All chemicals (reagent grade) were obtained from commercial sources and were used as supplied, without further purification.
Melting points were determined with an MPM-H1 Electrothermal melting point meter using the open glass capillary method and are uncorrected. The reaction progress and purity of the synthesized compounds were monitored by analytical thin layer chromatography (TLC) using Merck precoated Silica Gel 60F254 sheets (Darmstadt, Germany), heptane–ethyl-acetate 3:7 elution system and UV light for visualization. Elemental analysis was registered with a Vario El CHNS instrument (Hanau, Germany) and the results obtained for all synthesized compounds were in agreement with the calculated values within ±0.4% range. MS analyses were performed at 70 eV with an Agilent gas chromatograph 6890 (Darmstadt, Germany) equipped with an apolar Macherey Nagel Permabond SE 52 capillary column (Dueren, Germany) and with an LC-MS Shimadzu Mass Spectrometer (Shimadzu Corporation, Torrance, CA, USA). IR spectra were recorded using the ATR technique (Attenuated Total Reflectance) on a JASCO FT-IR—4100 spectrometer (Cremella, Italy). Nuclear magnetic resonance (1H NMR) spectra were recorded on a Bruker Avance NMR spectrometer (Karlsruhe, Germany) operating at 400 and 500 MHz, at room temperature, using tetramethylsilane (TMS) as