
Glycerin-Induced Conformational Changes in Bombyx mori Silk Fibroin Film Monitored by 13C CP/MAS NMR and 1H DQMAS NMR

Abstract
:
1. Introduction
2. Results and Discussion
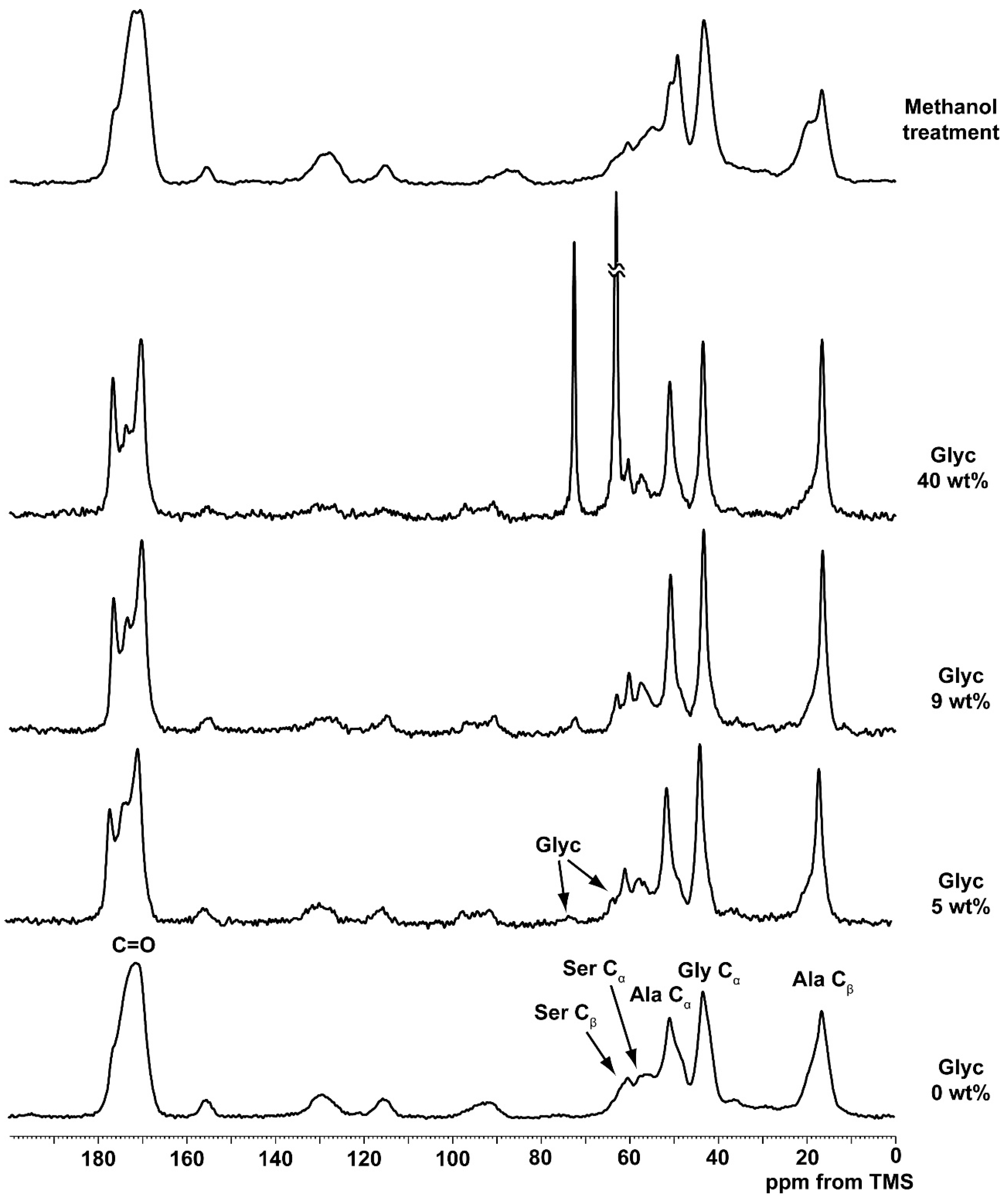
2.1. 13C Cross Polarization/Magic Angle Spinning Nuclear Magnetic Resonance (CP/MAS NMR) Spectra of Silk Fibroin (SF) and Glycerin (Glyc)-Blended SF Films
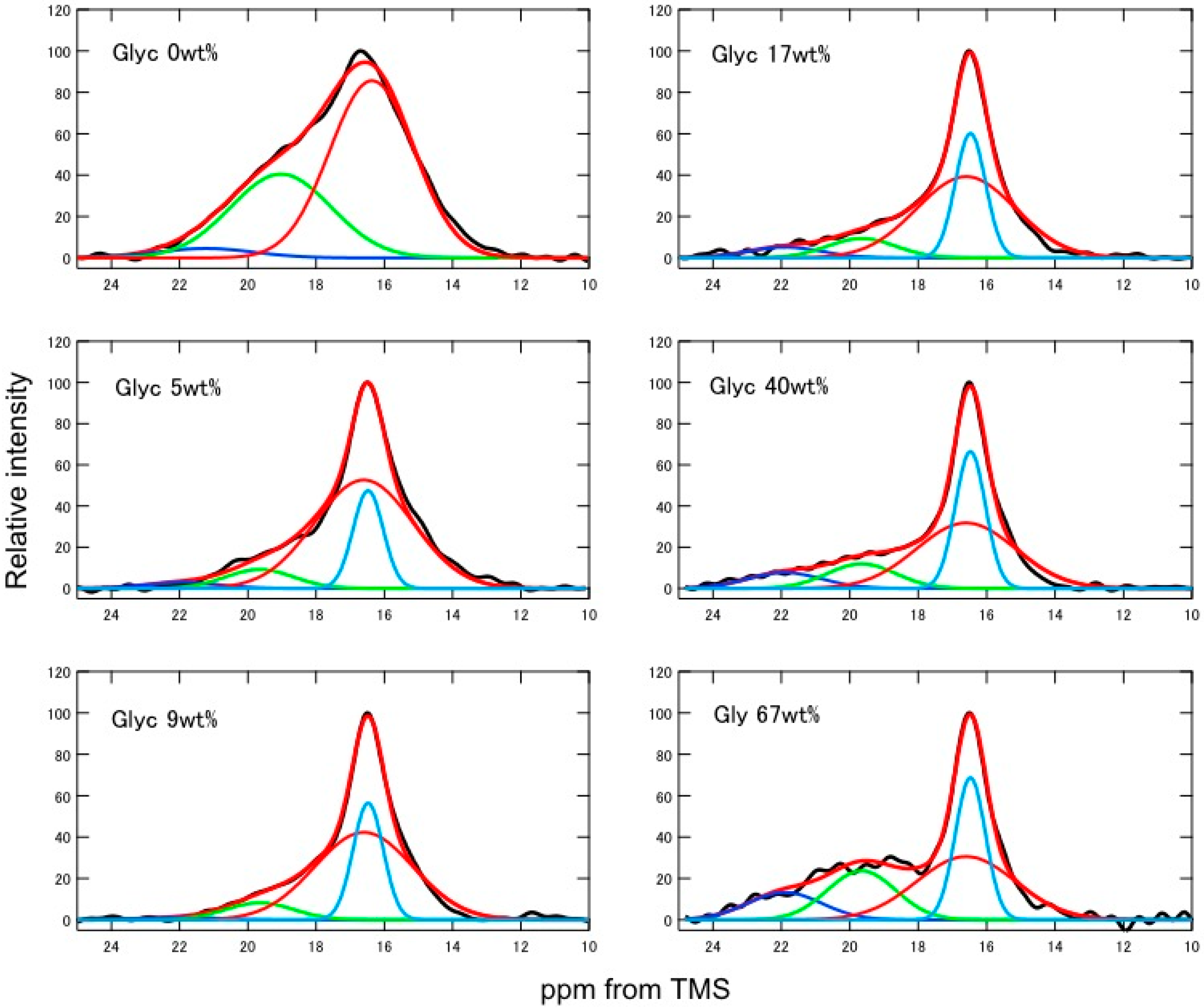
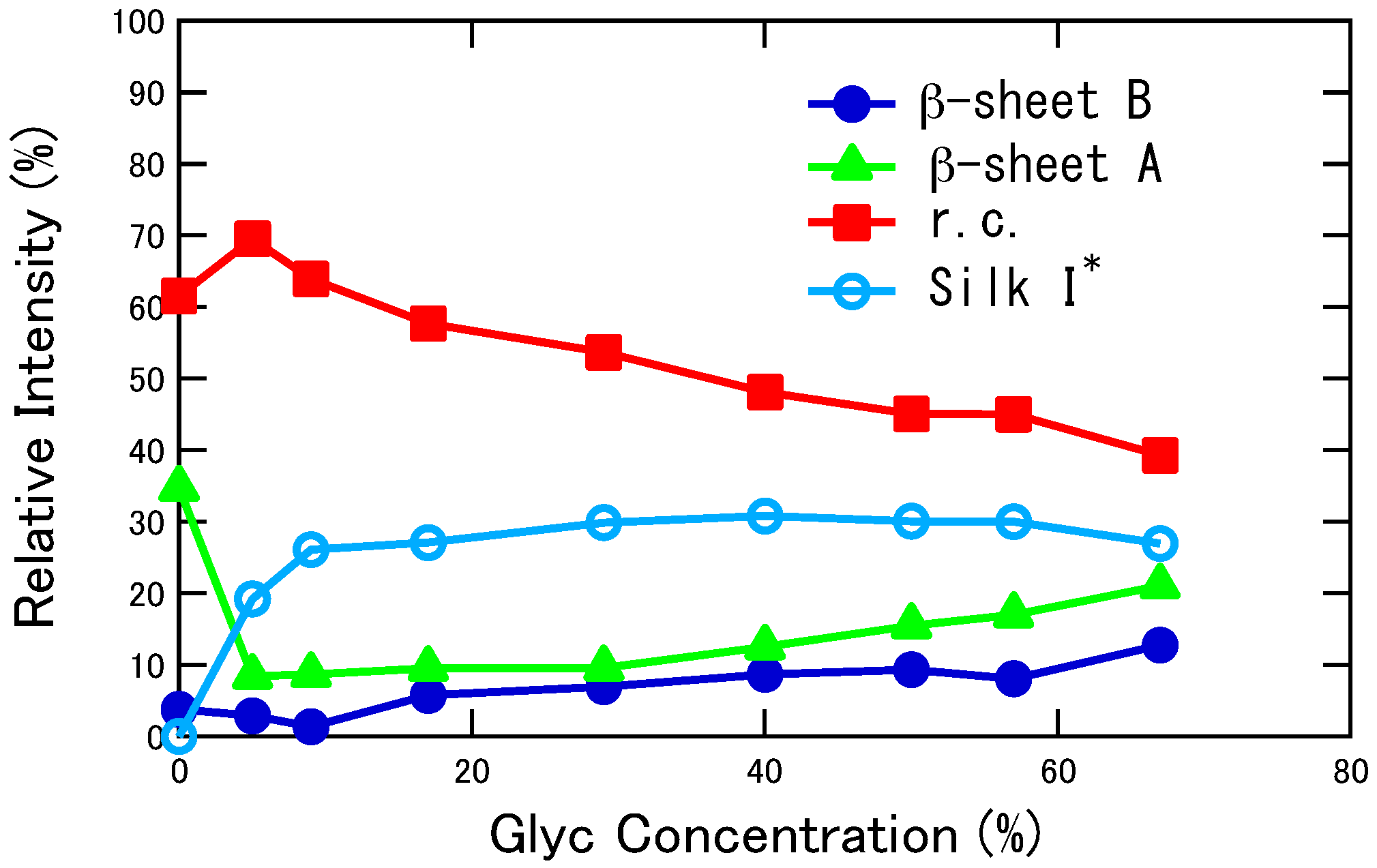
2.2. Quantitative Conformational Analysis of SF and Glyc-Blended SF from the Ala Cβ Peaks of the 13C CP/MAS NMR Spectra
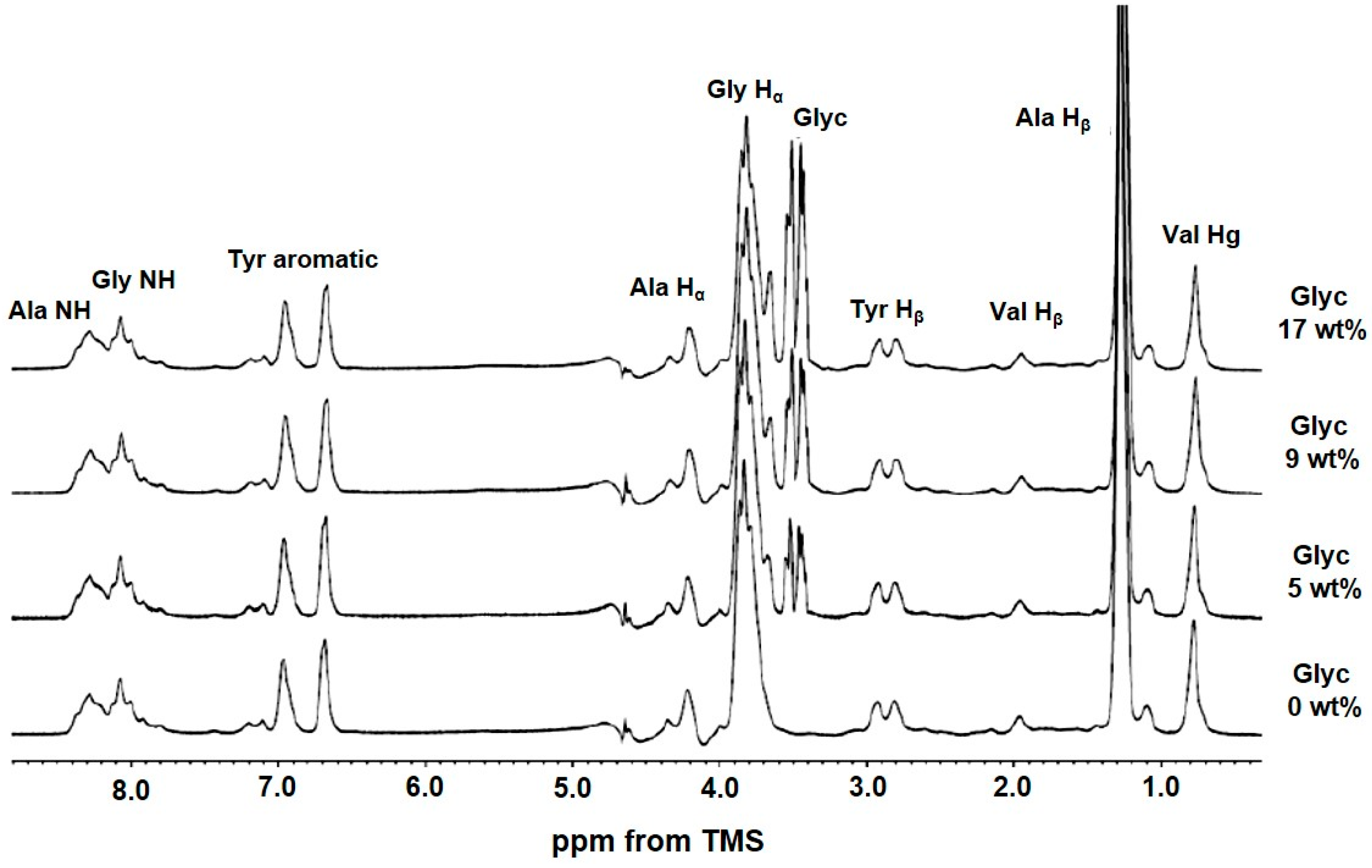
2.3. 1H Solution NMR Spectra of Regenerated SF Aqueous Solution as a Function of Glyc Concentration
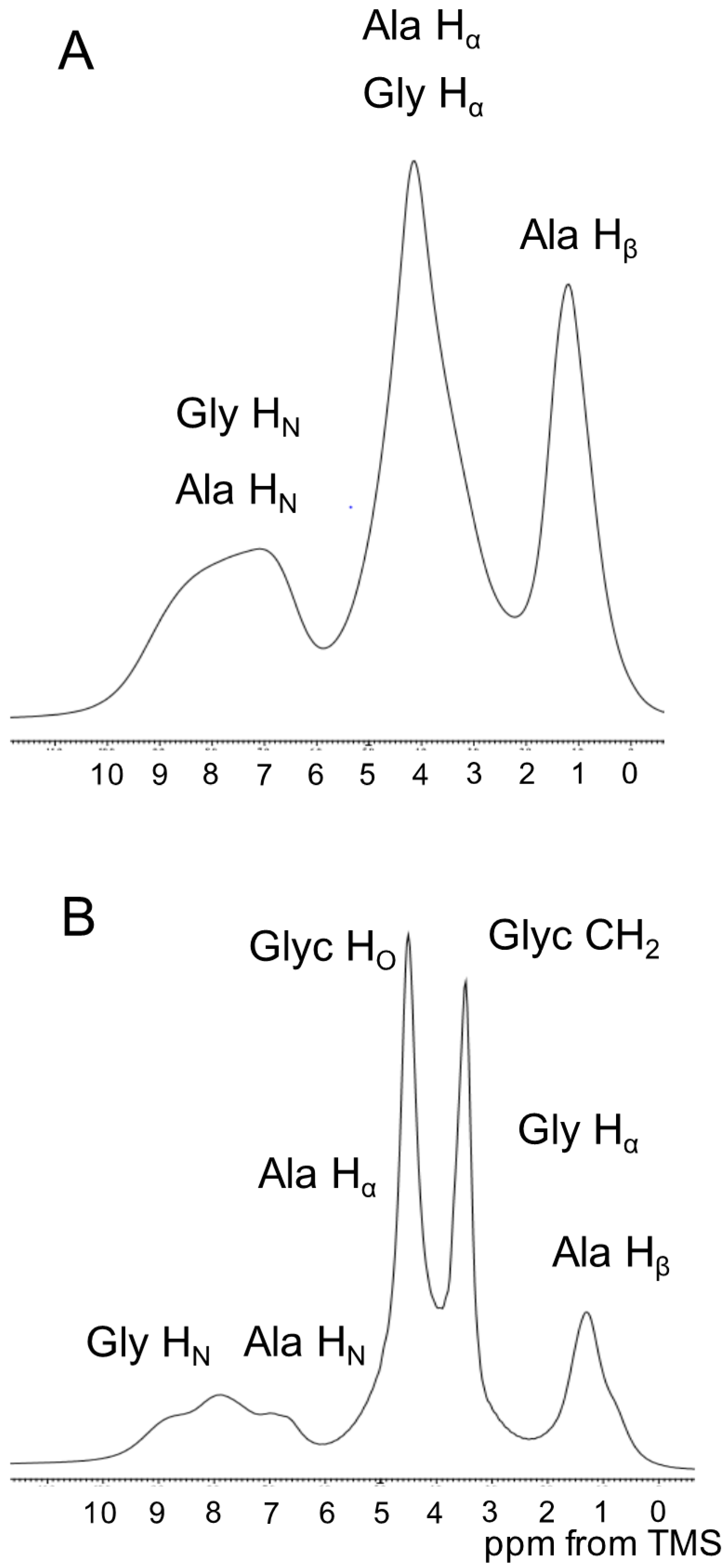
2.4. 1H Solid State NMR Spectra of SF and Glyc (29 wt %)-Blended SF Films
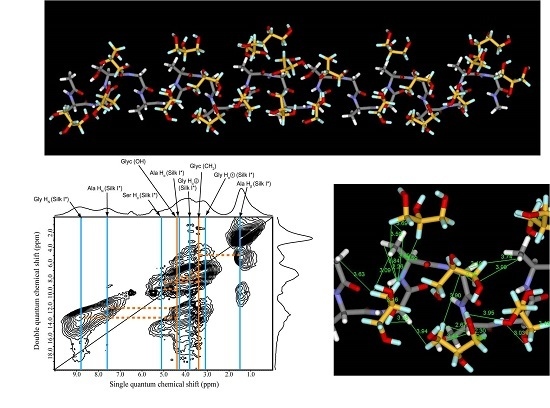
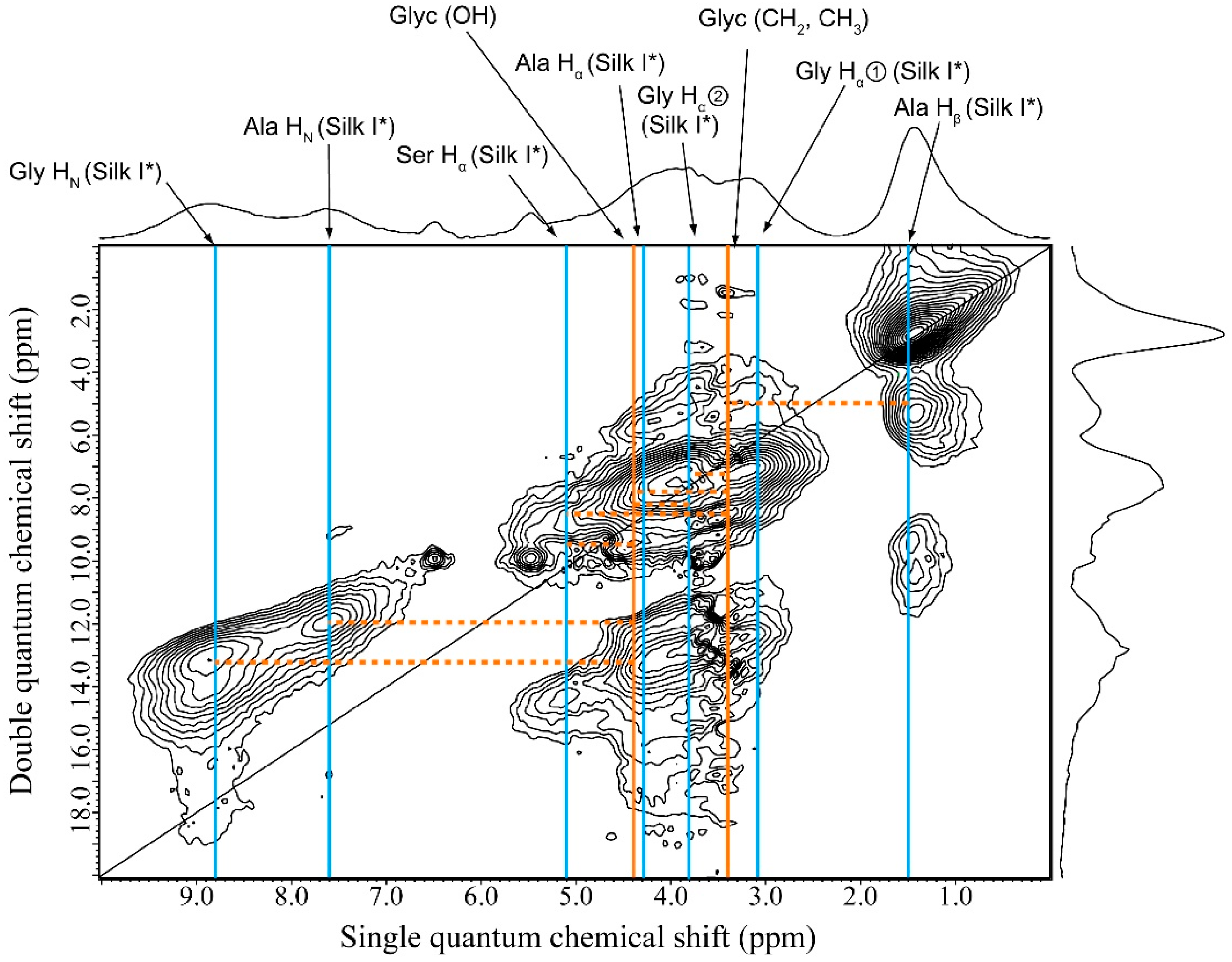
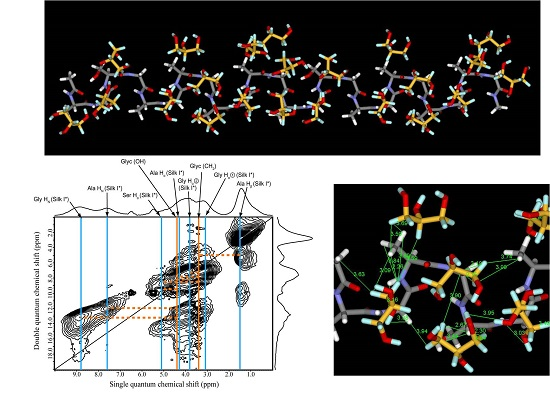
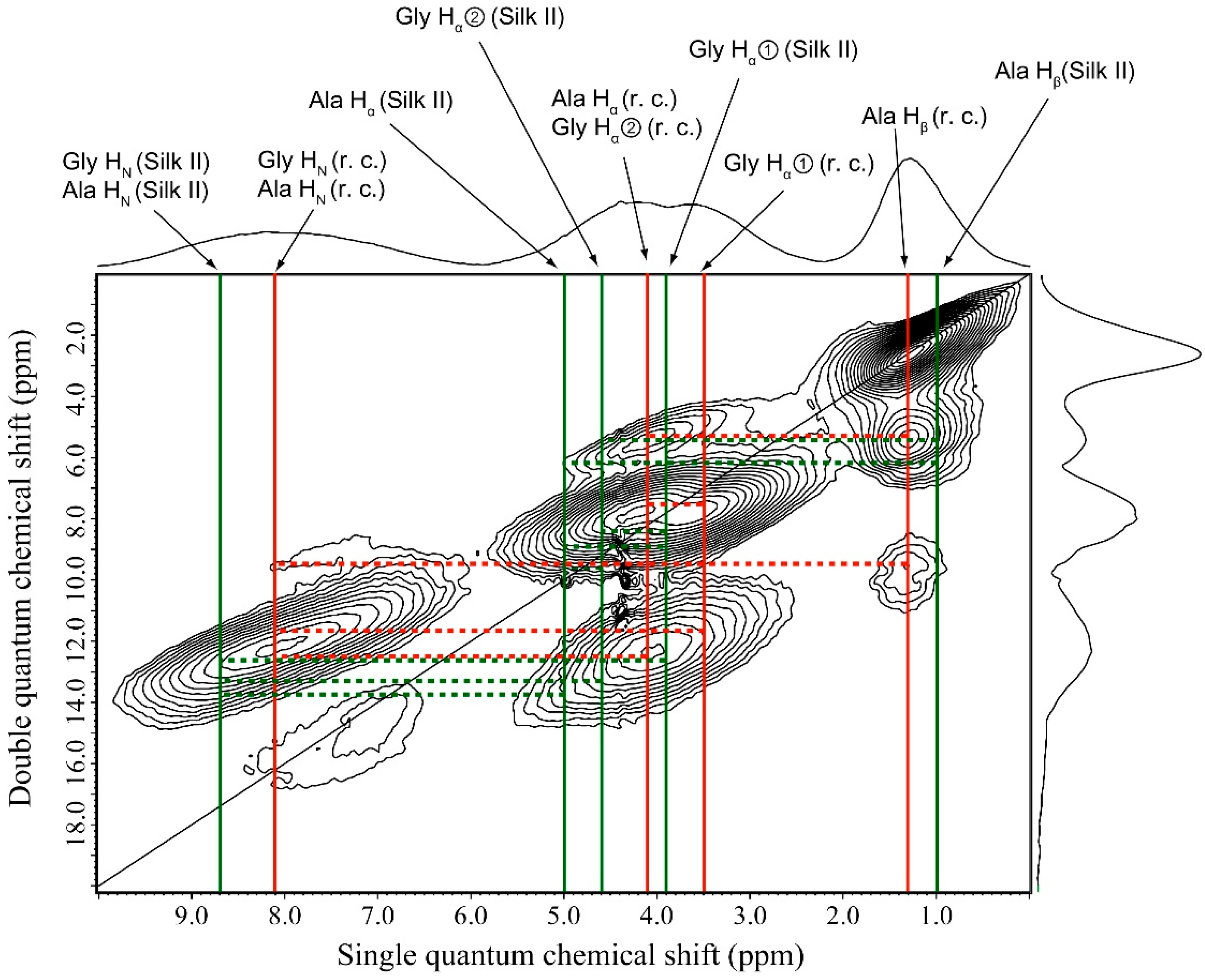
2.5. 1H Double-Quantum Magic Angle Spinning (DQMAS) NMR Spectrum of SF Film
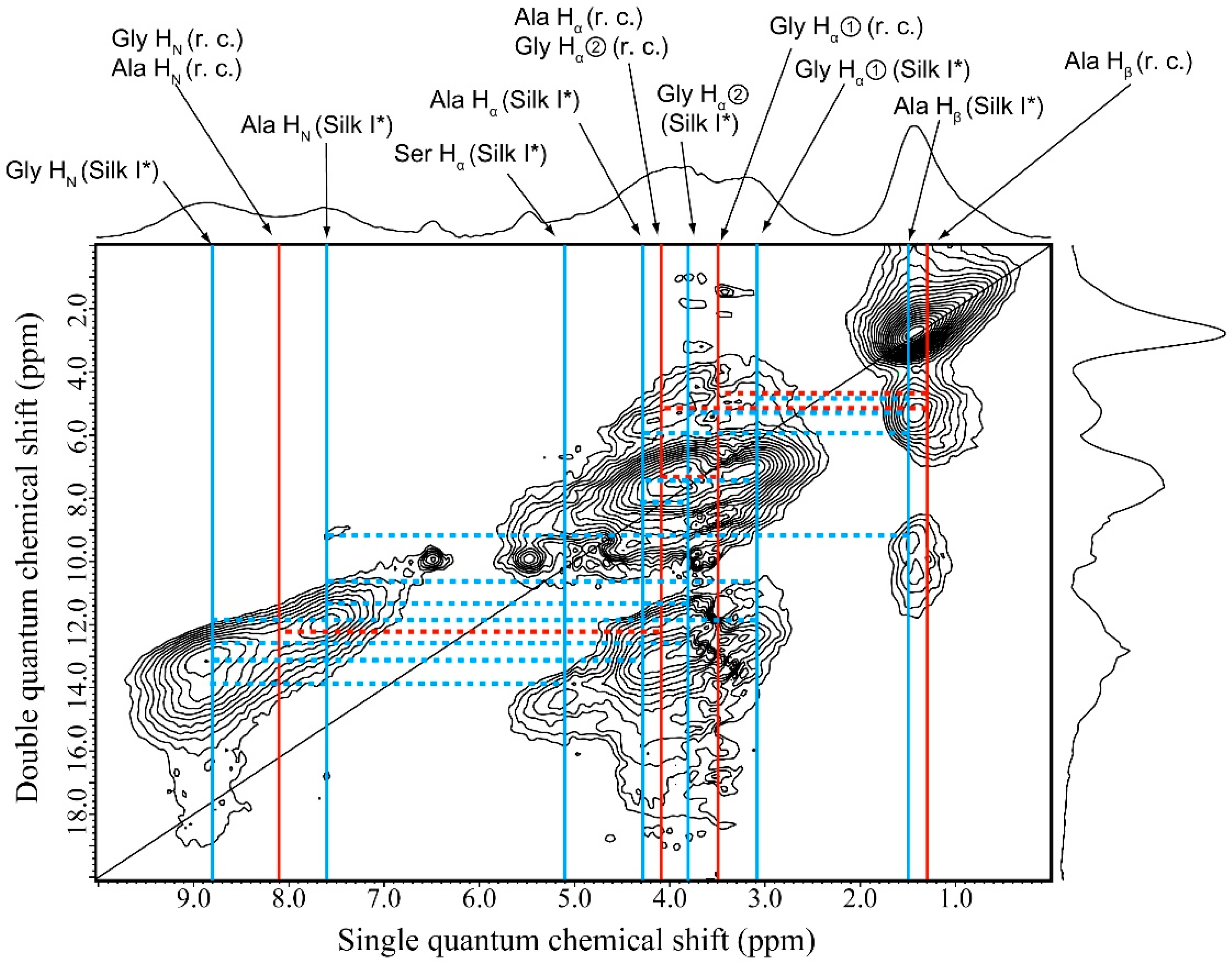
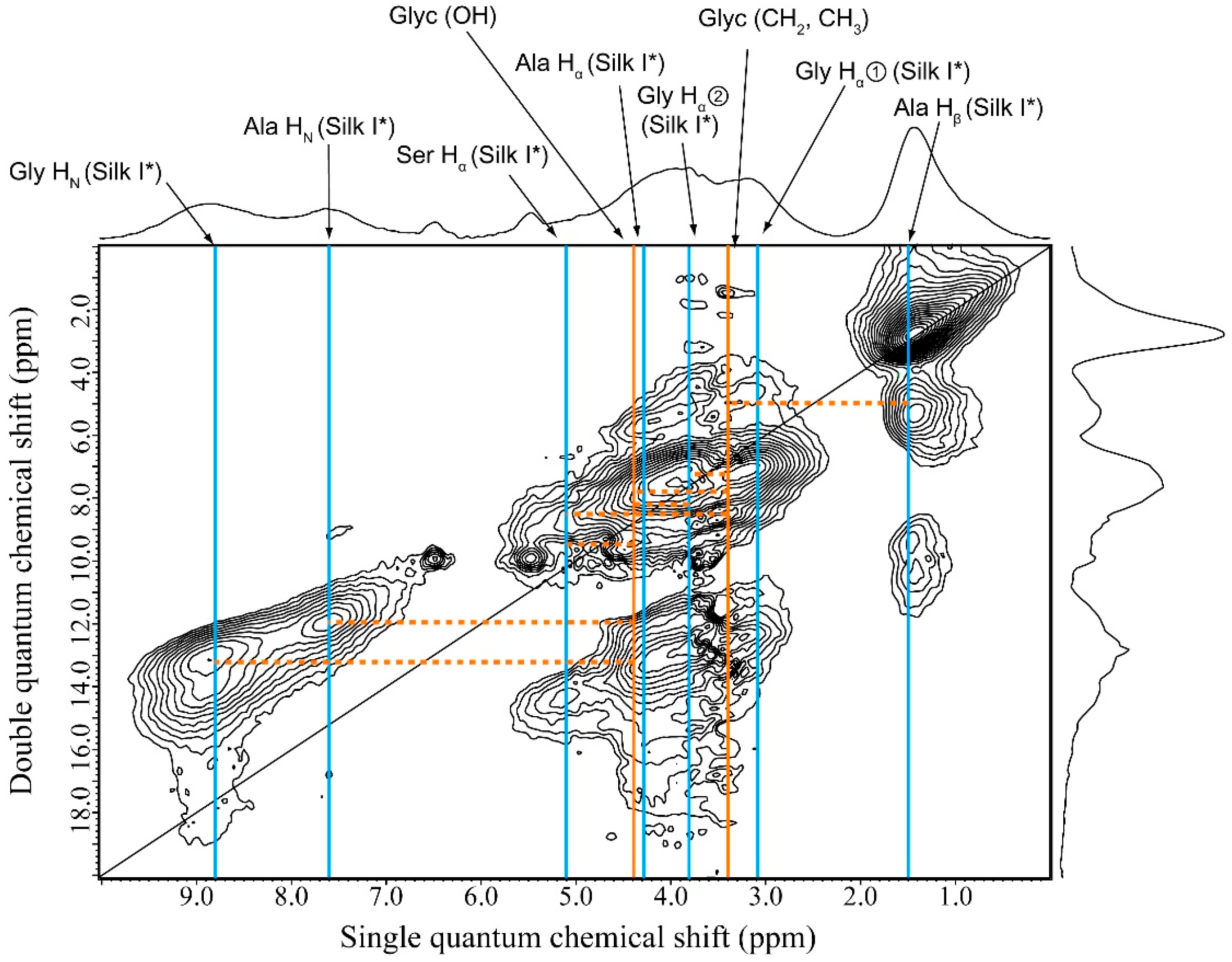
2.6. 1H DQMAS NMR Spectrum of Glyc (29 wt %)-Blended SF Film
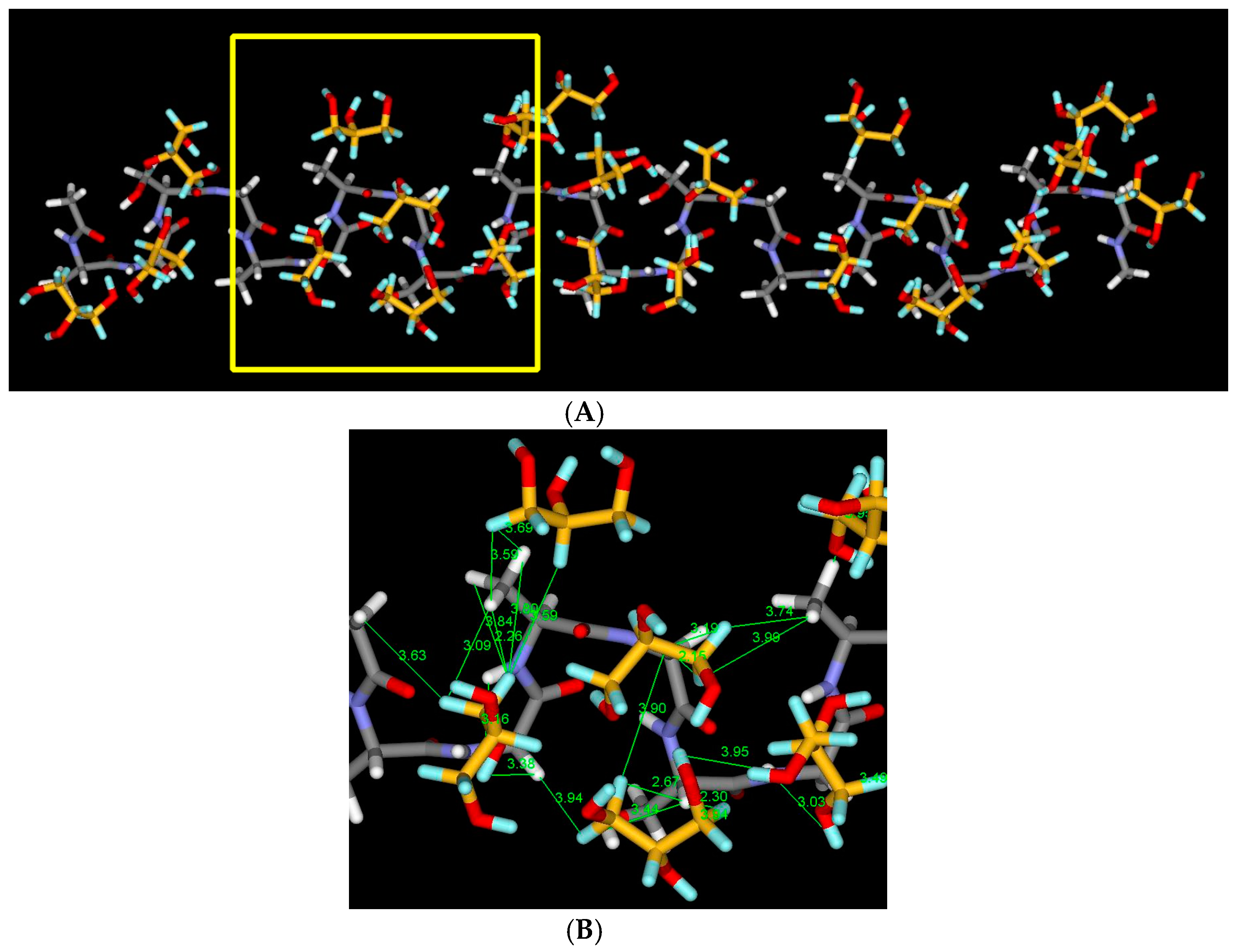
2.7. Structural Model of Glyc-SF Complex Having Silk I* Form
3. Materials and Methods
3.1. Preparation of Glyc-Blended SF Films
3.2. 13C CP/MAS NMR of Glyc-Blended SF Films
3.3. Deconvolution Analysis of 13C CP/MAS NMR Spectra
3.4. Solid State DQMAS 1H NMR
3.5. 1H Solution NMR of Regenerated SF Aqueous Solution
3.6. Model Building of Glyc-SF with Silk I* Form by Molecular Dynamics (MD) Simulation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Asakura, T.; Miller, T. Biotechnology of Silk; Springer: Dordrecht, The Netherlands, 2014. [Google Scholar]
- Vollrath, F.; Porter, D. Spider silk as archetypal protein elastomer. Soft Matter 2006, 2, 377–385. [Google Scholar] [CrossRef]
