
In Vitro Assessment of the Expression and T Cell Immunogenicity of the Tumor-Associated Antigens BORIS, MUC1, hTERT, MAGE-A3 and Sp17 in Uterine Cancer

Abstract
:
1. Introduction
2. Results
2.1. TAA Expression in Normal Human Tissues and Tumor Cell Lines at mRNA Level
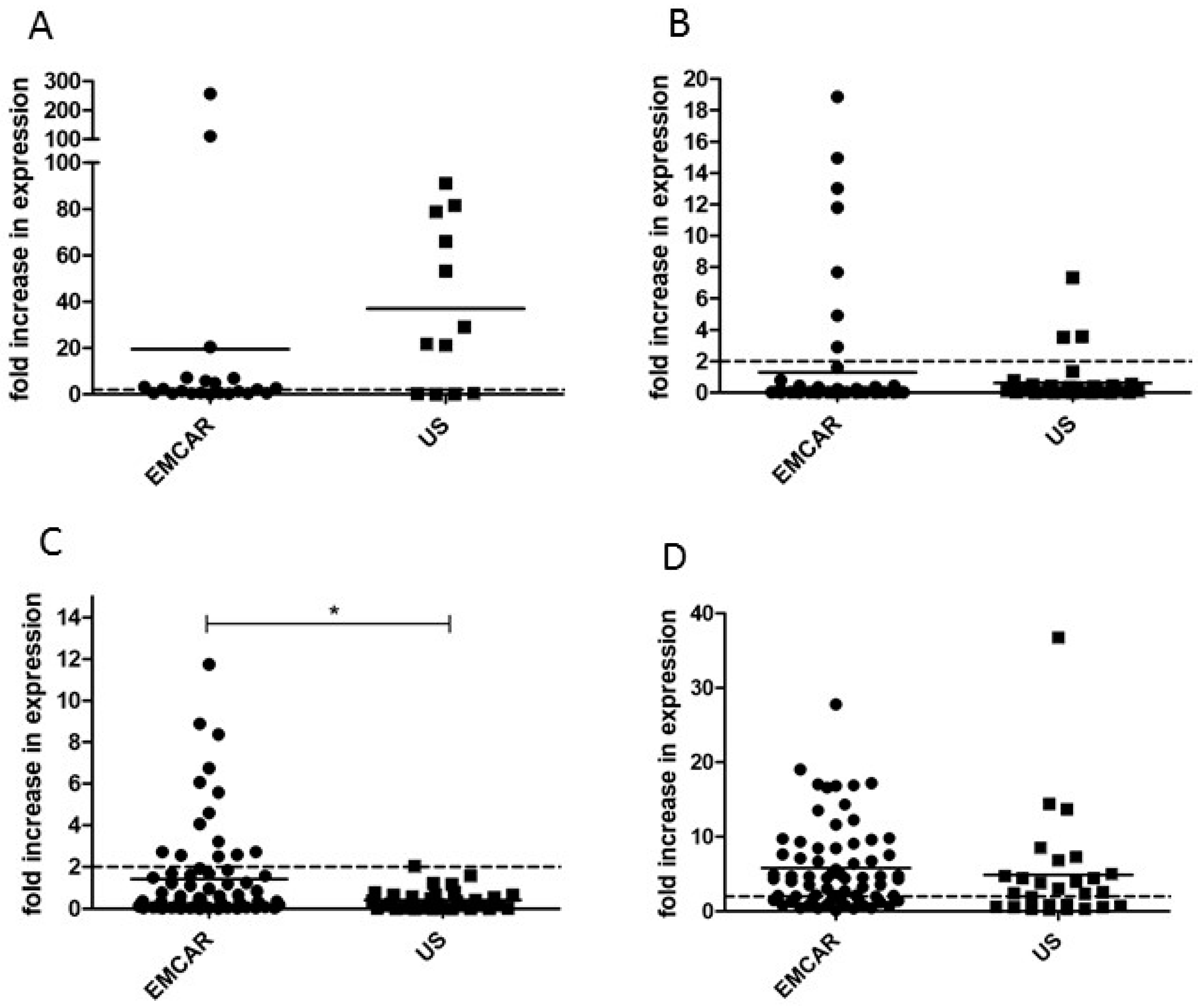
2.2. TAA Expression in Uterine Cancer at mRNA Level
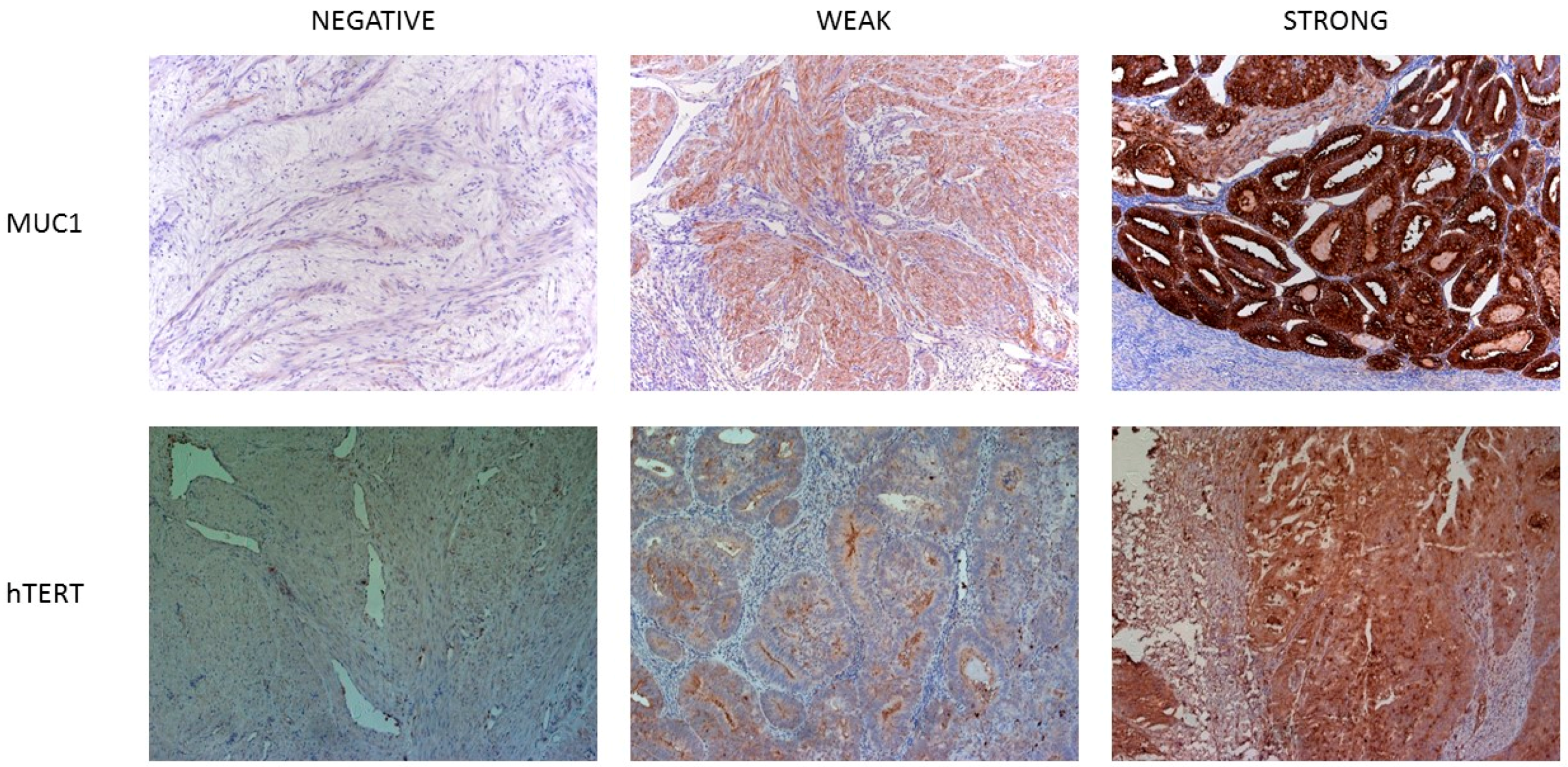
2.3. TAA Expression in Normal Human Tissues at Protein Level
2.4. TAA Expression in Uterine Cancer at Protein Level
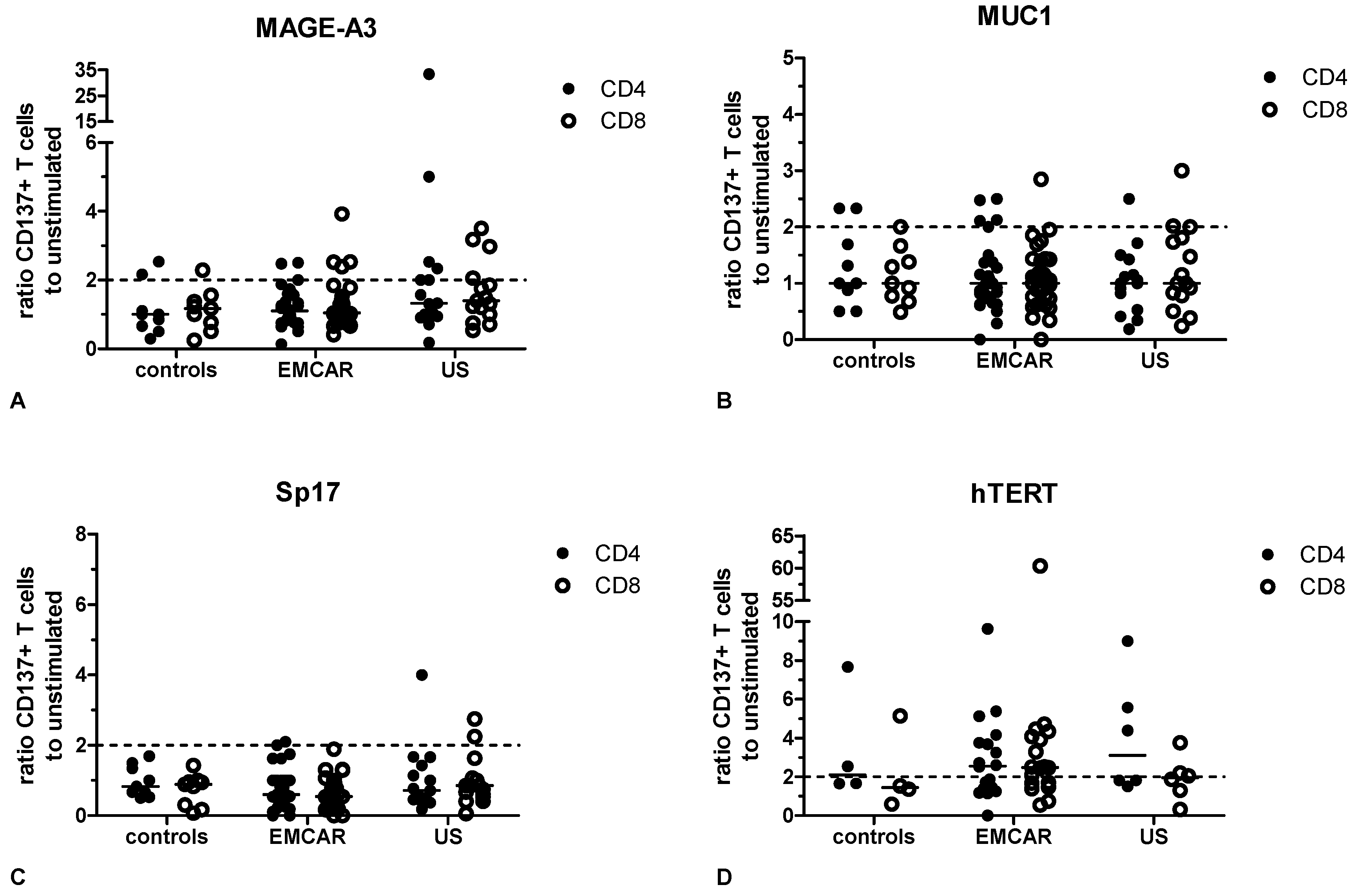
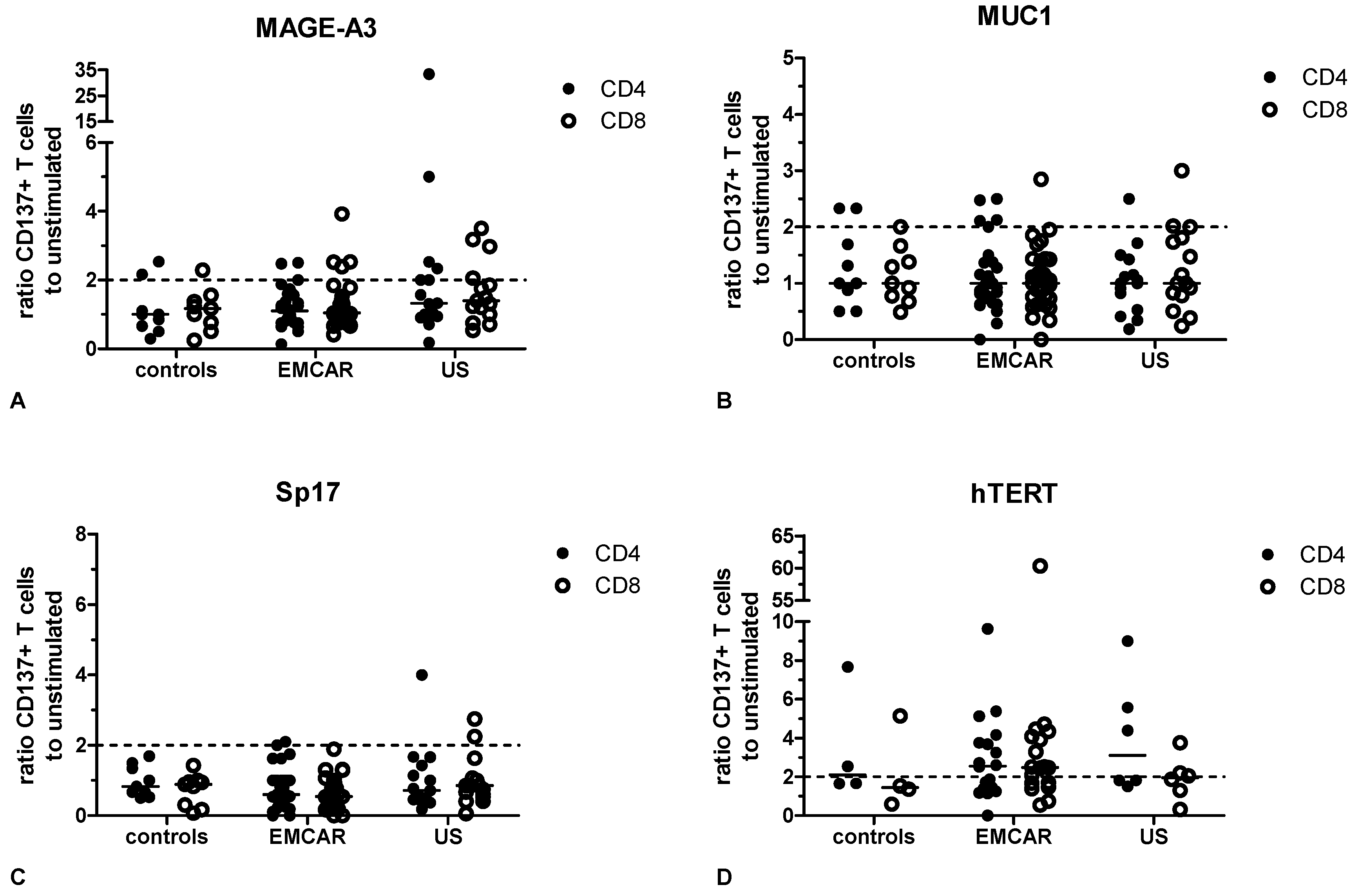
2.5. Endogenous T Cell Responses to TAA
3. Discussion
4. Experimental Section
4.1. Patient Samples, Cell Lines and Reagents
4.2. RNA Isolation and Reverse Transcription Reaction
4.3. Quantitative Real-Time PCR
4.4. Immunohistochemistry
4.5. Slide Scoring
4.6. PBMC Isolation
4.7. Detection of TAA-Specific T Cells
4.8. Flow Cytometry Staining and Analysis
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Vitale, I.; Eggermont, A.; Fridman, W.H.; Fucikova, J.; Cremer, I.; Galon, J.; Tartour, E.; Zitvogel, L.; Kroemer, G.; et al. Trial watch: Dendritic cell-based interventions for cancer therapy. Oncoimmunology 2013, 2, e25771. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, J.; van Pel, A.; Neyns, B.; Lethe, B.; Brasseur, F.; Renkvist, N.; van der Bruggen, P.; van Baren, N.; Paulus, R.; Thielemans, K.; et al. Vaccination of a melanoma patient with mature dendritic cells pulsed with MAGE-3 peptides triggers the activity of nonvaccine anti-tumor cells. J. Immunol. 2008, 180, 3585–3593. [Google Scholar] [CrossRef] [PubMed]
- Dillman, R.O.; Selvan, S.R.; Schiltz, P.M.; McClay, E.F.; Barth, N.M.; DePriest, C.; de Leon, C.; Mayorga, C.; Cornforth, A.N.; Allen, K. Phase II trial of dendritic cells loaded with antigens from self-renewing, proliferating autologous tumor cells as patient-specific antitumor vaccines in patients with metastatic melanoma: Final report. Cancer Biother. Radiopharm. 2009, 24, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Kyo, S.; Myojo, S.; Dohi, S.; Ishizaki, J.; Miyamoto, K.; Morita, S.; Sakamoto, J.; Enomoto, T.; Kimura, T.; et al. Wilms’ tumor 1 (WT1) peptide immunotherapy for gynecological malignancy. Anticancer Res. 2009, 29, 4779–4784. [Google Scholar] [PubMed]
- Wilgenhof, S.; van Nuffel, A.M.; Corthals, J.; Heirman, C.; Tuyaerts, S.; Benteyn, D.; de Coninck, A.; van Riet, I.; Verfaillie, G.; Vandeloo, J.; et al. Therapeutic vaccination with an autologous mRNA electroporated dendritic cell vaccine in patients with advanced melanoma. J. Immunother. 2011, 34, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Heimberger, A.B.; Archer, G.E.; Aldape, K.D.; Friedman, A.H.; Friedman, H.S.; Gilbert, M.R.; Herndon, J.E., 2nd; McLendon, R.E.; Mitchell, D.A.; et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2010, 28, 4722–4729. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, C.J.; Black, K.L.; Liu, G.; Mazer, M.; Zhang, X.X.; Pepkowitz, S.; Goldfinger, D.; Ng, H.; Irvin, D.; Yu, J.S. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res. 2008, 68, 5955–5964. [Google Scholar] [CrossRef] [PubMed]
