
Escaping Antiangiogenic Therapy: Strategies Employed by Cancer Cells

 ,
,
Abstract
:1. Tumor Angiogenesis and Antiangiogenesis
2. Mechanisms for Escape/Resistance to Antiangiogenic Therapy
3. Upregulation of Alternative/Compensatory Pathways
4. Vasculogenic Mimicry (VM)
5. Vessel (or Vascular) Co-Option
6. Future Directions for Antiangiogenic Therapies
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ferrara, N. VEGF and intraocular neovascularization: From discovery to therapy. Trans. Vis. Sci. Technol. 2016, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, M.; Shubi, P. Tumor angiogenesis: Transfilter diffusion studies in the hamster by the transparent chamber technique. J. Natl. Cancer Inst. 1968, 41, 111–124. [Google Scholar] [PubMed]
- Ehrmann, R.L.; Knoth, M. Choriocarcinoma. Transfilter stimulation of vasoproliferation in the hamster cheek pouch. Studied by light and electron microscopy. J. Natl. Cancer Inst. 1968, 41, 1329–1341. [Google Scholar] [PubMed]
- Ribatti, D.; Vacca, A.; Dammacco, F. The role of the vascular phase in solid tumor growth: A historical review. Neoplasia 1999, 1, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J.; Long, D.M., Jr.; Becker, F.F. Growth and metastasis of tumor in organ culture. Cancer 1963, 16, 453–467. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Finley, S.D.; Popel, A.S. Predicting the effects of antiangiogenic agents targeting specific VEGF isoforms. AAPS J. 2012, 14, 500–509. [Google Scholar] [CrossRef] [PubMed]
- McFee, R.M.; Rozell, T.G.; Cupp, A.S. The balance of proangiogenic and antiangiogenic VEGF-A isoforms regulate follicle development. Cell Tissue Res. 2012, 349, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Mohamedali, K.A.; Li, Z.G.; Starbuck, M.W.; Wan, X.; Yang, J.; Kim, S.; Zhang, W.; Rosenblum, M.G.; Navone, N.M. Inhibition of prostate cancer osteoblastic progression with VEGF121/RGEL, a single agent targeting osteoblasts, osteoclasts, and tumor neovasculature. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 2328–2338. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.E.; Fan, T.P. Suppression of VEGF-induced angiogenesis by the protein tyrosine kinase inhibitor, lavendustin a. Br. J. Pharmacol. 1995, 114, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L.S. Clinical experience with angiogenesis signaling inhibitors: Focus on vascular endothelial growth factor (VEGF) blockers. Cancer Control J. Moffitt Cancer Center 2002, 9, 36–44. [Google Scholar]
- Jackson, D.B.; Sood, A.K. Personalized cancer medicine-advances and socio-economic challenges. Nat. Rev. Clin. Oncol. 2011, 8, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Allegra, C.J.; Yothers, G.; O’Connell, M.J.; Sharif, S.; Petrelli, N.J.; Colangelo, L.H.; Atkins, J.N.; Seay, T.E.; Fehrenbacher, L.; Goldberg, R.M.; et al. Phase III trial assessing bevacizumab in stages II and III carcinoma of the colon: Results of NSABP protocol C-08. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 11–16. [Google Scholar] [CrossRef] [PubMed]
- De Gramont, A.; van Cutsem, E.; Schmoll, H.J.; Tabernero, J.; Clarke, S.; Moore, M.J.; Cunningham, D.; Cartwright, T.H.; Hecht, J.R.; Rivera, F.; et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): A phase 3 randomised controlled trial. Lancet Oncol. 2012, 13, 1225–1233. [Google Scholar] [CrossRef]
- Miller, K.; Wang, M.; Gralow, J.; Dickler, M.; Cobleigh, M.; Perez, E.A.; Shenkier, T.; Cella, D.; Davidson, N.E. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N. Engl. J. Med. 2007, 357, 2666–2676. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.A. The Biology of Cancer; Garland Science: New York, NY, USA, 2014. [Google Scholar]
- Bergers, G.; Hanahan, D. Modes of resistance to antiangiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.P.; Badtke, M.M.; Dudevoir, M.L.; Harrell, J.C.; Jacobsen, B.M.; Horwitz, K.B. Vascular endothelial growth factor secreted by activated stroma enhances angiogenesis and hormone-independent growth of estrogen receptor-positive breast cancer. Cancer Res. 2010, 70, 2655–2664. [Google Scholar] [CrossRef] [PubMed]
- Van Beijnum, J.R.; Nowak-Sliwinska, P.; Huijbers, E.J.; Thijssen, V.L.; Griffioen, A.W. The great escape; the hallmarks of resistance to antiangiogenic therapy. Pharmacol. Rev. 2015, 67, 441–461. [Google Scholar] [CrossRef] [PubMed]
- Vasudev, N.S.; Reynolds, A.R. Antiangiogenic therapy for cancer: Current progress, unresolved questions and future directions. Angiogenesis 2014, 17, 471–494. [Google Scholar] [CrossRef] [PubMed]
- Bridges, E.M.; Harris, A.L. The angiogenic process as a therapeutic target in cancer. Biochem. Pharmacol. 2011, 81, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Lieu, C.; Heymach, J.; Overman, M.; Tran, H.; Kopetz, S. Beyond VEGF: Inhibition of the fibroblast growth factor pathway and antiangiogenesis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 6130–6139. [Google Scholar] [CrossRef] [PubMed]