- Altman, G.H.; Diaz, F.; Jakuba, C.; Calabro, T.; Horan, R.L.; Chen, J.; Lu, H.; Richmond, J.; Kaplan, D.L. Silk-based biomaterials. Biomaterials 2003, 24, 401–416. [Google Scholar] [CrossRef]
- Gronau, G.; Krishnaji, S.T.; Kinahan, M.E.; Giesa, T.; Wong, J.Y.; Kaplan, D.L.; Buehler, M.J. A review of combined experimental and computational procedures for assessing biopolymer structure-process-property relationships. Biomaterials 2012, 33, 8240–8255. [Google Scholar] [CrossRef] [PubMed]
- Koh, L.D.; Cheng, Y.; Teng, C.P.; Khin, Y.W.; Loh, X.J.; Tee, S.Y.; Low, M.; Ye, E.; Yu, H.D.; Zhang, Y.W.; et al. Structures, mechanical properties and applications of silk fibroin materials. Prog. Polym. Sci. 2015, 46, 86–100. [Google Scholar] [CrossRef]
- Asakura, T.; Kaplan, D.L. Silk Production and Processing. In Encyclopedia of Agricultural Science; Arutzen, C.J., Ed.; Academic Press: New York, NY, USA, 1994; Volume 4, pp. 1–11. [Google Scholar]
- Cao, Y.; Wang, B. Biodegradation of Silk Biomaterials. Int. J. Mol. Sci. 2009, 10, 1514–1524. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, Y.; Furukawa, K.; Yamamoto, T. Self-Expansion Behavior of Silk Fibroin Film. Macromol. Mater. Eng. 2006, 291, 458–462. [Google Scholar] [CrossRef]
- Lu, S.; Wang, X.; Lu, Q.; Zhang, X.; Kluge, J.A.; Uppal, N.; Omenetto, F.; Kaplan, D.L. Insoluble and Flexible Silk Films Containing Glycerol. Biomacromolecules 2010, 11, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Liu, X.; Liu, S.; Lu, Q.; Liu, J.; Kaplan, D.L.; Zhu, H. A mild process to design silk scaffolds with reduced β-sheet structure and various topographies at the nanometer scale. Acta Biomater. 2015, 13, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Asakura, T.; Suzuki, Y.; Nakazawa, Y.; Holland, G.P.; Yarger, J.L. Elucidating silk structure using solid-state NMR. Soft Matter 2013, 9, 11440–11450. [Google Scholar] [CrossRef]
- Asakura, T.; Okushita, K.; Williamson, M.P. Analysis of the Structure of Bombyx mori Silk Fibroin by NMR. Macromolecules 2015, 48, 2345–2357. [Google Scholar] [CrossRef]