- Herbert, N.; Haferkamp, A.; Schmitz-Winnenthal, H.F.; Zoller, M. Concomitant tumor and autoantigen vaccination supports renal cell carcinoma rejection. J. Immunol. 2010, 185, 902–916. [Google Scholar] [CrossRef] [PubMed]
- Van Tendeloo, V.F.; van de Velde, A.; van Driessche, A.; Cools, N.; Anguille, S.; Ladell, K.; Gostick, E.; Vermeulen, K.; Pieters, K.; Nijs, G.; et al. Induction of complete and molecular remissions in acute myeloid leukemia by wilms’ tumor 1 antigen-targeted dendritic cell vaccination. Proc. Natl. Acad. Sci. USA 2010, 107, 13824–13829. [Google Scholar] [CrossRef] [PubMed]
- Bijen, C.B.; Bantema-Joppe, E.J.; de Jong, R.A.; Leffers, N.; Mourits, M.J.; Eggink, H.F.; van der Zee, A.G.; Hollema, H.; de Bock, G.H.; Nijman, H.W. The prognostic role of classical and nonclassical MHC class I expression in endometrial cancer. Int. J. Cancer 2010, 126, 1417–1427. [Google Scholar] [CrossRef] [PubMed]
- Vanderstraeten, A.; Tuyaerts, S.; Amant, F. The immune system in the normal uterus and its implication in uterine tumor development. J. Reprod. Immunol. 2015, 109, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Coosemans, A.; Wolfl, M.; Berneman, Z.N.; van Tendeloo, V.; Vergote, I.; Amant, F.; van Gool, S.W. Immunological response after therapeutic vaccination with WT1 mRNA-loaded dendritic cells in end-stage endometrial carcinoma. Anticancer Res. 2010, 30, 3709–3714. [Google Scholar] [PubMed]
- Coosemans, A.; Vanderstraeten, A.; Tuyaerts, S.; Verschuere, T.; Moerman, P.; Berneman, Z.N.; Vergote, I.; Amant, F.; van Gool, S.W. Wilms’ tumor gene 1 (WT1)-loaded dendritic cell immunotherapy in patients with uterine tumors: A phase I/II clinical trial. Anticancer Res. 2013, 33, 5495–5500. [Google Scholar] [PubMed]
- Hernando, J.J.; Park, T.W.; Kubler, K.; Offergeld, R.; Schlebusch, H.; Bauknecht, T. Vaccination with autologous tumour antigen-pulsed dendritic cells in advanced gynaecological malignancies: Clinical and immunological evaluation of a phase I trial. Cancer Immunol. Immunother. 2002, 51, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Santin, A.D.; Bellone, S.; Ravaggi, A.; Roman, J.J.; Pecorelli, S.; Parham, G.P.; Cannon, M.J. Induction of tumour-specific CD8+ cytotoxic T lymphocytes by tumour lysate-pulsed autologous dendritic cells in patients with uterine serous papillary cancer. Br. J. Cancer 2002, 86, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.F.; Le Blanc, K.; Brodin, B. Concise review: Cancer/testis antigens, stem cells, and cancer. Stem Cells 2007, 25, 707–711. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, V.; Pore, N.; Docquier, F.; Abdullaev, Z.K.; Chernukhin, I.; Kita, G.X.; Rai, S.; Smart, M.; Farrar, D.; Pack, S.; et al. BORIS, a paralogue of the transcription factor, CTCF, is aberrantly expressed in breast tumours. Br. J. Cancer 2008, 98, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Hines, W.C.; Bazarov, A.V.; Mukhopadhyay, R.; Yaswen, P. BORIS (CTCFL) is not expressed in most human breast cell lines and high grade breast carcinomas. PLoS ONE 2010, 5, e9738. [Google Scholar] [CrossRef] [PubMed]
- Lladser, A.; Sanhueza, C.; Kiessling, R.; Quest, A.F. Is survivin the potential achilles’ heel of cancer? Adv. Cancer Res. 2011, 111, 1–37. [Google Scholar] [PubMed]
- Patel, K.P.; Vonderheide, R.H. Telomerase as a tumor-associated antigen for cancer immunotherapy. Cytotechnology 2004, 45, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Beatson, R.E.; Taylor-Papadimitriou, J.; Burchell, J.M. MUC1 immunotherapy. Immunotherapy 2010, 2, 305–327. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.P.; Gubin, M.M.; Schreiber, R.D. The role of neoantigens in naturally occurring and therapeutically induced immune responses to cancer. Adv. Immunol. 2016, 130, 25–74. [Google Scholar] [PubMed]
- Lu, Y.C.; Robbins, P.F. Cancer immunotherapy targeting neoantigens. Semin. Immunol. 2016, 28, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Howitt, B.E.; Shukla, S.A.; Sholl, L.M.; Ritterhouse, L.L.; Watkins, J.C.; Rodig, S.; Stover, E.; Strickland, K.C.; D’Andrea, A.D.; Wu, C.J.; et al. Association of polymerase e-mutated and microsatellite-instable endometrial cancers with neoantigen load, number of tumor-infiltrating lymphocytes, and expression of PD-1 and PD-L1. JAMA Oncol. 2015, 1, 1319–1323. [Google Scholar] [CrossRef] [PubMed]
- Maki, R.G. Soft tissue sarcoma as a model disease to examine cancer immunotherapy. Curr. Opin. Oncol. 2001, 13, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Ten Bosch, G.J.; Toornvliet, A.C.; Friede, T.; Melief, C.J.; Leeksma, O.C. Recognition of peptides corresponding to the joining region of p210BCR-ABL protein by human T cells. Leukemia 1995, 9, 1344–1348. [Google Scholar] [PubMed]
- Pawelec, G.; Kalbacher, H.; Bruserud, O. Tumor-specific antigens revisited: Presentation to the immune system of fusion peptides resulting solely from tumor-specific chromosomal translocations. Oncol. Res. 1992, 4, 315–320. [Google Scholar] [PubMed]
