-F.; Long, H.-B.; Peng, D.-L. Exploring the host parasitism of the migratory plant-parasitic nematode Ditylenchus destuctor by expressed sequence tags analysis. PLoS ONE 2013, 8, e69579. [Google Scholar] [CrossRef] [PubMed]
- Lacomme, C.; Santa, C.S. Bax-induced cell death in tobacco is similar to the hypersensitive response. Proc. Natl. Acad. Sci. USA 1999, 96, 7956–7961. [Google Scholar] [CrossRef] [PubMed]
- Abramovitch, R.B.; Kim, Y.J.; Chen, S.; Dickman, M.B.; Martin, G.B. Pseudomonas type III effector AvrPtoB induces plant disease susceptibility by inhibition of host programmed cell death. EMBO J. 2003, 22, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Jamir, Y.; Guo, M.; Oh, H.S.; Petnicki-Ocwieja, T.; Chen, S.; Tang, X.; Dickman, M.B.; Collmer, A.; Alfano, J.R. Identification of Pseudomonas syringae type III effectors that can suppress programmed cell death in plants and yeast. Plant J. Cell Mol. Biol. 2004, 37, 554–565. [Google Scholar] [CrossRef]
- Chronis, D.; Chen, S.; Lu, S.; Hewezi, T.; Carpenter, S.C.; Loria, R.; Baum, T.J.; Wang, X. A ubiquitin carboxyl extension protein secreted from a plant-parasitic nematode Globodera rostochiensis is cleaved in planta to promote plant parasitism. Plant J. 2013, 74, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Hewezi, T.; Baum, T.J. Manipulation of plant cells by cyst and root-knot nematode effectors. Mol. Plant. Microbe Interact. 2013, 26, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Postma, W.J.; Slootweg, E.J.; Rehman, S.; Finkers-Tomczak, A.; Tytgat, T.O.; van Gelderen, K.; Lozano-Torres, J.L.; Roosien, J.; Pomp, R.; van Schaik, C. The effector SPRYSEC-19 of Globodera rostochiensis suppresses CC-NB-LRR-mediated disease resistance in plants. Plant Physiol. 2012, 160, 944–954. [Google Scholar] [CrossRef] [PubMed]
- Jaouannet, M.; Magliano, M.; Arguel, M.; Gourgues, M.; Evangelisti, E.; Abad, P.; Rosso, M. The root-knot nematode calreticulin Mi-CRT is a key effector in plant defense suppression. Mol. Plant. Microbe Interact. 2013, 26, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Dong, R.; Maier, T.; Allen, R.; Davis, E.L.; Baum, T.J.; Hussey, R.S. Use of solid-phase subtractive hybridization for the identification of parasitism gene candidates from the root-knot nematode Meloidogyne incognita. Mol. Plant Pathol. 2004, 5, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Eisenback, J.D. Diagnostic characters useful in the identification of the four most common species of root-knot nematodes (Meloidogyne spp.). In An Advance Treatise on Meloidogyne; North Carolina State University Graphics: Raleigh, NC, USA, 1985. [Google Scholar]
- Rammah, A.; Hirschmann, H. Meloidogyne mayaguensis n. sp. (Meloidogynidae), a root-knot nematode from puerto rico. J. Nematol. 1988, 20, 58–69. [Google Scholar] [PubMed]
- Tigano, M.; Siqueira, K.D.; Castagnone-Sereno, P.; Mulet, K.; Queiroz, P.; Santos, M.D.; Teixeira, C.; Almeida, M.; Silva, J.; Carneiro, R. Genetic diversity of the root-knot nematode Meloidogyne enterolobii and development of a scar marker for this guava-damaging species. Plant Pathol. 2010, 59, 1054–1061. [Google Scholar] [CrossRef]
- Ding, X.; Shields, J.; Allen, R.; Hussey, R.S. A secretory cellulose-binding protein cdna cloned from the root-knot nematode (Meloidogyne incognita). Mol. Plant Microbe Interact. 1998, 11, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Grossmann, S.; Vingron, M.; Robinson, P.N. Ontologizer 2.0—A multifunctional tool for go term enrichment analysis and data exploration. Bioinformatics 2008, 24, 1650–1651. [Google Scholar] [CrossRef] [PubMed]