- Presta, M.; Dell'Era, P.; Mitola, S.; Moroni, E.; Ronca, R.; Rusnati, M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 159–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pepper, M.S.; Ferrara, N.; Orci, L.; Montesano, R. Potent synergism between vascular endothelial growth factor and basic fibroblast growth factor in the induction of angiogenesis in vitro. Biochem. Biophys. Res. Commun. 1992, 189, 824–831. [Google Scholar] [CrossRef]
- Tille, J.C.; Wood, J.; Mandriota, S.J.; Schnell, C.; Ferrari, S.; Mestan, J.; Zhu, Z.; Witte, L.; Pepper, M.S. Vascular endothelial growth factor (VEGF) receptor-2 antagonists inhibit VEGF- and basic fibroblast growth factor-induced angiogenesis in vivo and in vitro. J. Pharmacol. Exp. Ther. 2001, 299, 1073–1085. [Google Scholar] [PubMed]
- Alessi, P.; Leali, D.; Camozzi, M.; Cantelmo, A.; Albini, A.; Presta, M. Anti-FGF2 approaches as a strategy to compensate resistance to anti-VEGF therapy: Long-pentraxin 3 as a novel antiangiogenic FGF2-antagonist. Eur. Cytokine Netw. 2009, 20, 225–234. [Google Scholar] [PubMed]
- Andre, F.; Bachelot, T.; Campone, M.; Dalenc, F.; Perez-Garcia, J.M.; Hurvitz, S.A.; Turner, N.; Rugo, H.; Smith, J.W.; Deudon, S.; et al. Targeting FGFR with dovitinib (TKI258): Preclinical and clinical data in breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 3693–3702. [Google Scholar] [CrossRef] [PubMed]
- Hilberg, F.; Roth, G.J.; Krssak, M.; Kautschitsch, S.; Sommergruber, W.; Tontsch-Grunt, U.; Garin-Chesa, P.; Bader, G.; Zoephel, A.; Quant, J.; et al. BIBF 1120: Triple angiokinase inhibitor with sustained receptor blockade and good antitumor efficacy. Cancer Res. 2008, 68, 4774–4782. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Xiang, J.J.; Li, D.; Deng, N.; Wang, H.; Gong, Y.P. Selection and characterization of a human neutralizing antibody to human fibroblast growth factor-2. Biochem. Biophys. Res. Commun. 2010, 394, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Park, H.; Chhim, S.; Ding, Y.; Jiang, W.; Queen, C.; Kim, K.J. A novel monoclonal antibody to fibroblast growth factor 2 effectively inhibits growth of hepatocellular carcinoma xenografts. Mol. Cancer Ther. 2012, 11, 864–872. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wei, X.; Xie, K.; Chen, K.; Li, J.; Fang, J. A novel decoy receptor fusion protein for FGF-2 potently inhibits tumour growth. Br. J. Cancer 2014, 111, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Hoff, P.M.; Morris, J.S.; Wolff, R.A.; Eng, C.; Glover, K.Y.; Adinin, R.; Overman, M.J.; Valero, V.; Wen, S.; et al. Phase II trial of infusional fluorouracil, irinotecan, and bevacizumab for metastatic colorectal cancer: Efficacy and circulating angiogenic biomarkers associated with therapeutic resistance. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Lee, J.H.; Simmons, B.H.; Wong, A.; Esparza, C.O.; Plumlee, P.A.; Feng, J.; Stewart, A.E.; Hu-Lowe, D.D.; Christensen, J.G. HGF/c-met acts as an alternative angiogenic pathway in sunitinib-resistant tumors. Cancer Res. 2010, 70, 10090–10100. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Adjei, A.A. In the clinic: Ongoing clinical trials evaluating c-met-inhibiting drugs. Ther. Adv. Med. Oncol. 2011, 3, S37–S50. [Google Scholar] [CrossRef] [PubMed]
- De Falco, S. The discovery of placenta growth factor and its biological activity. Exp. Mol. Med. 2012, 44, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Mazzone, M.; Jonckx, B.; Carmeliet, P. FLT1 and its ligands VEGFB and PLGF: Drug targets for antiangiogenic therapy? Nat. Rev. Cancer 2008, 8, 942–956. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, E.M.; Hosaka, K.; Zhong, Z.; Cao, R.; Cao, Y. Malignant cell-derived PLGF promotes normalization and remodeling of the tumor vasculature. Proc. Natl Acad. Sci. USA 2009, 106, 17505–17510. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Moons, L.; Luttun, A.; Vincenti, V.; Compernolle, V.; de Mol, M.; Wu, Y.; Bono, F.; Devy, L.; Beck, H.; et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 2001, 7, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Mac Gabhann, F.; Popel, A.S. Model of competitive binding of vascular endothelial growth factor and placental growth factor to VEGF receptors on endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H153–H164. [Google Scholar] [CrossRef] [PubMed]
- Holash, J.; Davis, S.; Papadopoulos, N.; Croll, S.D.; Ho, L.; Russell, M.; Boland, P.; Leidich, R.; Hylton, D.; Burova, E.; et al. VEGF-trap: A VEGF blocker with potent antitumor effects. Proc. Natl. Acad. Sci. USA 2002, 99, 11393–11398. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Tabernero, J.; Lakomy, R.; Prenen, H.; Prausova, J.; Macarulla, T.; Ruff, P.; van Hazel, G.A.; Moiseyenko, V.; Ferry, D.; et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 3499–3506. [Google Scholar] [CrossRef] [PubMed]