- Asakura, T.; Kuzuhara, A.; Tabeta, R.; Saito, H. Conformational characterization of Bombyx mori silk fibroin in the solid state by high-frequency carbon-13 cross polarization-magic angle spinning NMR, X-ray diffraction, and infrared spectroscopy. Macromolecules 1985, 18, 1841–1845. [Google Scholar] [CrossRef]
- Saito, H.; Tabeta, R.; Asakura, T.; Iwanaga, Y.; Shoji, A.; Ozaki, T.; Ando, I. High-resolution carbon-13 NMR study of silk fibroin in the solid state by the cross-polarization-magic angle spinning method. Conformational characterization of silk I and silk II type forms of Bombyx mori fibroin by the conformation-dependent carbon-13 chemical shifts. Macromolecules 1984, 17, 1405–1412. [Google Scholar]
- Van Beek, J.D.; Beaulieu, L.; Schäfer, H.; Demura, M.; Asakura, T.; Meier, B.H. Solid-state NMR determination of the secondary structure of Samia cynthia ricini silk. Nature 2000, 405, 1077–1079. [Google Scholar] [PubMed]
- Spera, S.; Bax, A. Empirical correlation between protein backbone conformation and Cα and Cβ 13C nuclear magnetic resonance chemical shifts. J. Am. Chem. Soc. 1991, 113, 5490–5492. [Google Scholar] [CrossRef]
- Asakura, T.; Iwadate, M.; Demura, M.; Williamson, M.P. Structural analysis of silk with 13C NMR chemical shift contour plots. Int. J. Biol. Macromol. 1999, 24, 167–171. [Google Scholar] [CrossRef]
- Wishart, D.S.; Sykes, B.D.; Richards, F.M. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 1991, 222, 311–333. [Google Scholar] [CrossRef]
- Asakura, T.; Taoka, K.; Demura, M.; Williamson, M.P. The relationship between amide proton chemical shifts and secondary structure in proteins. J. Biomol. NMR 1995, 6, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Asakura, T.; Ashida, J.; Yamane, T.; Kameda, T.; Nakazawa, Y.; Ohgo, K.; Komatsu, K. A repeated β-turn structure in poly(Ala-Gly) as a model for silk I of Bombyx mori silk fibroin studied with two-dimensional spin-diffusion NMR under off magic angle spinning and rotational echo double resonance. J. Mol. Biol. 2001, 306, 291–305. [Google Scholar] [CrossRef] [PubMed]