- Gremel, G.; Liew, M.; Hamzei, F.; Hardell, E.; Selling, J.; Ghaderi, M.; Stemme, S.; Ponten, F.; Carlson, J.W. A prognosis based classification of undifferentiated uterine sarcomas: Identification of mitotic index, hormone receptors and YWHAE-FAM22 translocation status as predictors of survival. Int. J. Cancer 2015, 136, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- Risinger, J.I.; Chandramouli, G.V.; Maxwell, G.L.; Custer, M.; Pack, S.; Loukinov, D.; Aprelikova, O.; Litzi, T.; Schrump, D.S.; Murphy, S.K.; et al. Global expression analysis of cancer/testis genes in uterine cancers reveals a high incidence of BORIS expression. Clin. Cancer Res. 2007, 13, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Nagorsen, D.; Scheibenbogen, C.; Marincola, F.M.; Letsch, A.; Keilholz, U. Natural T cell immunity against cancer. Clin. Cancer Res. 2003, 9, 4296–4303. [Google Scholar] [PubMed]
- Martin-Kleiner, I. Boris in human cancers—A review. Eur. J. Cancer 2012, 48, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Hoivik, E.A.; Kusonmano, K.; Halle, M.K.; Berg, A.; Wik, E.; Werner, H.M.; Petersen, K.; Oyan, A.M.; Kalland, K.H.; Krakstad, C.; et al. Hypomethylation of the CTCFL/BORIS promoter and aberrant expression during endometrial cancer progression suggests a role as an Epi-driver gene. Oncotarget 2014, 5, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Mkrtichyan, M.; Ghochikyan, A.; Davtyan, H.; Movsesyan, N.; Loukinov, D.; Lobanenkov, V.; Cribbs, D.H.; Laust, A.K.; Nelson, E.L.; Agadjanyan, M.G. Cancer-testis antigen, BORIS based vaccine delivered by dendritic cells is extremely effective against a very aggressive and highly metastatic mouse mammary carcinoma. Cell. Immunol. 2011, 270, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Chitale, D.A.; Jungbluth, A.A.; Marshall, D.S.; Leitao, M.M.; Hedvat, C.V.; Kolb, D.; Spagnoli, G.C.; Iversen, K.; Soslow, R.A. Expression of cancer-testis antigens in endometrial carcinomas using a tissue microarray. Mod. Pathol. 2005, 18, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Inokuma, M.; dela Rosa, C.; Schmitt, C.; Haaland, P.; Siebert, J.; Petry, D.; Tang, M.; Suni, M.A.; Ghanekar, S.A.; Gladding, D.; et al. Functional T cell responses to tumor antigens in breast cancer patients have a distinct phenotype and cytokine signature. J. Immunol. 2007, 179, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Esfandiary, A.; Ghafouri-Fard, S. MAGE-A3: An immunogenic target used in clinical practice. Immunotherapy 2015, 7, 683–704. [Google Scholar] [CrossRef] [PubMed]
- Li, F.Q.; Liu, Q.; Han, Y.L.; Wu, B.; Yin, H.L. Sperm protein 17 is highly expressed in endometrial and cervical cancers. BMC Cancer 2010. [Google Scholar] [CrossRef] [PubMed]
- Nakazato, T.; Kanuma, T.; Tamura, T.; Faried, L.S.; Aoki, H.; Minegishi, T. Sperm protein 17 influences the tissue-specific malignancy of clear cell adenocarcinoma in human epithelial ovarian cancer. Int. J. Gynecol. Cancer 2007, 17, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Straughn, J.M., Jr.; Shaw, D.R.; Guerrero, A.; Bhoola, S.M.; Racelis, A.; Wang, Z.; Chiriva-Internati, M.; Grizzle, W.E.; Alvarez, R.D.; Lim, S.H.; et al. Expression of sperm protein 17 (Sp17) in ovarian