- Folprecht, G.; Pericay, C.; Saunders, M.P.; Thomas, A.; Lopez Lopez, R.; Roh, J.K.; Chistyakov, V.; Hohler, T.; Kim, J.S.; Hofheinz, R.D.; et al. Oxaliplatin and 5-FU/folinic acid (modified FOLFOX6) with or without aflibercept in first-line treatment of patients with metastatic colorectal cancer: The affirm study. Ann. Oncol. 2016, 27, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Scartozzi, M.; Vincent, L.; Chiron, M.; Cascinu, S. Aflibercept, a new way to target angiogenesis in the second line treatment of metastatic colorectal cancer (MCRC). Target. Oncol. 2016, 11, 489–500. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Joulain, F.; Hoff, P.M.; Mitchell, E.; Ruff, P.; Lakomy, R.; Prausova, J.; Moiseyenko, V.M.; van Hazel, G.; Cunningham, D.; et al. Aflibercept plus folfiri vs. Placebo plus folfiri in second-line metastatic colorectal cancer: A post hoc analysis of survival from the phase III velour study subsequent to exclusion of patients who had recurrence during or within 6 months of completing adjuvant oxaliplatin-based therapy. Target. Oncol. 2016, 11, 383–400. [Google Scholar] [PubMed]
- Hamdan, R.; Zhou, Z.; Kleinerman, E.S. SDF-1α induces PDGF-b expression and the differentiation of bone marrow cells into pericytes. Mol. Cancer Res. MCR 2011, 9, 1462–1470. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Thaker, P.H.; Lin, Y.G.; Spannuth, W.; Landen, C.N.; Merritt, W.M.; Jennings, N.B.; Langley, R.R.; Gershenson, D.M.; Yancopoulos, G.D.; et al. Impact of vessel maturation on antiangiogenic therapy in ovarian cancer. Am. J. Obstet. Gynecol. 2008, 198, 477 e1–477 e10. [Google Scholar] [CrossRef] [PubMed]
- Crawford, Y.; Kasman, I.; Yu, L.; Zhong, C.; Wu, X.; Modrusan, Z.; Kaminker, J.; Ferrara, N. PDGF-c mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell 2009, 15, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.P.; Dye, W.W.; Jacobsen, B.M.; Horwitz, K.B. Malignant stroma increases luminal breast cancer cell proliferation and angiogenesis through platelet-derived growth factor signaling. BMC Cancer 2014, 14, 735. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.; Luo, M.; Wang, C.; Fan, L.; Yang, S.N.; Cardenas, M.; Geng, H.; Leonard, J.P.; Melnick, A.; Cerchietti, L.; et al. Imatinib disrupts lymphoma angiogenesis by targeting vascular pericytes. Blood 2013, 121, 5192–5202. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Tugues, S.; Li, X.; Gualandi, L.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 2011, 437, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Mamluk, R.; Gechtman, Z.; Kutcher, M.E.; Gasiunas, N.; Gallagher, J.; Klagsbrun, M. Neuropilin-1 binds vascular endothelial growth factor 165, placenta growth factor-2, and heparin via its b1b2 domain. J. Biol. Chem. 2002, 277, 24818–24825. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, C.; Fantin, A.; Lampropoulou, A.; Denti, L.; Chikh, A.; Ruhrberg, C. Imatinib inhibits VEGF-independent angiogenesis by targeting neuropilin 1-dependent ABL1 activation in endothelial cells. J. Exp. Med. 2014, 211, 1167–1183. [Google Scholar] [CrossRef] [PubMed]
- Blanke, C.D.; Rankin, C.; Corless, C.; Eary, J.F.; Mulder, K.; Okuno, S.H.; George, S.; Heinrich, M. S0502: A SWOG phase III randomized study of imatinib, with or without bevacizumab, in patients with untreated metastatic or unresectable gastrointestinal stromal tumors. Oncologist 2015, 20, 1353–1354. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Hamilton, B.K.; Rosen, M.A.; Amaravadi, R.K.; Schuchter, L.M.; Gallagher, M.; Chen, H.; Sehgal, C.; O’Dwyer, P.J. Phase I/II trial of imatinib and bevacizumab in patients with advanced melanoma and other advanced cancers. Oncologist 2015, 20, 952–959. [Google Scholar] [CrossRef] [PubMed]
- Shim, W.S.; Ho, I.A.; Wong, P.E. Angiopoietin: A TIE(d) balance in tumor angiogenesis. Mol. Cancer Res. MCR 2007, 5, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Poveda, A.; Vergote, I.; Raspagliesi, F.; Fujiwara, K.; Bae, D.S.; Oaknin, A.; Ray-Coquard, I.; Provencher, D.M.; Karlan, B.Y.; et al. Anti-angiopoietin therapy with trebananib for recurrent ovarian cancer (TRINOVA-1): A randomised, multicentre, double-blind, placebo-controlled phase 3 trial. Lancet Oncol. 2014, 15, 799–808. [Google Scholar] [CrossRef]
- Clarke, J.M.; Hurwitz, H.I. Understanding and targeting resistance to antiangiogenic therapies. J. Gastrointest. Oncol. 2013, 4, 253–263. [Google Scholar] [PubMed]