- Asakura, T.; Ohgo, K.; Komatsu, K.; Kanenari, M.; Okuyama, K. Refinement of Repeated β-turn Structure for Silk I Conformation of Bombyx mori Silk Fibroin Using 13C Solid-State NMR and X-ray Diffraction Methods. Macromolecules 2005, 38, 7397–7403. [Google Scholar] [CrossRef]
- Asakura, T.; Ohata, T.; Kametani, S.; Okushita, K.; Yazawa, K.; Nishiyama, Y.; Nishimura, K.; Aoki, A.; Suzuki, F.; Kaji, H.; et al. Intermolecular Packing in B. mori Silk Fibroin: Multinuclear NMR Study of the Model Peptide (Ala-Gly)15 Defines a Heterogeneous Antiparallel Antipolar Mode of Assembly in the Silk II Form. Macromolecules 2015, 48, 28–36. [Google Scholar] [CrossRef]
- Jenkins, J.E.; Creager, M.S.; Lewis, R.V.; Holland, G.P.; Yarger, J.L. Quantitative Correlation between the Protein Primary Sequences and Secondary Structures in Spider Dragline Silks. Biomacromolecules 2010, 11, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Schnell, I.; Brown, S.P.; Low, H.Y.; Ishida, H.; Spiess, H.W. An Investigation of Hydrogen Bonding in Benzoxazine Dimers by Fast Magic-Angle Spinning and Double-Quantum 1H NMR Spectroscopy. J. Am. Chem. Soc. 1998, 120, 11784–11795. [Google Scholar] [CrossRef]
- Yates, J.R.; Pham, T.N.; Pickard, C.J.; Mauri, F.; Amado, A.M.; Gil, A.M.; Brown, S.P. An Investigation of Weak CH···O Hydrogen Bonds in Maltose Anomers by a Combination of Calculation and Experimental Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2005, 127, 10216–10220. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R.; Pickard, C.J.; Mauri, F. Calculation of NMR chemical shifts for extended systems using ultrasoft pseudopotentials. Phys. Rev. B 2007, 76, 024401. [Google Scholar] [CrossRef]
- Brown, S.P. Probing proton–proton proximities in the solid state. Prog. Nucl. Magn. Reson. Spectrosc. 2007, 50, 199–251. [Google Scholar] [CrossRef]