cancer. Int. J. Cancer 2004, 108, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Chiriva-Internati, M.; Wang, Z.; Salati, E.; Bumm, K.; Barlogie, B.; Lim, S.H. Sperm protein 17 (Sp17) is a suitable target for immunotherapy of multiple myeloma. Blood 2002, 100, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Chiriva-Internati, M.; Wang, Z.; Salati, E.; Timmins, P.; Lim, S.H. Tumor vaccine for ovarian carcinoma targeting sperm protein 17. Cancer 2002, 94, 2447–2453. [Google Scholar] [CrossRef] [PubMed]
- Chiriva-Internati, M.; Wang, Z.; Salati, E.; Wroblewski, D.; Lim, S.H. Successful generation of sperm protein 17 (Sp17)-specific cytotoxic T lymphocytes from normal donors: Implication for tumour-specific adoptive immunotherapy following allogeneic stem cell transplantation for Sp17-positive multiple myeloma. Scand. J. Immunol. 2002, 56, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tian, X.; Jiang, S.; Ren, X.; Liu, F.; Yang, J.; Chen, Y.; Jiang, Y. Umbilical cord blood-derived dendritic cells infected by adenovirus for Sp17 expression induce antigen-specific cytotoxic T cells against nsclc cells. Cell. Immunol. 2015, 298, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Sivridis, E.; Giatromanolaki, A.; Koukourakis, M.I.; Georgiou, L.; Anastasiadis, P. Patterns of episialin/MUC1 expression in endometrial carcinomas and prognostic relevance. Histopathology 2002, 40, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; McConnell, M.J.; Yu, B.; Li, J.; Balko, J.M.; Black, E.P.; Johnson, J.O.; Lloyd, M.C.; Altiok, S.; Haura, E.B. MUC1 is a downstream target of STAT3 and regulates lung cancer cell survival and invasion. Int. J. Oncol. 2009, 35, 337–345. [Google Scholar] [PubMed]
- Suzuki, Y.; Sutoh, M.; Hatakeyama, S.; Mori, K.; Yamamoto, H.; Koie, T.; Saitoh, H.; Yamaya, K.; Funyu, T.; Habuchi, T.; et al. MUC1 carrying core 2 O-glycans functions as a molecular shield against NK cell attack, promoting bladder tumor metastasis. Int. J. Oncol. 2012, 40, 1831–1838. [Google Scholar] [PubMed]
- Ye, Q.; Yan, Z.; Liao, X.; Li, Y.; Yang, J.; Sun, J.; Kawano, T.; Wang, X.; Cao, Z.; Wang, Z.; et al. MUC1 induces metastasis in esophageal squamous cell carcinoma by upregulating matrix metalloproteinase 13. Lab. Investig. 2011, 91, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Guckel, B.; Rentzsch, C.; Nastke, M.D.; Marme, A.; Gruber, I.; Stevanovic, S.; Kayser, S.; Wallwiener, D. Pre-existing T-cell immunity against mucin-1 in breast cancer patients and healthy volunteers. J. Cancer Res. Clin. Oncol. 2006, 132, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Rivalland, G.; Loveland, B.; Mitchell, P. Update on mucin-1 immunotherapy in cancer: A clinical perspective. Expert Opin. Biol. Ther. 2015, 15, 1773–1787. [Google Scholar] [CrossRef] [PubMed]