- Thurston, G.; Noguera-Troise, I.; Yancopoulos, G.D. The delta paradox: Dll-4 blockade leads to more tumour vessels but less tumour growth. Nat. Rev. Cancer 2007, 7, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Li, J.L.; Sainson, R.C.; Shi, W.; Leek, R.; Harrington, L.S.; Preusser, M.; Biswas, S.; Turley, H.; Heikamp, E.; Hainfellner, J.A.; et al. Delta-like 4 Notch ligand regulates tumor angiogenesis, improves tumor vascular function, and promotes tumor growth in vivo. Cancer Res. 2007, 67, 11244–11253. [Google Scholar] [CrossRef] [PubMed]
- Noguera-Troise, I.; Daly, C.; Papadopoulos, N.J.; Coetzee, S.; Boland, P.; Gale, N.W.; Lin, H.C.; Yancopoulos, G.D.; Thurston, G. Blockade of Dll-4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 2006, 444, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Ridgway, J.; Zhang, G.; Wu, Y.; Stawicki, S.; Liang, W.C.; Chanthery, Y.; Kowalski, J.; Watts, R.J.; Callahan, C.; Kasman, I.; et al. Inhibition of Dll-4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 2006, 444, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Brzozowa, M.; Wojnicz, R.; Kowalczyk-Ziomek, G.; Helewski, K. The Notch ligand delta-like 4 (Dll-4) as a target in angiogenesis-based cancer therapy? Contemp. Oncol. 2013, 17, 234–237. [Google Scholar]
- Andersson, E.R.; Lendahl, U. Therapeutic modulation of notch signalling—Are we there yet? Nat. Rev. Drug Discov. 2014, 13, 357–378. [Google Scholar] [CrossRef] [PubMed]
- Ceradini, D.J.; Kulkarni, A.R.; Callaghan, M.J.; Tepper, O.M.; Bastidas, N.; Kleinman, M.E.; Capla, J.M.; Galiano, R.D.; Levine, J.P.; Gurtner, G.C. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat. Med. 2004, 10, 858–864. [Google Scholar] [CrossRef] [PubMed]
- De Falco, E.; Porcelli, D.; Torella, A.R.; Straino, S.; Iachininoto, M.G.; Orlandi, A.; Truffa, S.; Biglioli, P.; Napolitano, M.; Capogrossi, M.C.; et al. SDF-1 involvement in endothelial phenotype and ischemia-induced recruitment of bone marrow progenitor cells. Blood 2004, 104, 3472–3482. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Song, S.; Meyer-Morse, N.; Bergsland, E.; Hanahan, D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J. Clin. Investig. 2003, 111, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Antonelli-Orlidge, A.; Saunders, K.B.; Smith, S.R.; D’Amore, P.A. An activated form of transforming growth factor β is produced by cocultures of endothelial cells and pericytes. Proc. Natl. Acad. Sci. USA 1989, 86, 4544–4548. [Google Scholar] [CrossRef] [PubMed]
- Franco, M.; Roswall, P.; Cortez, E.; Hanahan, D.; Pietras, K. Pericytes promote endothelial cell survival through induction of autocrine VEGF-A signaling and BCL-W expression. Blood 2011, 118, 2906–2917. [Google Scholar] [CrossRef] [PubMed]
- Folberg, R.; Hendrix, M.J.; Maniotis, A.J. Vasculogenic mimicry and tumor angiogenesis. Am. J. Pathol. 2000, 156, 361–381. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Folberg, R.; Maniotis, A.J. Vasculogenic mimicry. APMIS 2004, 112, 508–525. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Zhang, D.; Zhao, N.; Zhao, X. Epithelial-to-endothelial transition and cancer stem cells: Two cornerstones of vasculogenic mimicry in malignant tumors. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fisher, S.J.; Janatpour, M.; Genbacev, O.; Dejana, E.; Wheelock, M.; Damsky, C.H. Human cytotrophoblasts adopt a vascular phenotype as they differentiate. A strategy for successful endovascular invasion? J. Clin. Investig. 1997, 99, 2139–2151. [Google Scholar] [CrossRef] [PubMed]
- Auguste, P.; Lemiere, S.; Larrieu-Lahargue, F.; Bikfalvi, A. Molecular mechanisms of tumor vascularization. Crit. Rev. Oncol. Hematol. 2005, 54, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, J.; Yao, W.Y.; Ben, Q.W.; Chen, D.F.; He, X.Y.; Li, L.; Yuan, Y.Z. The origins of vacularization in tumors. Front. Biosci. (Landmark Ed) 2012, 17, 2559–2565. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, D.; Wang, Y.; Zhao, W.; Guo, H.; Zhao, X.; Sun, B. Morphologic research of microcirculation patterns in human and animal melanoma. Med. Oncol. 2006, 23, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Salnikov, A.V.; Liu, L.; Platen, M.; Gladkich, J.; Salnikova, O.; Ryschich, E.; Mattern, J.; Moldenhauer, G.; Werner, J.; Schemmer, P.; et al. Hypoxia induces EMT in low and highly aggressive pancreatic tumor cells but only cells with cancer stem cell characteristics acquire pronounced migratory potential. PLoS ONE 2012, 7, e46391. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Sun, B.; Liu, T.; Zhao, X.; Che, N.; Gu, Q.; Dong, X.; Yao, Z.; Li, R.; Li, J.; et al. Slug promoted vasculogenic mimicry in hepatocellular carcinoma. J. Cell. Mol. Med. 2013, 17, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.S.; Gao, X.L.; Liu, X.; Gao, S.Y.; Fan, Y.L.; Jiang, Y.P.; Ma, X.R.; Jiang, J.; Feng, H.; Chen, Q.M.; et al. CD133+ cancer stem-like cells promote migration and invasion of salivary adenoid cystic carcinoma by inducing vasculogenic mimicry formation. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Bao, M.; Miele, L.; Sarkar, F.H.; Wang, Z.; Zhou, Q. Tumour vasculogenic mimicry is associated with poor prognosis of human cancer patients: A systemic review and meta-analysis. Eur. J. Cancer 2013, 49, 3914–3923. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Yao, N.; Wang, G.; Tian, L.; Ma, J.; Shi, X.; Zhang, L.; Zhang, J.; Zhou, X.; Zhou, G.; et al. Vasculogenic mimicry: A new prognostic sign of human osteosarcoma. Hum. Pathol. 2014, 45, 2120–2129. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Yang, B.; Cao, Q.; Wu, X. Association of vasculogenic mimicry formation and CD133 expression with poor prognosis in ovarian cancer. Gynecol. Obstet. Investig. 2016. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhao, X.L.; Gu, Q.; Wang, J.Y.; Zhang, S.W.; Zhang, D.F.; Wang, X.H.; Zhao, N.; Gao, Y.T.; Sun, B.C. Correlation of vasculogenic mimicry with clinicopathologic features and prognosis of ovarian carcinoma. Zhonghua Bing Li Xue Za Zhi 2009, 38, 585–589. [Google Scholar] [PubMed]
- Li, M.; Gu, Y.; Zhang, Z.; Zhang, S.; Zhang, D.; Saleem, A.F.; Zhao, X.; Sun, B. Vasculogenic mimicry: A new prognostic sign of gastric adenocarcinoma. Pathol. Oncol. Res. POR 2010, 16, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Seftor, R.E.; Hess, A.R.; Seftor, E.A.; Kirschmann, D.A.; Hardy, K.M.; Margaryan, N.V.; Hendrix, M.J. Tumor cell vasculogenic mimicry: From controversy to therapeutic promise. Am. J. Pathol. 2012, 181, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Kleinman, H.K.; Martin, G.R. Matrigel: Basement membrane matrix with biological activity. Semin. Cancer Biol. 2005, 15, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Sun, B.; Zhao, X.; Du, J.; Gu, Q.; Liu, Y.; Cheng, R.; Dong, X. Wnt5a promotes vasculogenic mimicry and epithelial-mesenchymal transition via protein kinase Cα in epithelial ovarian cancer. Oncol. Rep. 2014, 32, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Zhang, D.; Zhao, X.; Sun, B.; Liu, Y.; Gu, Q.; Zhang, Y.; Zhao, X.; Che, N.; Zheng, Y.; et al. Dickkopf-1-promoted vasculogenic mimicry in non-small cell lung cancer is associated with EMT and development of a cancer stem-like cell phenotype. J. Cell. Mol. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Sun, B.; Zhao, X.; Ma, Y.; Ji, R.; Gu, Q.; Dong, X.; Li, J.; Liu, F.; Jia, X.; et al. Twist1 expression induced by sunitinib accelerates tumor cell vasculogenic mimicry by increasing the population of CD133+ cells in triple-negative breast cancer. Mol. Cancer 2014, 13, 207. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Sun, B.; Zhao, X.; Gu, Q.; Dong, X.; Mo, J.; Sun, T.; Wang, J.; Sun, R.; Liu, Y. Hypoxia promotes vasculogenic mimicry formation by inducing epithelial-mesenchymal transition in ovarian carcinoma. Gynecol. Oncol. 2014, 133, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Yu, L.; Wang, D.; Zhou, L.; Cheng, Z.; Chai, D.; Ma, L.; Tao, Y. Aberrant expression of CD133 in non-small cell lung cancer and its relationship to vasculogenic mimicry. BMC Cancer 2012, 12, 535. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Sun, B.; Zhao, X.; Zhang, D.; Zhao, X.; Gu, Q.; Dong, X.; Zhao, N.; Liu, P.; Liu, Y. Doxycycline as an inhibitor of the epithelial-to-mesenchymal transition and vasculogenic mimicry in hepatocellular carcinoma. Mol. Cancer Ther. 2014, 13, 3107–3122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.G.; Li, X.Y.; Wang, Y.Z.; Zhang, Q.D.; Gu, S.Y.; Wu, X.; Zhu, G.H.; Li, Q.; Liu, G.L. Rock is involved in vasculogenic mimicry formation in hepatocellular carcinoma cell line. PLoS ONE 2014, 9, e107661. [Google Scholar] [CrossRef] [PubMed]
- Kirschmann, D.A.; Seftor, E.A.; Hardy, K.M.; Seftor, R.E.; Hendrix, M.J. Molecular pathways: Vasculogenic mimicry in tumor cells: Diagnostic and therapeutic implications. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 2726–2732. [Google Scholar] [CrossRef] [PubMed]
- Francescone, R.; Scully, S.; Bentley, B.; Yan, W.; Taylor, S.L.; Oh, D.; Moral, L.; Shao, R. Glioblastoma-derived tumor cells induce vasculogenic mimicry through FLK-1 protein activation. J. Biol. Chem. 2012, 287, 24821–24831. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Ping, Y.; Liu, Y.; Chen, K.; Yoshimura, T.; Liu, M.; Gong, W.; Chen, C.; Niu, Q.; Guo, D.; et al. Vascular endothelial growth factor receptor 2 (VEGFR-2) plays a key role in vasculogenic mimicry formation, neovascularization and tumor initiation by glioma stem-like cells. PLoS ONE 2013, 8, e57188. [Google Scholar] [CrossRef]