internal standard. Chemical shifts were reported in ppm (δ). The samples were prepared by dissolving the compounds in DMSO-d6 (δH = 2.51 ppm) as solvent and the spectra were recorded using a single excitation pulse of 12 μs (1H NMR). Spin multiplets are given as s (singlet), d (doublet), t (triplet) and m (multiplet). 13C NMR spectra were recorded on Bruker Avance NMR spectrometer (Karlsruhe, Germany) operating at 125 MHz in DMSO-d6, using a waltz-16 decoupling scheme. All spectral analyses data were in accordance with the assigned structures.
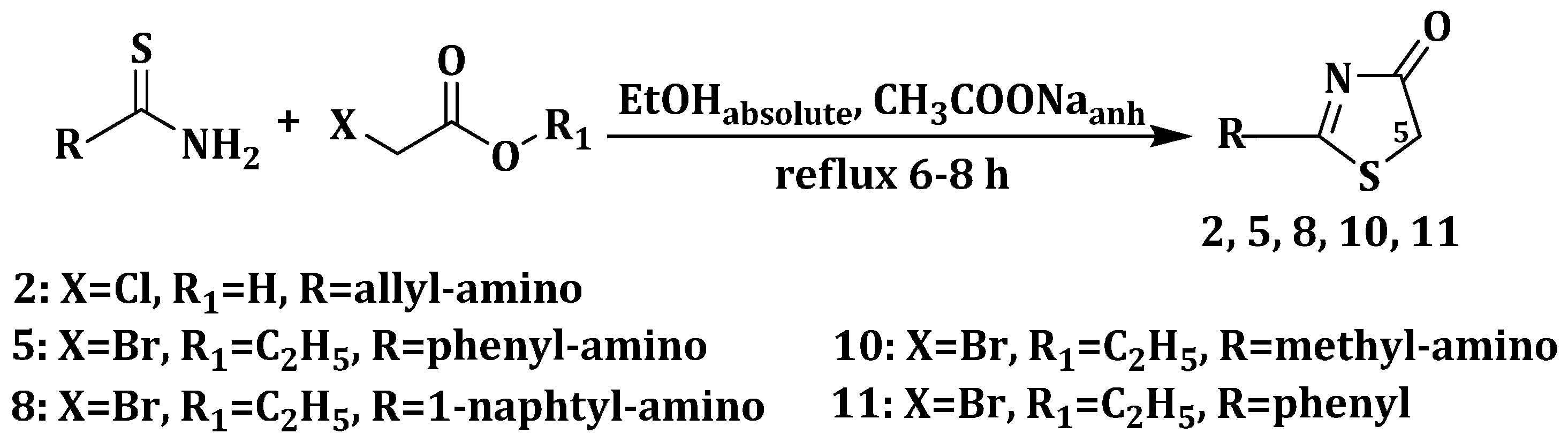
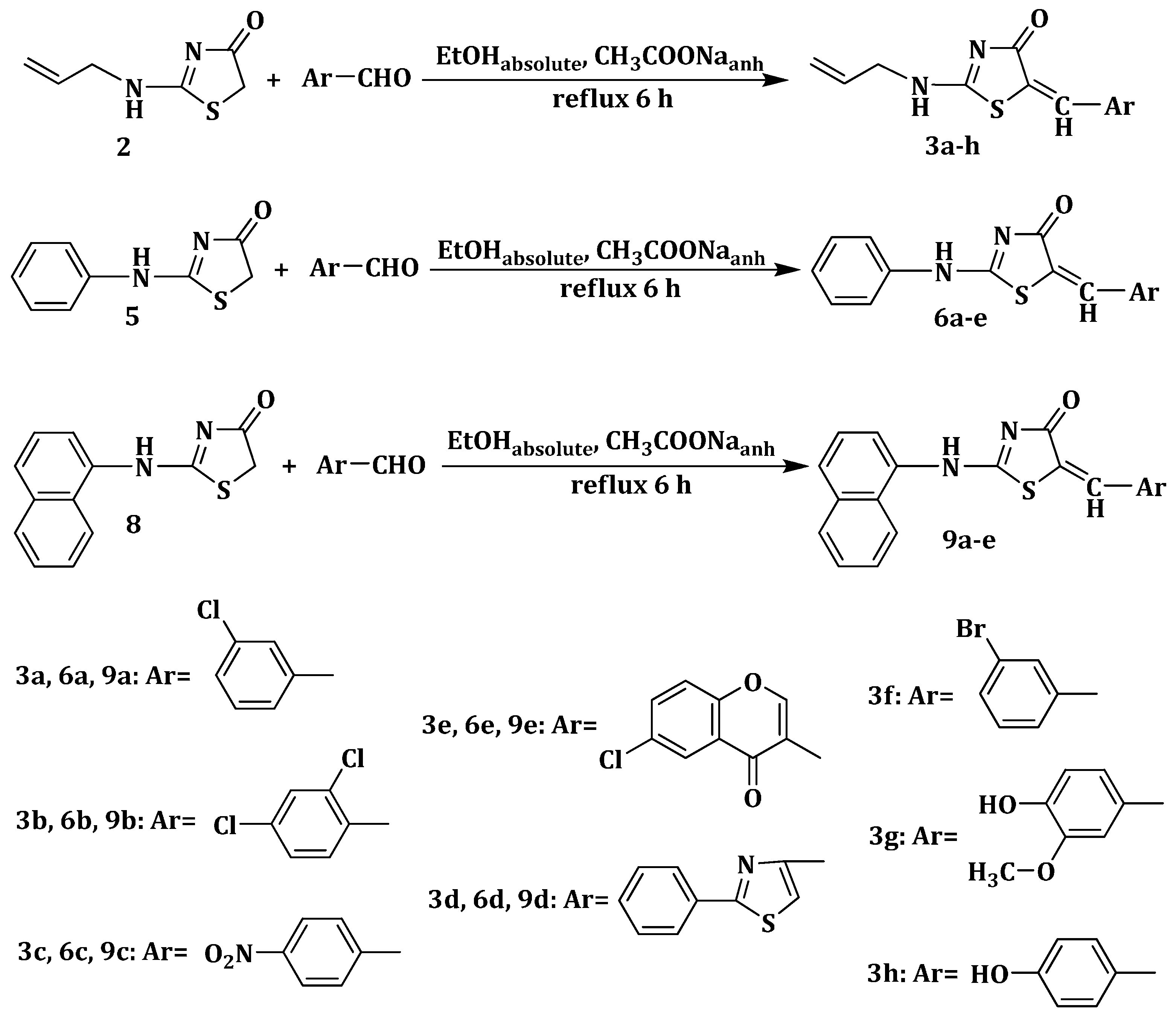
3.1.2. General Procedure for the Synthesis of 2-(Allyl/aryl-amino)-5-arylidene-thiazolin-4-ones 3a–h, 6a–e and 9a–e
2 mmol of the appropriate 2-(allyl/aryl-amino)-thiazolin-4-one were suspended in 8 mL of absolute ethanol and then, to the obtained suspension were added 8 mmol (0.656 g) of anhydrous sodium acetate and 2 mmol of the corresponding aromatic aldehyde. The reaction mixture was refluxed for 6 h, and, after the reaction was completed, it was dropwise poured into ice-cold water. The resulting precipitate was filtered off and washed on the filter with distilled water and ethanol. The obtained compound was recrystallized from an appropriate solvent (absolute ethanol or absolute methanol).