- Kyo, S.; Kanaya, T.; Takakura, M.; Tanaka, M.; Inoue, M. Human telomerase reverse transcriptase as a critical determinant of telomerase activity in normal and malignant endometrial tissues. Int. J. Cancer 1999, 80, 60–63. [Google Scholar] [CrossRef]
- Lehner, R.; Enomoto, T.; McGregor, J.A.; Shroyer, A.L.; Haugen, B.R.; Pugazhenthi, U.; Shroyer, K.R. Quantitative analysis of telomerase htert mRNA and telomerase activity in endometrioid adenocarcinoma and in normal endometrium. Gynecol. Oncol. 2002, 84, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Hiyama, E.; Hiyama, K.; Yokoyama, T.; Shay, J.W. Immunohistochemical detection of telomerase (hTERT) protein in human cancer tissues and a subset of cells in normal tissues. Neoplasia 2001, 3, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Kyo, S.; Masutomi, K.; Maida, Y.; Kanaya, T.; Yatabe, N.; Nakamura, M.; Tanaka, M.; Takarada, M.; Sugawara, I.; Murakami, S.; et al. Significance of immunological detection of human telomerase reverse transcriptase: Re-evaluation of expression and localization of human telomerase reverse transcriptase. Am. J. Pathol. 2003, 163, 859–867. [Google Scholar] [CrossRef]
- Minev, B.; Hipp, J.; Firat, H.; Schmidt, J.D.; Langlade-Demoyen, P.; Zanetti, M. Cytotoxic t cell immunity against telomerase reverse transcriptase in humans. Proc. Natl. Acad. Sci. USA 2000, 97, 4796–4801. [Google Scholar] [CrossRef] [PubMed]
- Ayyoub, M.; Migliaccio, M.; Guillaume, P.; Lienard, D.; Cerottini, J.C.; Romero, P.; Levy, F.; Speiser, D.E.; Valmori, D. Lack of tumor recognition by hTERT peptide 540–548-specific CD8+ T cells from melanoma patients reveals inefficient antigen processing. Eur. J. Immunol. 2001, 31, 2642–2651. [Google Scholar] [CrossRef]
- Maecker, B.; von Bergwelt-Baildon, M.S.; Anderson, K.S.; Vonderheide, R.H.; Anderson, K.C.; Nadler, L.M.; Schultze, J.L. Rare naturally occurring immune responses to three epitopes from the widely expressed tumour antigens hTERT and CYP1B1 in multiple myeloma patients. Clin. Exp. Immunol. 2005, 141, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H.; Schultze, J.L.; Anderson, K.S.; Maecker, B.; Butler, M.O.; Xia, Z.; Kuroda, M.J.; von Bergwelt-Baildon, M.S.; Bedor, M.M.; Hoar, K.M.; et al. Equivalent induction of telomerase-specific cytotoxic T lymphocytes from tumor-bearing patients and healthy individuals. Cancer Res. 2001, 61, 8366–8370. [Google Scholar] [PubMed]
- Amarnath, S.M.; Dyer, C.E.; Ramesh, A.; Iwuagwu, O.; Drew, P.J.; Greenman, J. In vitro quantification of the cytotoxic T lymphocyte response against human telomerase reverse transcriptase in breast cancer. Int. J. Oncol. 2004, 25, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Titu, L.V.; Loveday, R.L.; Madden, L.A.; Cawkwell, L.; Monson, J.R.; Greenman, J. Cytotoxic T-cell immunity against telomerase reverse transcriptase in colorectal cancer patients. Oncol. Rep. 2004, 12, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Babiak, A.; Steinhauser, M.; Gotz, M.; Herbst, C.; Dohner, H.; Greiner, J. Frequent T cell responses against immunogenic targets in lung cancer patients for targeted immunotherapy. Oncol. Rep. 2014, 31, 384–390. [Google Scholar] [PubMed]