- Bradley, J.P.; Tripon, C.; Filip, C.; Brown, S.P. Determining relative proton–proton proximities from the build-up of two-dimensional correlation peaks in 1H double-quantum MAS NMR: Insight from multi-spin density-matrix simulations. Phys. Chem. Chem. Phys. 2009, 11, 6941–6952. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.K.; Hodgkinson, P.; Zorin, V.; Dumez, J.N.; Herrmann, B.E.; Emsley, L.; Salager, E.; Stein, R.S. Computation and NMR crystallography of terbutaline sulfate. Magn. Reson. Chem. 2010, 48, S103–S112. [Google Scholar] [CrossRef] [PubMed]
- Yazawa, K.; Suzuki, F.; Nishiyama, Y.; Ohata, T.; Aoki, A.; Nishimura, K.; Kaji, H.; Shimizu, T.; Asakura, T. Determination of accurate 1H positions of an alanine tripeptide with anti-parallel and parallel β-sheet structures by high resolution 1H solid state NMR and GIPAW chemical shift calculation. Chem. Commun. 2012, 48, 11199–11201. [Google Scholar] [CrossRef] [PubMed]
- Asakura, T.; Yazawa, K.; Horiguchi, K.; Suzuki, F.; Nishiyama, Y.; Nishimura, K.; Kaji, H. Difference in the structures of alanine tri- and tetra-peptides with antiparallel β-sheet assessed by X-ray diffraction, solid-state NMR and chemical shift calculations by GIPAW. Biopolymers 2014, 10, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Asakura, T.; Suzuki, Y.; Yazawa, K.; Aoki, A.; Nishiyama, Y.; Nishimura, K.; Suzuki, F.; Kaji, H. Determination of Accurate 1H Positions of (Ala-Gly)n as a Sequential Peptide Model of Bombyx mori Silk Fibroin before Spinning (Silk I). Macromolecules 2013, 46, 8046–8050. [Google Scholar] [CrossRef]
- Asakura, T.; Demura, M.; Date, T.; Miyashita, N.; Ogawa, K.; Williamson, M.P. NMR study of silk I structure of Bombyx mori silk fibroin with 15N- and 13C-NMR chemical shift contour plots. Biopolymers 1996, 41, 193–203. [Google Scholar] [CrossRef]
- Asakura, T.; Sato, Y.; Aoki, A. Stretching-Induced Conformational Transition of the Crystalline and Noncrystalline Domains of 13C-Labeled Bombyx mori Silk Fibroin Monitored by Solid State NMR. Macromolecules 2015, 48, 5761–5769. [Google Scholar] [CrossRef]