2-(Allylamino)-5-(3-chlorobenzylidene)thiazol-4(5H)-one (3a). Yield 87% (0.485 g); light yellow powder; m.p. 198 °C; IR (ATR, ν (cm−1)): 3437 (N–Hamine), 1726 (C=OTZ), 1155 (C–Cl); 1H NMR (500 MHz, DMSO-d6, δ/ppm): 7.74 (t, 1H, NH), 7.50 (s, 1H, –CH=), 7.36–7.50 (m, 4H, Ar–H), 6.03 (m, 1H, CH=), 5.23 (d, 2H, =CH2), 3.98 (m, 2H, CH2); 13C NMR (125 MHz, DMSO-d6, δ/ppm): 168.12 (C=O), 157.23 (C), 139.54 (CH), 136.75 (C), 135.62 (C), 133.81 (C), 131.05 (CH), 130.55 (CH), 129.11 (CH), 128.33 (CH), 126.27 (CH), 118.44 (CH2), 50.15 (CH2); MS (EI, 70 eV) m/z (%): 279 (M + 1, 100), 280 (M + 2, 10.6), 281 (M + 3, 36.8), 252 (29.0), 226 (6.7), 209 (22.6), 195 (100), 176 (19.0); Anal. Calcd. for C13H11ClN2OS (278.76): C, 56.01; H, 3.98; N, 10.05; S, 11.50; Found: C, 56.32; H, 3.79; N, 10.12; S, 11.52.
2-(Allylamino)-5-(2,4-dichlorobenzylidene)thiazol-4(5H)-one (3b). Yield 65% (0.407 g); white powder; m.p. 238 °C; IR (ATR, ν (cm−1)): 3438 (N–Hamine), 1714 (C=OTZ), 1158 (C–Cl), 1093 (C–Cl); 1H NMR (500 MHz, DMSO-d6, δ/ppm): 7.93 (t, 1H, NH), 7.56 (s, 1H, –CH=), 7.10–7.12 (m, 3H, Ar–H), 6.31 (m, 1H, CH=), 5.34 (d, 2H, =CH2), 3.66 (m, 2H, CH2); 13C NMR (125 MHz, DMSO-d6, δ/ppm): 166.20 (C=O), 158.42 (C), 138.94 (CH), 137.77 (C), 135.31 (C), 133.15 (C), 131.56 (C), 130.99 (CH), 129.78 (CH), 126.63 (CH), 124.88 (CH), 117.08 (CH2), 49.94 (CH2); MS (EI, 70 eV) m/z (%): 313 (M + 1, 100), 314 (M + 2, 13.3), 315 (M + 3, 95.5), 316 (M + 4, 17.3), 317 (M + 5, 19.2), 277 (100), 256 (7.2), 208 (14.1), 185 (5.9), 149 (5.1); Anal. Calcd. for C13H10Cl2N2OS (313.20): C, 49.85; H, 3.22; N, 8.94; S, 10.24; Found: C, 49.93; H, 3.35; N, 8.98; S, 10.36.
2-(Allylamino)-5-(4-nitrobenzylidene)thiazol-4(5H)-one (3c). Yield 89% (0.514 g); pale yellow powder; m.p. 265 °C; IR (ATR, ν (cm−1)): 3431 (N–Hamine), 1705 (C=OTZ), 1523 (NO2 asymmetric), 1326 (NO2 symmetric); 1H NMR (500 MHz, DMSO-d6, δ/ppm): 7.98 (t, 1H, NH), 7.62 (s, 1H, –CH=), 7.13–7.84 (m, 4H, Ar–H), 5.98 (m, 1H, CH=), 5.25 (d, 2H, =CH2), 3.84 (m, 2H, CH2); 13C NMR (125 MHz, DMSO-d6, δ/ppm): 175.30 (C=O), 161.57 (C), 148.11 (C), 142.59 (C), 138.72 (CH), 134.09 (C), 131.25 (2CH), 129.44 (CH), 125.22 (2CH), 117.45 (CH2), 50.11 (CH2); MS (EI, 70 eV) m/z (%): 290 (M + 1, 100), 291 (M + 2, 17.2), 292 (M + 3, 6.3), 248 (97.0), 245 (100), 203 (7.3), 174 (10.2), 141 (17.9); Anal. Calcd. for C13H11N3O3S (289.31): C, 53.97; H, 3.83; N, 14.52; S, 11.08; Found: C, 53.88; H, 3.97; N, 14.67; S, 11.23.