- Terashima, T.; Mizukoshi, E.; Arai, K.; Yamashita, T.; Yoshida, M.; Ota, H.; Onishi, I.; Kayahara, M.; Ohtsubo, K.; Kagaya, T.; et al. P53, hTERT, WT-1, and VEGFR2 are the most suitable targets for cancer vaccine therapy in HLA-A24 positive pancreatic adenocarcinoma. Cancer Immunol. Immunother. 2014, 63, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Vonderheide, R.H. Telomerase as a universal tumor antigen for cancer vaccines. Expert Rev. Vaccines 2008, 7, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Ugel, S.; Scarselli, E.; Iezzi, M.; Mennuni, C.; Pannellini, T.; Calvaruso, F.; Cipriani, B.; de Palma, R.; Ricci-Vitiani, L.; Peranzoni, E.; et al. Autoimmune B-cell lymphopenia after successful adoptive therapy with telomerase-specific t lymphocytes. Blood 2010, 115, 1374–1384. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, M. A second chance for telomerase reverse transcriptase in anticancer immunotherapy. Nat. Rev. Clin. Oncol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ophir, E.; Bobisse, S.; Coukos, G.; Harari, A.; Kandalaft, L.E. Personalized approaches to active immunotherapy in cancer. Biochim. Biophys. Acta 2016, 1865, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Prickett, T.D.; Crystal, J.S.; Cohen, C.J.; Pasetto, A.; Parkhurst, M.R.; Gartner, J.J.; Yao, X.; Wang, R.; Gros, A.; Li, Y.F.; et al. Durable complete response from metastatic melanoma after transfer of autologous T cells recognizing 10 mutated tumor antigens. Cancer Immunol. Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.F.; Lu, Y.C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Vanderstraeten, A.; Everaert, T.; van Bree, R.; Verbist, G.; Luyten, C.; Amant, F.; Tuyaerts, S. In vitro validation of survivin as target tumor-associated antigen for immunotherapy in uterine cancer. J. Immunother. 2015, 38, 239–249. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TAA 1 | Samples | Healthy Controls | Endometrial Carcinoma | Uterine Sarcoma |
|---|---|---|---|---|
| BORIS 2 | # 3 samples tested (% 4) | 9 (100) | 49 (100) | 15 (100) |
| # evaluable samples (%) 5 | 5 (56) | 43 (88) | 13 (87) | |
| # undetectable samples (%) 6 | 2 (40) | 21 (50) | 1 (8) | |
| % >2-fold change 7 (%) | 11 (26) | 8 (62) | ||
| MAGE-A3 8 | # samples tested (%) | 12 (100) | 71 (100) | 35 (100) |
| # evaluable samples (%) 5 | 10 (83) | 70 (99) | 34 (97) | |
| # undetectable samples (%) 6 | 0 (0) | 7 (10) | 3 (9) | |
| % >2-fold change 7 | 7 (10) | 3 (9) | ||
| Sp17 9 | # samples tested (%) | 12 (100) | 71 (100) | 34 (100) |