- Van der Schaft, D.W.; Seftor, R.E.; Seftor, E.A.; Hess, A.R.; Gruman, L.M.; Kirschmann, D.A.; Yokoyama, Y.; Griffioen, A.W.; Hendrix, M.J. Effects of angiogenesis inhibitors on vascular network formation by human endothelial and melanoma cells. J. Natl. Cancer Inst. 2004, 96, 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, Q.; Li, X.Y.; Yang, Q.Y.; Xu, W.W.; Liu, G.L. Short-term anti-vascular endothelial growth factor treatment elicits vasculogenic mimicry formation of tumors to accelerate metastasis. J. Exp. Clin. Cancer Res. CR 2012, 31, 16. [Google Scholar] [CrossRef] [PubMed]
- Dunleavey, J.M.; Xiao, L.; Thompson, J.; Kim, M.M.; Shields, J.M.; Shelton, S.E.; Irvin, D.M.; Brings, V.E.; Ollila, D.W.; Brekken, R.A.; et al. Vascular channels formed by subpopulations of PECAM1+ melanoma cells. Nat. Commun. 2014, 5, 5200. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.L.; Zhang, Q.; Ye, L.Y.; Liang, F.; Sun, X.; Chen, Y.; Hu, Q.D.; Fu, Q.H.; Su, W.; Chen, Z.; et al. Myocyte enhancer factor 2c regulation of hepatocellular carcinoma via vascular endothelial growth factor and Wnt/β-catenin signaling. Oncogene 2015, 34, 4089–4097. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Sun, B.; Zhao, X.; Zhao, X.; Gu, Q.; Dong, X.; Zheng, Y.; Sun, J.; Cheng, R.; Qi, H.; et al. Overexpression of wnt5a promotes angiogenesis in nsclc. BioMed Res. Int. 2014, 2014, 832562. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Song, W.; Liu, Z.; Zhao, X.; Cao, W.; Sun, B. Wnt3a promotes the vasculogenic mimicry formation of colon cancer via Wnt/β-catenin signaling. Int. J. Mol. Sci. 2015, 16, 18564–18579. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Sun, B.; Liu, Z.; Li, H.; Gao, J.; Leng, X. Dickkopf-1 inhibits epithelial-mesenchymal transition of colon cancer cells and contributes to colon cancer suppression. Cancer Sci. 2012, 103, 828–835. [Google Scholar] [CrossRef] [PubMed]
- La Porta, C. AQP1 is not only a water channel: It contributes to cell migration through Lin7/β-catenin. Cell Adhes. Migr. 2010, 4, 204–206. [Google Scholar] [CrossRef]
- Clapp, C.; Martinez de la Escalera, G. Aquaporin-1: A novel promoter of tumor angiogenesis. Trends Endocrinol. Metab. 2006, 17, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Holash, J.; Maisonpierre, P.C.; Compton, D.; Boland, P.; Alexander, C.R.; Zagzag, D.; Yancopoulos, G.D.; Wiegand, S.J. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science 1999, 284, 1994–1998. [Google Scholar] [CrossRef] [PubMed]
- Carbonell, W.S.; Ansorge, O.; Sibson, N.; Muschel, R. The vascular basement membrane as “soil” in brain metastasis. PLoS ONE 2009, 4, e5857. [Google Scholar] [CrossRef] [PubMed]
- Valiente, M.; Obenauf, A.C.; Jin, X.; Chen, Q.; Zhang, X.H.; Lee, D.J.; Chaft, J.E.; Kris, M.G.; Huse, J.T.; Brogi, E.; et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 2014, 156, 1002–1016. [Google Scholar] [CrossRef] [PubMed]
- Sardari Nia, P.; Hendriks, J.; Friedel, G.; van Schil, P.; van Marck, E. Distinct angiogenic and non-angiogenic growth patterns of lung metastases from renal cell carcinoma. Histopathology 2007, 51, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Stessels, F.; van den Eynden, G.; van der Auwera, I.; Salgado, R.; van den Heuvel, E.; Harris, A.L.; Jackson, D.G.; Colpaert, C.G.; van Marck, E.A.; Dirix, L.Y.; et al. Breast adenocarcinoma liver metastases, in contrast to colorectal cancer liver metastases, display a non-angiogenic growth pattern that preserves the stroma and lacks hypoxia. Br. J. Cancer 2004, 90, 1429–1436. [Google Scholar] [CrossRef] [PubMed]
- Weisshardt, P.; Trarbach, T.; Durig, J.; Paul, A.; Reis, H.; Tilki, D.; Miroschnik, I.; Ergun, S.; Klein, D. Tumor vessel stabilization and remodeling by antiangiogenic therapy with bevacizumab. Histochem. Cell Biol. 2012, 137, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Arjaans, M.; Oude Munnink, T.H.; Oosting, S.F.; Terwisscha van Scheltinga, A.G.; Gietema, J.A.; Garbacik, E.T.; Timmer-Bosscha, H.; Lub-de Hooge, M.N.; Schroder, C.P.; de Vries, E.G. Bevacizumab-induced normalization of blood vessels in tumors hampers antibody uptake. Cancer Res. 2013, 73, 3347–3355. [Google Scholar] [CrossRef] [PubMed]
- Franco, M.; Paez-Ribes, M.; Cortez, E.; Casanovas, O.; Pietras, K. Use of a mouse model of pancreatic neuroendocrine tumors to find pericyte biomarkers of resistance to antiangiogenic therapy. Horm. Metab. Res. 2011, 43, 884–889. [Google Scholar] [PubMed]