2-(Allylamino)-5-((2-phenylthiazol-4-yl)methylene)thiazol-4(5H)-one (3d). Yield 80% (0.523 g); white powder; m.p. 339 °C; IR (ATR, ν (cm−1)): 3435 (N–Hamine), 1728 (C=OTZ); 1H NMR (500 MHz, DMSO-d6, δ/ppm): 8.02 (s, 1H, NH), 8.00 (s, 1H, C5-thiazole-H), 7.70 (s, 1H, –CH=), 7.42–7.71 (m, 5H, Ar–H), 6.05 (m, 1H, CH=), 5.32 (d, 2H, =CH2), 4.15 (m, 2H, CH2); 13C NMR (125 MHz, DMSO-d6, δ/ppm): 172.20 (C=O), 169.85 (C), 158.33 (C), 150.08 (C), 140.88 (CH), 138.63 (C), 134.66 (CH), 133.19 (C), 130.97 (2CH), 129.14 (2CH), 128.93 (CH), 127.51 (CH), 117.68 (CH2), 50.42 (CH2); MS (EI, 70 eV) m/z (%): 328 (M + 1, 100), 329 (M + 2, 11.7), 330 (M + 3, 11.5), 301 (5.1), 287 (28.7), 261 (22.5), 246 (39.6), 218 (100), 155 (14.1); Anal. Calcd. for C16H13N3OS2 (327.42): C, 58.69; H, 4.00; N, 12.83; S, 19.59; Found: C, 58.82; H, 4.21; N, 12.99; S, 19.68.
2-(Allylamino)-5-((6-chloro-4-oxo-4H-chromen-3-yl)methylene)thiazol-4(5H)-one (3e). Yield 91% (0.631 g); pale pink powder; m.p. 299–301 °C; IR (ATR, ν (cm−1)): 3432 (N–Hamine), 1684 (C=OTZ), 1647 (C=Ochromone), 1273 (C–Ochromone), 1167 (C–Cl), 1045 (C–Ochromone); 1H NMR (500 MHz, DMSO-d6, δ/ppm): 7.93 (t, 1H, NH), 7.74 (s, 1H, –CH=), 7.11–8.74 (m, 4H, Ar–H), 6.27 (m, 1H, CH=), 5.27 (d, 2H, =CH2), 3.73 (m, 2H, CH2); 13C NMR (125 MHz, DMSO-d6, δ/ppm): 178.55 (C=O), 167.64 (C=O), 159.21 (C), 155.82 (C), 153.03 (CH), 149.43 (CH), 136.97 (C), 136.16 (CH), 134.68 (CH), 130.74 (CH), 128.42 (C), 125.55 (C), 119.25 (C), 119.01 (CH), 118.14 (CH2), 51.26 (CH2); MS (EI, 70 eV) m/z (%): 347 (M + 1, 100), 348 (M + 2, 19.9), 349 (M + 3, 33.8), 330 (9.8), 305 (36.5), 290 (14.8), 277 (20.3), 265 (74.2), 237 (100), 213 (1.9), 205 (1.8), 155 (26.7); Anal. Calcd. for C16H11ClN2O3S (346.79): C, 55.41; H, 3.20; N, 8.08; S, 9.25; Found: C, 55.65; H, 3.33; N, 8.31; S, 9.34.
2-(Allylamino)-5-(3-bromobenzylidene)thiazol-4(5H)-one (3f). Yield 75% (0.485 g); white powder; m.p. 330 °C; IR (ATR, ν (cm−1)): 3429 (N–Hamine), 1689 (C=OTZ),1035 (C–Br); 1H NMR (500 MHz, DMSO-d6, δ/ppm): 7.96 (t, 1H, NH), 7.58 (s, 1H, –CH=), 7.44–7.86 (m, 4H, Ar–H), 6.30 (m, 1H, CH=), 5.29 (d, 2H, =CH2), 3.86 (m, 2H, CH2); 13C NMR (125 MHz, DMSO-d6, δ/ppm): 168.74 (C=O), 158.62 (C), 138.42 (CH), 136.33 (C), 135.10 (C), 133.96 (C), 131.14 (CH), 130.27 (CH), 129.19 (CH), 128.45 (CH), 126.47 (CH), 118.21 (CH2), 51.17 (CH2); MS (EI, 70 eV) m/z (%): 323 (M + 1, 100), 324 (M + 2, 52.1), 325 (M + 3, 50.1), 306 (9.0), 279 (17.6), 259 (13.6), 240 (20.8), 235 (11.0), 177 (17.7), 161 (22.6), 145 (9.3), 121 (12.7); Anal. Calcd. for C13H11BrN2OS (323.21): C, 48.31; H, 3.43; N, 8.67; S, 9.92; Found: C, 48.45; H, 3.55; N, 8.54; S, 9.85.