| # evaluable samples (%) 5 | 10 (83) | 70 (99) | 34 (100) | |
| # undetectable samples (%) 6 | 0 (0) | 0 (0) | 0 (0) | |
| % >2-fold change 7 | 14 (20) | 1 (3) | ||
| hTERT 10 | # samples tested (%) | 9 (100) | 70 (100) | 34 (100) |
| # evaluable samples (%) 5 | 9 (100) | 70 (100) | 33 (97) | |
| # undetectable samples (%) 6 | 6 (67) | 0 (0) | 26 (79) | |
| % >2-fold change 3 | 50 (71) | 16 (48) |
| Tumor Type | MUC1 | hTERT | ||
|---|---|---|---|---|
| #1 Samples | %2 Positive | # Samples | % Positive | |
| Normal tissue | 17 | 82.4 | 14 | 92.8 |
| Primary carcinoma | 48 | 91.6 | 30 | 66.7 |
| Recurrent carcinoma | 11 | 100 | 10 | 90 |
| Metastatic carcinoma | 10 | 90 | 9 | 66.7 |
| Primary sarcoma | 39 | 33.3 | 22 | 50 |
| Recurrent sarcoma | 15 | 26.7 | 12 | 41.7 |
| Metastatic sarcoma | 13 | 69.2 | 13 | 76.9 |
| TAA | Primers and Probe |
|---|---|
| BORIS | Human CTCFL Taqman gene expression assay from Applied Biosystems (Life Technologies) |
| MAGE-A3 | Sense primer, 5′-GTCGTCGGAAATTGGCAGTAT-3′ |
| Antisense primer, 5′-GCAGGTGGCAAAGATGTACAA-3′ | |
| Probe, 5′-6FAM-AAAGCTTCCAGTTCCTT-MGB-3′ | |
| Sp17 | Human SPA17 Taqman gene expression assay from Applied Biosystems (Life Technologies) |
| hTERT | Human TERT Taqman gene expression assay from Applied Biosystems (Life Technologies) |
| β-actin | Human ACTB (beta actin) Endogenous Control from Applied Biosystems (Life Technologies) |
| β-glucuronidase | Human GUSB (beta glucuronidase) Endogenous Control from Applied Biosystems (Life Technologies) |
| Percent Positive Tumor Cells | Staining Intensity | Assigned Score |
|---|---|---|
| 1–25 | 1 | 0 |
| 1–25 | 2 | 0 |
| 1–25 | 3 | 0 |
| 25–50 | 1 | 0 |
| 25–50 | 2 | 1+ |
| 25–50 | 3 | 2+ |
| >50 | 1 | 0 |
| >50 | 2 | 3+ |
| >50 | 3 | 4+ |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanderstraeten, A.; Tuyaerts, S.; Everaert, T.; Van Bree, R.; Verbist, G.; Luyten, C.; Amant, F. In Vitro Assessment of the Expression and T Cell Immunogenicity of the Tumor-Associated Antigens BORIS, MUC1, hTERT, MAGE-A3 and Sp17 in Uterine Cancer. Int. J. Mol. Sci. 2016, 17, 1525. https://doi.org/10.3390/ijms17091525
Vanderstraeten A, Tuyaerts S, Everaert T, Van Bree R, Verbist G, Luyten C, Amant F. In Vitro Assessment of the Expression and T Cell Immunogenicity of the Tumor-Associated Antigens BORIS, MUC1, hTERT, MAGE-A3 and Sp17 in Uterine Cancer. International Journal of Molecular Sciences. 2016; 17(9):1525. https://doi.org/10.3390/ijms17091525
Chicago/Turabian StyleVanderstraeten, Anke, Sandra Tuyaerts, Tina Everaert, Rieta Van Bree, Godelieve Verbist, Cathérine Luyten, and Frederic Amant. 2016. "In Vitro Assessment of the Expression and T Cell Immunogenicity of the Tumor-Associated Antigens BORIS, MUC1, hTERT, MAGE-A3 and Sp17 in Uterine Cancer" International Journal of Molecular Sciences 17, no. 9: 1525. https://doi.org/10.3390/ijms17091525