- Asakura, T.; Suzuki, H.; Watanabe, Y. Conformational characterization of silk fibroin in intact Bombyx mori and Philosamia cynthia ricini silkworms by carbon-13 NMR spectroscopy. Macromolecules 1983, 16, 1024–1026. [Google Scholar] [CrossRef]
- Suzuki, Y.; Yamazaki, T.; Aoki, A.; Shindo, H.; Asakura, T. NMR Study of the Structures of Repeated Sequences, GAGXGA (X = S, Y, V), in Bombyx mori Liquid Silk. Biomacromolecules 2014, 15, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.Z.; Confalonieri, F.; Jacquet, M.; Perasso, R.; Li, Z.G.; Janin, J. Silk fibroin: Structural implications of a remarkable amino acid sequence. Proteins 2001, 44, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Percot, A.; Colomban, P.; Paris, C.; Dinh, H.M.; Wojcieszak, M.; Mauchamp, B. Water dependent structural changes of silk from Bombyx mori gland to fibre as evidenced by Raman and IR spectroscopies. Vib. Spectrosc. 2014, 73, 79–89. [Google Scholar] [CrossRef]
- Colomban, P.; Dinh, H.M.; Riand, J.; Prinsloo, L.C.; Mauchamp, B. Nanomechanics of single silkworm and spider fibres: A Raman and micro-mechanical in situ study of the conformation change with stress. J. Raman Spectrosc. 2008, 39, 1749–1764. [Google Scholar] [CrossRef]
- Asakura, T. Structure of Bombyx mori silk fibroin in aqueous solution. Makromol. Chem. Rapid Commun. 1986, 7, 755–759. [Google Scholar] [CrossRef]
- Asakura, T.; Ashida, J.; Yamane, T. Structure of Bombyx mori Silk Fibroin before Spinning in Silkworm. In NMR Spectroscopy of Polymers in Solution and in the Solid State; Cheng, H.N., English, A.D., Eds.; American Chemical Society: Washington, DC, USA, 2004; pp. 71–82. [Google Scholar]
- Takahashi, Y.; Gehoh, M.; Yuzuriha, K. Structure refinement and diffuse streak scattering of silk (Bombyx mori). Int. J. Biol. Macromol. 1999, 24, 127–138. [Google Scholar] [CrossRef]
- Asakura, T.; Watanabe, Y.; Uchida, A.; Minagawa, H. NMR of silk fibroin. Carbon-13 NMR study of the chain dynamics and solution structure of Bombyx mori silk fibroin. Macromolecules 1984, 17, 1075–1081. [Google Scholar] [CrossRef]