2-(Allylamino)-5-(4-hydroxy-3-methoxybenzylidene)thiazol-4(5H)-one (3g). Yield 77% (0.447 g); pale yellow powder; m.p. 240 °C; IR (ATR, ν (cm−1)): 3435 (N–Hamine), 3285 (O–Hphenol), 1705 (C=OTZ), 1336 (C–Ophenol), 1046 (C–Omethoxy), 1019 (C–Omethoxy); 1H NMR (500 MHz, DMSO-d6, δ/ppm): 9.31 (s, 1H, OH), 8.06 (t, 1H, NH), 7.72 (s, 1H, –CH=), 7.13–7.74 (m, 3H, Ar–H), 6.31 (m, 1H, CH=), 5.34 (d, 2H, =CH2), 4.01 (m, 2H, CH2), 3.83 (s, 3H, –OCH3); 13C NMR (125 MHz, DMSO-d6, δ/ppm): 187.08 (C=O), 174.77 (C), 159.42 (C), 152.06 (CH), 148.20 (C), 134.12 (CH), 132.66 (C), 129.01 (C), 122.81 (CH), 117.30 (CH2), 116.57 (CH), 111.96 (CH), 55.13 (CH3), 50.56 (CH2); MS (EI, 70 eV) m/z (%): 291 (M + 1, 100), 292 (M + 2, 12.8), 293 (M + 3, 13.2), 265 (5.1), 238 (3.1), 180 (6.6), 138 (10.6); Anal. Calcd. for C14H14N2O3S (290.34): C, 57.92; H, 4.86; N, 9.65; S, 11.04; Found: C, 57.87; H, 4.83; N, 9.61; S, 11.17.
2-(Allylamino)-5-(4-hydroxybenzylidene)thiazol-4(5H)-one (3h). Yield 59% (0.307 g); pale yellow powder; m.p. 338 °C; IR (ATR, ν (cm−1)): 3428 (N–Hamine), 3289 (O–Hphenol), 1695 (C=OTZ), 1331 (C–Ophenol); 1H NMR (500 MHz, DMSO-d6, δ/ppm): 9.35 (s, 1H, OH), 7.88 (t, 1H, NH), 7.67 (s, 1H, –CH=), 7.35–7.71 (m, 4H, Ar–H), 6.29 (m, 1H, CH=), 5.31 (d, 2H, =CH2), 3.85 (m, 2H, CH2); 13C NMR (125 MHz, DMSO-d6, δ/ppm): 184.14 (C=O), 162.56 (C), 159.13 (C), 152.47 (CH), 134.75 (CH), 132.86 (C), 129.91 (2CH), 127.50 (C), 117.49 (CH2), 116.04 (2CH), 50.32 (CH2); MS (EI, 70 eV) m/z (%): 261 (M + 1, 100), 262 (M + 2, 8.7), 263 (M + 3, 3.3), 209 (1.9), 183 (1.4), 158 (1.3), 138 (5.0); Anal. Calcd. for C13H12N2O2S (260.31): C, 59.98; H, 4.65; N, 10.76; S, 12.32; Found: C, 59.79; H, 4.76; N, 10.93; S, 12.36.
5-(3-Chlorobenzylidene)-2-(phenylamino)thiazol-4(5H)-one (6a). Yield 74% (0.466 g); light brown powder; m.p. 278–280 °C; IR (ATR, ν (cm−1)): 3427 (N–Hamine), 1721 (C=OTZ), 1157 (C–Cl); 1H NMR (500 MHz, DMSO-d6, δ/ppm): 11.95 (s, 1H, NH), 7.57 (s, 1H, –CH=), 7.04–7.84 (m, 9H, Ar–H); 13C NMR (125 MHz, DMSO-d6, δ/ppm): 187.52 (C=O), 176.43 (C), 142.15 (CH), 139.42 (C), 136.01 (C), 134.28 (C), 132.98 (C), 130.21 (CH), 129.68 (2CH), 128.77 (CH), 126.79 (CH), 126.30 (CH), 122.84 (CH), 120.71 (2CH); MS (EI, 70 eV) m/z (%): 315 (M + 1, 100), 316 (M + 2, 20.0), 317 (M + 3, 53.4), 286 (1.7), 196 (1.0), 169 (0.9), 118 (0.9); Anal. Calcd. for C16H11ClN2OS (314.79): C, 61.05; H, 3.52; N, 8.90; S, 10.19; Found: C, 61.09; H, 3.65; N, 8.94; S, 10.27.