- Ekseth, O.K.; Kuiper, M.; Mironov, V. Orthagogue: An agile tool for the rapid prediction of orthology relations. Bioinformatics 2014, 30, 734–736. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. Trimal: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.O.; Kumar, S.; Tamura, K.; Nei, M. Mega6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar]
- Eddy, S.R. Accelerated profile hmm searches. PLoS Comput. Biol. 2011, 7, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, J.M.; Griggs, A.D.; Smith, J.L.; Haas, B.J.; Wortman, J.R.; Zeng, Q. Kinannote, a computer program to identify and classify members of the eukaryotic protein kinase superfamily. Bioinformatics 2013, 29, 2387–2394. [Google Scholar] [CrossRef] [PubMed]
- McVeigh, P.; Leech, S.; Mair, G.R.; Marks, N.J.; Geary, T.G.; Maule, A.G. Analysis of fmrfamide-like peptide (FLP) diversity in phylum nematoda. Int. J. Parasitol. 2005, 35, 1043–1060. [Google Scholar] [CrossRef] [PubMed]
- McVeigh, P.; Geary, T.G.; Marks, N.J.; Maule, A.G. The FLP-side of nematodes. Trends Parasitol. 2006, 22, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Husson, S.J.; Lindemans, M.; Janssen, T.; Schoofs, L. Comparison of Caenorhabditis elegans NLP peptides with arthropod neuropeptides. Trends Parasitol. 2009, 25, 171–181. [Google Scholar] [CrossRef] [PubMed]
- McVeigh, P.; Alexander-Bowman, S.; Veal, E.; Mousley, A.; Marks, N.J.; Maule, A.G. Neuropeptide-like protein diversity in phylum nematoda. Int. J. Parasitol. 2008, 38, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Thomas Nordahl, P.; SøRen, B.; Gunnar, V.H.; Henrik, N. Signalp 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar]
- Wang, Q.; Han, C.; Ferreira, A.O.; Yu, X.; Ye, W.; Tripathy, S.; Kale, S.D.; Gu, B.; Sheng, Y.; Sui, Y. Transcriptional programming and functional interactions within the phytophthora sojae RXLR effector repertoire. Plant Cell 2011, 23, 2064–2086. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assembly Metric | Library | Contigs | Singletons |
|---|---|---|---|
| Total base pairs | 165,040,879 | – | – |
| Total reads | 408,663 | – | – |
| Reads in assembly | 355,760 | – | – |
| Average length (bp) | 403 | 1202 | 380 |
| EST number | – | 8193 | 31,860 |
| Length range (bp) | – | 62–18,104 | 50–1043 |
| Operon number | – | 8143 | 29,043 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Yang, D.; Niu, J.; Zhao, J.; Jian, H. De Novo Analysis of the Transcriptome of Meloidogyne enterolobii to Uncover Potential Target Genes for Biological Control. Int. J. Mol. Sci. 2016, 17, 1442. https://doi.org/10.3390/ijms17091442
Li X, Yang D, Niu J, Zhao J, Jian H. De Novo Analysis of the Transcriptome of Meloidogyne enterolobii to Uncover Potential Target Genes for Biological Control. International Journal of Molecular Sciences. 2016; 17(9):1442. https://doi.org/10.3390/ijms17091442
Chicago/Turabian StyleLi, Xiangyang, Dan Yang, Junhai Niu, Jianlong Zhao, and Heng Jian. 2016. "De Novo Analysis of the Transcriptome of Meloidogyne enterolobii to Uncover Potential Target Genes for Biological Control" International Journal of Molecular Sciences 17, no. 9: 1442. https://doi.org/10.3390/ijms17091442