- Yoshimizu, H.; Asakura, T. The structure of Bombyx mori silk fibroin membrane swollen by water studied with ESR, 13C-NMR, and FT-IR spectroscopies. J. Appl. Polym. Sci. 1990, 40, 1745–1756. [Google Scholar] [CrossRef]
- Asakura, T.; Yao, J.; Yamane, T.; Umemura, K.; Ulrich, A.S. Heterogeneous Structure of Silk Fibers from Bombyx mori Resolved by 13C Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2002, 124, 8794–8795. [Google Scholar] [CrossRef] [PubMed]
- Asakura, T.; Yao, J. 13C CP/MAS NMR study on structural heterogeneity in Bombyx mori silk fiber and their generation by stretching. Protein Sci. 2002, 11, 2706–2713. [Google Scholar] [CrossRef] [PubMed]
- Asakura, T.; Isobe, K.; Aoki, A.; Kametani, S. Conformation of Crystalline and Noncrystalline Domains of [3–13C]Ala-, [3–13C]Ser-, and [3–13C]Tyr-Bombyx mori Silk Fibroin in a Hydrated State Studied with 13C DD/MAS NMR. Macromolecules 2015, 48, 8062–8069. [Google Scholar] [CrossRef]
- Yamauchi, K.; Yamasaki, S.; Takahashi, R.; Asakura, T. Microscopic structural analysis of fractured silk fibers from Bombyx mori and Samia cynthia ricini using 13C CP/MAS NMR with a 1 mm microcoil MAS NMR probehead. Solid State Nucl. Mag. 2010, 38, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, M.; Fayon, F.; Cadars, S.; Rollet, A.L.; Massiot, D. 1H and 19F ultra-fast MAS double-quantum single-quantum NMR correlation experiments using three-spin terms of the dipolar homonuclear Hamiltonian. Phys. Chem. Chem. Phys. 2011, 13, 8024–8030. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 13C Chemical Shift | ||||||||
| Conformation | Ala Cβ | Ala Cα | Ala CO | Gly Cα | Gly CO | Ser Cβ | ||
| r.c. | 16.7 | 50.0 | 175.5 | 42.6 | 171.1–171.5 | - | ||
| Silk II | 19.6(A), 21.7(B) | 49.2 | 172.6 | 43.0 | 169.1 | - | ||
| Silk I* | 16.5 | 51.4 | 177.0 | 43.8 | 170.7 | 60.7 | ||
| 1H Chemical Shift | ||||||||
| Conformation | Ala Hβ | Ala Hα | Ala HN | Gly Hα① | Gly Hα② | Gly HN | Ser Hα | |
| r.c. | 1.3 | 4.1 | 8.1 | 3.5 | 4.1 | 8.1 | - | |