5-(2,4-Dichlorobenzylidene)-2-(phenylamino)thiazol-4(5H)-one (6b). Yield 61% (0.426 g); light brown powder; m.p. 204–206 °C; IR (ATR, ν (cm−1)): 3434 (N–Hamine), 1717 (C=OTZ), 1156 (C–Cl), 1096 (C–Cl); 1H NMR (500 MHz, DMSO-d6, δ/ppm): 11.97 (s, 1H, NH), 7.61 (s, 1H, –CH=), 7.06–7.80 (m, 8H, Ar–H); 13C NMR (125 MHz, DMSO-d6, δ/ppm): 188.02 (C=O), 177.67 (C), 140.30 (CH), 139.15 (C), 135.45 (C), 133.36 (C), 131.63 (C), 130.33 (CH), 129.52 (2CH), 128.82 (CH), 127.94 (CH), 125.66 (C), 123.74 (CH), 120.78 (2CH); MS (EI, 70 eV) m/z (%): 349 (M + 1, 100), 350 (M + 2, 13.2), 351 (M + 3, 87.6), 352 (15.8), 353 (27.0), 315 (12.0), 313 (100), 138 (0.9); Anal. Calcd. for C16H10Cl2N2OS (349.23): C, 55.03; H, 2.89; N, 8.02; S, 9.18; Found: C, 55.12; H, 2.91; N, 8.15; S, 9.34.
5-(4-Nitrobenzylidene)-2-(phenylamino)thiazol-4(5H)-one (6c). Yield 87% (0.566 g); yellow powder; m.p. 347 °C; IR (ATR, ν (cm−1)): 3436 (N–Hamine), 1710 (C=OTZ), 1519 (NO2 asymmetric), 1330 (NO2 symmetric); 1H NMR (500 MHz, DMSO-d6, δ/ppm): 11.99 (s, 1H, NH), 7.69 (s, 1H, –CH=), 7.23–7.91 (m, 9H, Ar–H); 13C NMR (125 MHz, DMSO-d6, δ/ppm): 187.65 (C=O), 176.13 (C), 148.62 (C), 143.23 (CH), 141.85 (C), 139.99 (C), 132.44 (C), 130.09 (2CH), 129.27 (2CH), 123.95 (2CH), 122.11 (CH), 120.74 (2CH); MS (EI, 70 eV) m/z (%): 326 (M + 1, 100), 327 (M + 2, 14.2), 328 (M + 3, 6.0), 313 (8.8), 281 (100), 280 (93.0), 180 (13.4), 119 (40.9); Anal. Calcd. for C16H11N3O3S (325.34): C, 59.07; H, 3.41; N, 12.92; S, 9.86; Found: C, 59.19; H, 3.53; N, 12.98; S, 9.89.
2-(Phenylamino)-5-((2-phenylthiazol-4-yl)methylene)thiazol-4(5H)-one (6d). Yield 89% (0.647 g); light brown powder; m.p. 287–289 °C; IR (ATR, ν (cm−1)): 3430 (N–Hamine), 1719 (C=OTZ); 1H NMR (500 MHz, DMSO-d6, δ/ppm): 11.92 (s, 1H, NH), 8.11 (s, 1H, C5-thiazole-H), 7.73 (s, 1H, –CH=), 7.34–8.01 (m, 10H, Ar–H); 13C NMR (125 MHz, DMSO-d6, δ/ppm): 187.24 (C=O), 171.76 (C), 156.61 (C), 149.98 (C), 143.85 (CH), 139.48 (C), 138.53 (C), 132.07 (C), 130.69 (2CH), 129.78 (2CH), 129.15 (2CH), 128.66 (CH), 126.31 (CH), 122.54 (CH), 120.35 (2CH); MS (EI, 70 eV) m/z (%): 364 (M + 1, 100), 365 (M + 2, 22.4), 366 (M + 3, 10.5), 338 (18.4), 261 (95.5), 243 (10.4), 246 (58.9), 219 (22.0), 218 (100), 201 (12.2); Anal. Calcd. for C19H13N3OS2 (363.46): C, 62.79; H, 3.61; N, 11.56; S, 17.64; Found: C, 62.84; H, 3.73; N, 11.67; S, 17.77.