- Di Tomaso, E.; Snuderl, M.; Kamoun, W.S.; Duda, D.G.; Auluck, P.K.; Fazlollahi, L.; Andronesi, O.C.; Frosch, M.P.; Wen, P.Y.; Plotkin, S.R.; et al. Glioblastoma recurrence after cediranib therapy in patients: Lack of “rebound” revascularization as mode of escape. Cancer Res. 2011, 71, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Van den Eynden, G.G.; Bird, N.C.; Majeed, A.W.; Van Laere, S.; Dirix, L.Y.; Vermeulen, P.B. The histological growth pattern of colorectal cancer liver metastases has prognostic value. Clin. Exp. Metastasis 2012, 29, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Kusters, B.; Leenders, W.P.; Wesseling, P.; Smits, D.; Verrijp, K.; Ruiter, D.J.; Peters, J.P.; van Der Kogel, A.J.; de Waal, R.M. Vascular endothelial growth factor-a(165) induces progression of melanoma brain metastases without induction of sprouting angiogenesis. Cancer Res. 2002, 62, 341–345. [Google Scholar] [PubMed]
- Passalidou, E.; Trivella, M.; Singh, N.; Ferguson, M.; Hu, J.; Cesario, A.; Granone, P.; Nicholson, A.G.; Goldstraw, P.; Ratcliffe, C.; et al. Vascular phenotype in angiogenic and non-angiogenic lung non-small cell carcinomas. Br. J. Cancer 2002, 86, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Yang, H.; Shi, H.; Wang, X.; Chen, X.; Yuan, Y.; Lin, S.; Wei, Y. Distinct contributions of angiogenesis and vascular co-option during the initiation of primary microtumors and micrometastases. Carcinogenesis 2011, 32, 1143–1150. [Google Scholar] [CrossRef] [PubMed]
- Auf, G.; Jabouille, A.; Guerit, S.; Pineau, R.; Delugin, M.; Bouchecareilh, M.; Magnin, N.; Favereaux, A.; Maitre, M.; Gaiser, T.; et al. Inositol-requiring enzyme 1α is a key regulator of angiogenesis and invasion in malignant glioma. Proc. Natl. Acad. Sci. USA 2010, 107, 15553–15558. [Google Scholar] [CrossRef] [PubMed]
- Connor, Y.; Tekleab, S.; Nandakumar, S.; Walls, C.; Tekleab, Y.; Husain, A.; Gadish, O.; Sabbisetti, V.; Kaushik, S.; Sehrawat, S.; et al. Physical nanoscale conduit-mediated communication between tumour cells and the endothelium modulates endothelial phenotype. Nat. Commun. 2015, 6, 8671. [Google Scholar] [CrossRef] [PubMed]
- Barnhill, R.L.; Lugassy, C. Angiotropic malignant melanoma and extravascular migratory metastasis: Description of 36 cases with emphasis on a new mechanism of tumour spread. Pathology 2004, 36, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Loges, S.; Mazzone, M.; Hohensinner, P.; Carmeliet, P. Silencing or fueling metastasis with VEGF inhibitors: Antiangiogenesis revisited. Cancer Cell 2009, 15, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Lu, K.V.; Petritsch, C.; Liu, P.; Ganss, R.; Passegue, E.; Song, H.; Vandenberg, S.; Johnson, R.S.; Werb, Z.; et al. HIF1α induces the recruitment of bone marrow-derived vascular modulatory cells to regulate tumor angiogenesis and invasion. Cancer Cell 2008, 13, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Paez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Vinals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 15, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.L.; Kim, J.; Ozawa, T.; Zhang, M.; Westphal, M.; Deen, D.F.; Shuman, M.A. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia 2000, 2, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Saidi, A.; Hagedorn, M.; Allain, N.; Verpelli, C.; Sala, C.; Bello, L.; Bikfalvi, A.; Javerzat, S. Combined targeting of interleukin-6 and vascular endothelial growth factor potently inhibits glioma growth and invasiveness. Int. J. Cancer 2009, 125, 1054–1064. [Google Scholar] [CrossRef] [PubMed]
- Blouw, B.; Song, H.; Tihan, T.; Bosze, J.; Ferrara, N.; Gerber, H.P.; Johnson, R.S.; Bergers, G. The hypoxic response of tumors is dependent on their microenvironment. Cancer Cell 2003, 4, 133–146. [Google Scholar] [CrossRef]
- Lamszus, K.; Brockmann, M.A.; Eckerich, C.; Bohlen, P.; May, C.; Mangold, U.; Fillbrandt, R.; Westphal, M. Inhibition of glioblastoma angiogenesis and invasion by combined treatments directed against vascular endothelial growth factor receptor-2, epidermal growth factor receptor, and vascular endothelial-cadherin. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 4934–4940. [Google Scholar] [CrossRef] [PubMed]
- Kuczynski, E.A.; Yin, M.; Bar-Zion, A.; Lee, C.R.; Butz, H.; Man, S.; Daley, F.; Vermeulen, P.B.; Yousef, G.M.; Foster, F.S.; et al. Co-option of liver vessels and not sprouting angiogenesis drives acquired sorafenib resistance in hepatocellular carcinoma. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Jabouille, A.; Delugin, M.; Pineau, R.; Dubrac, A.; Soulet, F.; Lhomond, S.; Pallares-Lupon, N.; Prats, H.; Bikfalvi, A.; Chevet, E.; et al. Glioblastoma invasion and cooption depend on IRE1Α endoribonuclease activity. Oncotarget 2015, 6, 24922–24934. [Google Scholar] [CrossRef] [PubMed]
- Saltz, L.B.; Lenz, H.J.; Kindler, H.L.; Hochster, H.S.; Wadler, S.; Hoff, P.M.; Kemeny, N.E.; Hollywood, E.M.; Gonen, M.; Quinones, M.; et al. Randomized phase II trial of cetuximab, bevacizumab, and irinotecan compared with cetuximab and bevacizumab alone in irinotecan-refractory colorectal cancer: The bond-2 study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 4557–4561. [Google Scholar] [CrossRef] [PubMed]