| Silk II | 1.0 | 5.0 | 8.7 | 3.9 | 4.6 | 8.7 | - | |
| Silk I* | 1.5 | 4.3 | 7.6 | 3.8 | 3.1 | 8.8 | 5.1 | |
| SF Film | |||
| r.c. | Silk II | ||
| Ala Hβ—Ala Hα/Gly Hα② | Ala Hβ—Gly Hα② | ||
| Ala Hβ—Gly Hα① | Ala Hβ—Ala Hα | ||
| Ala Hβ—Ala HN/Gly HN | Gly Hα①—Gly Hα② | ||
| Gly Hα①—Ala Hα/Gly Hα② | Gly Hα①—Ala Hα | ||
| Gly Hα①—Ala HN/Gly HN | Gly Hα①—Gly HN/Ala HN | ||
| Ala Hα/Gly Hα②—Ala HN/Gly HN | Ala Hα—Gly Hα② | ||
| - | Gly Hα②–Gly HN/Ala HN | ||
| - | Ala Hα—Gly HN/Ala HN | ||
| Glyc-Blend SF Film | |||
| r.c. | Silk I* | Glyc—Silk I* | |
| Ala Hβ—Ala Hα/Gly Hα② | Ala Hβ—Gly Hα② | Glyc (CH2)—Ala Hβ | |
| Ala Hβ—Gly Hα① | Ala Hβ—Gly Hα① | Glyc (CH2)—Gly Hα① | |
| Gly Hα①—Ala Hα/Gly Hα② | Ala Hβ—Ala Hα | Glyc (CH2)—Ser Hα | |
| Ala Hα—Ala HN/Gly HN | Ala Hβ—Ala HN | Glyc (OH)—Gly Hα① | |
| - | Gly Hα②—Ala Hα | Glyc (OH)—Ser Hα | |
| - | Gly Hα②—Ala HN | Glyc (OH)—Ala HN | |
| - | Gly Hα②—Gly HN | Glyc (OH)—Gly HN | |
| - | Gly Hα①—Ala Hα | - | |
| - | Gly Hα①—Ala HN | - | |
| - | Gly Hα①—Gly HN | - | |
| - | Ser Hα—Gly HN | - | |
| - | Ala Hα—Gly HN | - | |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asakura, T.; Endo, M.; Hirayama, M.; Arai, H.; Aoki, A.; Tasei, Y. Glycerin-Induced Conformational Changes in Bombyx mori Silk Fibroin Film Monitored by 13C CP/MAS NMR and 1H DQMAS NMR. Int. J. Mol. Sci. 2016, 17, 1517. https://doi.org/10.3390/ijms17091517
Asakura T, Endo M, Hirayama M, Arai H, Aoki A, Tasei Y. Glycerin-Induced Conformational Changes in Bombyx mori Silk Fibroin Film Monitored by 13C CP/MAS NMR and 1H DQMAS NMR. International Journal of Molecular Sciences. 2016; 17(9):1517. https://doi.org/10.3390/ijms17091517
Chicago/Turabian StyleAsakura, Tetsuo, Masanori Endo, Misaki Hirayama, Hiroki Arai, Akihiro Aoki, and Yugo Tasei. 2016. "Glycerin-Induced Conformational Changes in Bombyx mori Silk Fibroin Film Monitored by 13C CP/MAS NMR and 1H DQMAS NMR" International Journal of Molecular Sciences 17, no. 9: 1517. https://doi.org/10.3390/ijms17091517