5-((6-Chloro-4-oxo-4H-chromen-3-yl)methylene)-2-(phenylamino)thiazol-4(5H)-one (6e). Yield 94% (0.720 g); brown powder; m.p. 356 °C; IR (ATR, ν (cm−1)): 3429 (N–Hamine), 1699 (C=OTZ), 1653 (C=Ochromone), 1270 (C–Ochromone), 1173 (C–Cl), 1041 (C–Ochromone); 1H NMR (500 MHz, DMSO-d6, δ/ppm): 11.98 (s, 1H, NH), 7.71 (s, 1H, –CH=), 7.12–8.86 (m, 9H, Ar–H); 13C NMR (125 MHz, DMSO-d6, δ/ppm): 188.58 (C=O), 170.33 (C=O), 158.46 (C), 156.02 (C), 151.65 (CH), 149.39 (CH), 139.87 (C), 136.43 (C), 136.05 (CH), 130.58 (CH), 129.81 (2CH), 129.17 (C), 126.01 (C), 122.22 (CH), 120.75 (2CH), 119.30 (C), 119.12 (CH); MS (EI, 70 eV) m/z (%): 383 (M + 1, 100), 384 (M + 2, 20.2), 385 (M + 3, 31.9), 386 (M + 4, 11.3), 326 (30.03), 312 (12.7), 259 (7.8), 205 (5.9), 183 (5.5), 138 (4.8); Anal. Calcd. for C19H11ClN2O3S (382.82): C, 59.61; H, 2.90; N, 7.32; S, 8.38; Found: C, 59.66; H, 2.97; N, 7.41; S, 8.47.
5-(3-Chlorobenzylidene)-2-(naphthalen-1-ylamino)thiazol-4(5H)-one (9a). Yield 81% (0.591 g); yellow powder; m.p. 140 °C; IR (ATR, ν (cm−1)): 3430 (N–Hamine), 1686 (C=OTZ), 1151 (C–Cl); 1H NMR (500 MHz, DMSO-d6, δ/ppm): 12.01 (s, 1H, NH), 7.71 (s, 1H, –CH=), 7.05–7.92 (m, 11H, Ar–H); 13C NMR (125 MHz, DMSO-d6, δ/ppm): 175.20 (C=O), 161.05 (C), 154.43 (CH), 144.60 (C), 135.89 (C), 134.38 (C), 134.07 (C), 132.10 (C), 130.24 (CH), 128.41 (CH), 128.11 (CH), 127.57 (CH), 126.89 (CH), 126.45 (CH), 126.26 (CH), 124.81 (CH), 123.47 (C), 121.35 (CH), 116.43 (CH), 105.63 (CH); MS (EI, 70 eV) m/z (%): 365 (M + 1, 100), 366 (M + 2, 2.5), 367 (M + 3, 19.7), 368 (M + 4, 7.7), 369 (M + 5, 1.4), 336 (3.3), 322 (2.2), 252 (4.9), 210 (2.1), 197 (64.5), 171 (10.3), 169 (100), 134 (52.2); Anal. Calcd. for C20H13ClN2OS (364.85): C, 65.84; H, 3.59; N, 7.68; S, 8.79; Found: C, 65.90; H, 3.66; N, 7.63; S, 8.84.
5-(2,4-Dichlorobenzylidene)-2-(naphthalen-1-ylamino)thiazol-4(5H)-one (9b). Yield 63% (0.503 g); yellow powder; m.p. 185 °C; IR (ATR, ν (cm−1)): 3433 (N–Hamine), 1713 (C=OTZ), 1155 (C–Cl), 1099 (C–Cl); 1H NMR (500 MHz, DMSO-d6, δ/ppm): 12.06 (s, 1H, NH), 7.62 (s, 1H, –CH=), 7.11–8.08 (m, 10H, Ar–H); 13C NMR (125 MHz, DMSO-d6, δ/ppm): 175.74 (C=O), 161.42 (C), 154.35 (CH), 143.89 (C), 135.94 (C), 134.51 (C), 132.68 (C), 131.33 (C), 130.12 (CH), 128.60 (CH), 128.19 (CH), 127.76 (CH), 126.81 (CH), 126.40 (CH), 125.53 (C), 124.99 (CH), 123.93 (C), 121.17 (CH), 118.32 (CH), 105.25 (CH); MS (EI, 70 eV) m/z (%): 399 (M + 1, 90.5), 400 (M + 2, 22.2), 401 (M + 3, 100), 402 (M + 4, 17.0), 365 (31.6), 363 (100), 297 (1.0), 168 (13.2), 159 (3.4); Anal. Calcd. for C20H12Cl2N2OS (399.29): C, 60.16; H, 3.03; N, 7.02; S, 8.03; Found: C, 60.35; H, 3.08; N, 7.11; S, 8.05.