- Marslin, G.; Revina, A.M.; Khandelwal, V.K.; Balakumar, K.; Prakash, J.; Franklin, G.; Sheeba, C.J. Delivery as nanoparticles reduces imatinib mesylate-induced cardiotoxicity and improves anticancer activity. Int. J. Nanomed. 2015, 10, 3163–3170. [Google Scholar]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Zhang, S.; Zhong, T.; Ren, W.; Yao, X.; Guo, Y.; Duan, X.C.; Yin, Y.F.; Zhang, S.S.; Zhang, X. Multi-targeting NGR-modified liposomes recognizing glioma tumor cells and vasculogenic mimicry for improving anti-glioma therapy. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Ju, R.J.; Zeng, F.; Liu, L.; Mu, L.M.; Xie, H.J.; Zhao, Y.; Yan, Y.; Wu, J.S.; Hu, Y.J.; Lu, W.L. Destruction of vasculogenic mimicry channels by targeting epirubicin plus celecoxib liposomes in treatment of brain glioma. Int. J. Nanomed. 2016, 11, 1131–1146. [Google Scholar]
- Luo, F.; Yang, K.; Liu, R.L.; Meng, C.; Dang, R.F.; Xu, Y. Formation of vasculogenic mimicry in bone metastasis of prostate cancer: Correlation with cell apoptosis and senescence regulation pathways. Pathol. Res. Pract. 2014, 210, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Song, Z.; Liao, D.; Zhang, T.; Liu, F.; Zheng, W.; Luo, K.; Yang, L. Mir-503 inhibits cell proliferation and invasion in glioma by targeting L1CAM. Int. J. Clin. Exp. Med. 2015, 8, 18441–18447. [Google Scholar] [PubMed]
- Kurozumi, A.; Goto, Y.; Matsushita, R.; Fukumoto, I.; Kato, M.; Nishikawa, R.; Sakamoto, S.; Enokida, H.; Nakagawa, M.; Ichikawa, T.; et al. Tumor-suppressive microRNA-223 inhibits cancer cell migration and invasion by targeting ITGA3/ITGB1 signaling in prostate cancer. Cancer Sci. 2016, 107, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Dabrosin, C.; Yin, X.; Fuster, M.M.; Arreola, A.; Rathmell, W.K.; Generali, D.; Nagaraju, G.P.; El-Rayes, B.; Ribatti, D.; et al. Broad targeting of angiogenesis for cancer prevention and therapy. Semin. Cancer Biol. 2015, 35 (Suppl. S224–S243). [Google Scholar] [CrossRef] [PubMed]





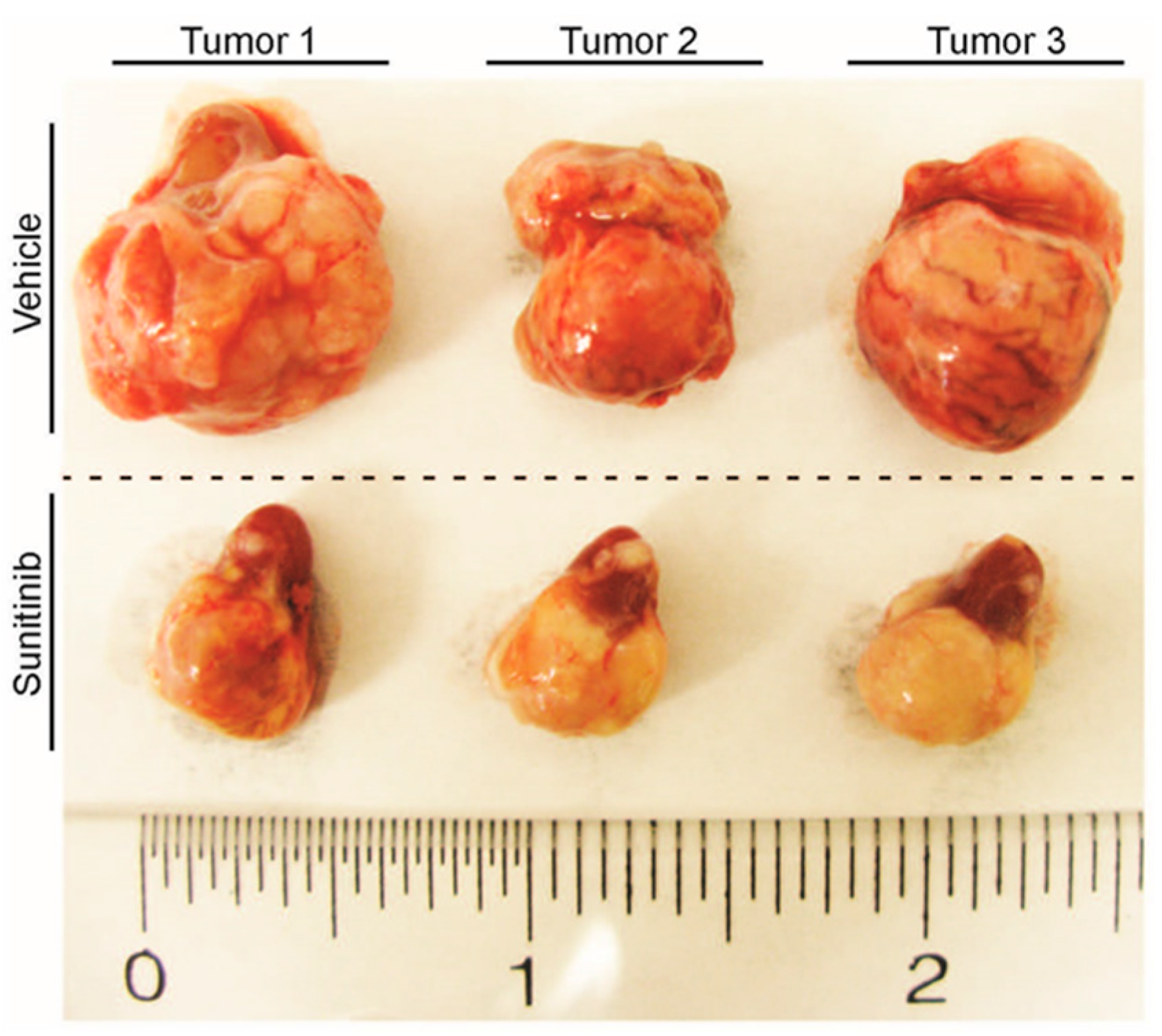{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name (Commercial) | Cancer Type (Recommended Use) | Main Mechanism of Action |
|---|---|---|
| Bevacizumab (Avastin) | Metastatic colorectal | VEGF antibody |
| Renal cell carcinoma (RCC) | ||
| Non-small cell lung cancer (NSCLC) | ||
| Ovarian | ||
| Ramucirumab (Cyramza) | Advanced stomach | VEGFR2 antibody |
| Gastroesophageal adenocarcinoma | ||
| NSCLC | ||
| Metastatic colorectal | ||
| Cetuximab (Erbitux) | CRC | EGFR antibody |
| Head and neck squamous cell carcinoma | ||
| NSCLC | ||
| Panitumumab (Vectibix) | CRC | EGFR antibody |
| Trastuzumab (Herceptin) | HER2+ breast cancer | EGFR antibody |
| Aflibercept (Zaltrap or Eylea) | Colorectal cancer (CRC) | VEGF-trap * |
| Sunitinib (Sotent) | RCC | TKI |
| Pancreatic neuroendocrine tumors (PNETs) | ||
| Gastrointestinal stromal tumors (GISTs) | ||
| Axitinib (Inlyta) | RCC | TKI |
| Vandetanib (Caprelsa) | Medullary carcinoma of thyroid | TKI |
| Lenvatinib (Lenvima) | Thyroid | TKI |
| Pazopanib (Votrient) | RCC | TKI |
| Soft tissue sarcoma | ||
| Cabozantinib (Cometriq) | Medullary thyroid cancer | TKI |
| Erlotinib (Tarceva) | Pancreatic cancer | TKI |
| Gefitinib (Iressa) | Lung cancer | TKI |
| Breast cancer | ||
| Imatinib (Glivec) | Chronic myeloid leukemia | TKI |
| Acute lymphoid leukemia | ||
| GISTs | ||
| Lapatinib (Tyverb) | HER2+ breast cancer | TKI |
| Sorafenib (Nexavar) | Hepatocellular carcinoma | TKI |
| RCC | ||
| Thyroid | ||
| Regorafenib (Stivarga) | Refractory metastatic colorectal | TKI |
| Advanced GISTs | ||
| Thalidomide (Thalomid) | Multiple myeloma | Immunomodulator |
| Lenalidomide (Revlimid) | Multiple myeloma | Immunomodulator |
| Non-Hodgkin lymphoma | ||
| Rapamycin (Sirolimus) | RCC | mTOR inhibitor |
| Temsirolimus (Torisel) | RCC | mTOR inhibitor |
| Everolimus (Afinitor) | RCC | mTOR inhibitor |
| Advanced breast | ||
| PNETs |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, M.P.; Sotomayor, P.; Carrasco-Avino, G.; Corvalan, A.H.; Owen, G.I. Escaping Antiangiogenic Therapy: Strategies Employed by Cancer Cells. Int. J. Mol. Sci. 2016, 17, 1489. https://doi.org/10.3390/ijms17091489
Pinto MP, Sotomayor P, Carrasco-Avino G, Corvalan AH, Owen GI. Escaping Antiangiogenic Therapy: Strategies Employed by Cancer Cells. International Journal of Molecular Sciences. 2016; 17(9):1489. https://doi.org/10.3390/ijms17091489
Chicago/Turabian StylePinto, Mauricio P., Paula Sotomayor, Gonzalo Carrasco-Avino, Alejandro H. Corvalan, and Gareth I. Owen. 2016. "Escaping Antiangiogenic Therapy: Strategies Employed by Cancer Cells" International Journal of Molecular Sciences 17, no. 9: 1489. https://doi.org/10.3390/ijms17091489