2-(Naphthalen-1-ylamino)-5-(4-nitrobenzylidene)thiazol-4(5H)-one (9c). Yield 72% (0.540 g); yellow powder; m.p. 152 °C; IR (ATR, ν (cm−1)): 3426 (N–Hamine), 1706 (C=OTZ), 1521 (NO2 asymmetric), 1328 (NO2 symmetric); 1H NMR (500 MHz, DMSO-d6, δ/ppm): 12.23 (s, 1H, NH), 7.68 (s, 1H, –CH=), 7.13–8.18 (m, 11H, Ar–H); 13C NMR (125 MHz, DMSO-d6, δ/ppm): 174.88 (C=O), 161.31 (C), 154.10 (CH), 147.66 (C), 141.41 (C), 140.73 (C), 134.56 (C), 132.46 (C), 129.41 (2CH), 128.57 (CH), 127.32 (CH), 126.18 (CH), 125.86 (CH), 124.91 (C), 123.77 (2CH), 121.05 (CH), 118.81 (CH), 105.95 (CH); MS (EI, 70 eV) m/z (%): 376 (M + 1, 100), 378 (M + 2, 16.4), 379 (M + 3, 9.3), 330 (30.6), 316 (5.7), 208 (10.7), 180 (21.3), 170 (22.3), 169 (100), 150 (3.8), 134 (37.7); Anal. Calcd. for C20H13N3O3S (375.40): C, 63.99; H, 3.49; N, 11.19; S, 8.54; Found: C, 63.78; H, 3.52; N, 11.24; S, 8.64.
2-(Naphthalen-1-ylamino)-5-((2-phenylthiazol-4-yl)methylene)thiazol-4(5H)-one (9d). Yield 86% (0.711 g); pale yellow powder; m.p. 312 °C; IR (ATR, ν (cm−1)): 3429 (N–Hamine), 1728 (C=OTZ); 1H NMR (500 MHz, DMSO-d6, δ/ppm): 12.31 (s, 1H, NH), 7.69 (s, 1H, –CH=), 7.12–8.91 (m, 13H, Ar–H + C5-thiazole-H); 13C NMR (125 MHz, DMSO-d6, δ/ppm): 174.67 (C=O), 169.54 (C), 161.11 (C), 149.69 (C), 142.33 (CH), 140.61 (C), 138.89 (C), 134.54 (C), 131.26 (C), 130.97 (2CH), 129.38 (2CH), 128.88 (CH), 128.79 (CH), 127.64 (CH), 126.78 (CH), 125.99 (CH), 125.07 (CH), 124.76 (C), 121.13 (CH), 118.92 (CH), 105.86 (CH); MS (EI, 70 eV) m/z (%): 414 (M + 1, 100), 415 (M + 2, 22.2), 416 (M + 3, 15.7), 387 (4.1), 261 (30.1), 246 (10.5), 218 (100), 213 (6.8), 174 (4.5), 158 (7.9), 130 (2.5); Anal. Calcd. for C23H15N3OS2 (413.51): C, 66.80; H, 3.66; N, 10.16; S, 15.51; Found: C, 66.66; H, 3.74; N, 10.19; S, 15.44.
5-((6-Chloro-4-oxo-4H-chromen-3-yl)methylene)-2-(naphthalen-1-ylamino)thiazol-4(5H)-one (9e). Yield 90% (0.779 g); orange powder; m.p. 266–268 °C; IR (ATR, ν (cm−1)): 3434 (N–Hamine), 1701 (C=OTZ), 1649 (C=Ochromone), 1271 (C–Ochromone), 1170 (C–Cl), 1044 (C–Ochromone); 1H NMR (500 MHz, DMSO-d6, δ/ppm): 12.51 (s, 1H, NH), 7.70 (s, 1H, –CH=), 7.11–8.74 (m, 11H, Ar–H); 13C NMR (125 MHz, DMSO-d6, δ/ppm): 174.21 (C=O), 160.63 (C=O), 154.38 (2C), 135.15 (CH), 134.36 (C), 131.09 (CH), 128.45 (2C), 127.49 (CH), 127.03 (C), 126.56 (CH), 126.49 (C), 125.06 (C), 124.95 (2CH), 124.49 (CH), 123.48 (2CH), 121.78 (C), 121.48 (CH), 118.76 (CH), 116.60 (CH); MS (EI, 70 eV) m/z (%): 433 (M + 1, 100), 434 (M + 2, 17.8), 435 (M + 3, 52.6), 436 (M + 4, 10.9), 413 (5.6), 401 (100), 376 (28.4), 290 (34.2), 299 (3.5), 267 (32.8), 265 (100), 237 (85.8), 205 (22.2), 165 (21.1); Anal. Calcd. for C23H13ClN2O3S (432.88): C, 63.82; H, 3.03; N, 6.47; S, 7.41; Found: C, 63.70; H, 3.01; N, 6.50; S, 7.53.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









