
Dmp1 Promoter-Driven Diphtheria Toxin Receptor Transgene Expression Directs Unforeseen Effects in Multiple Tissues

 , and
, and 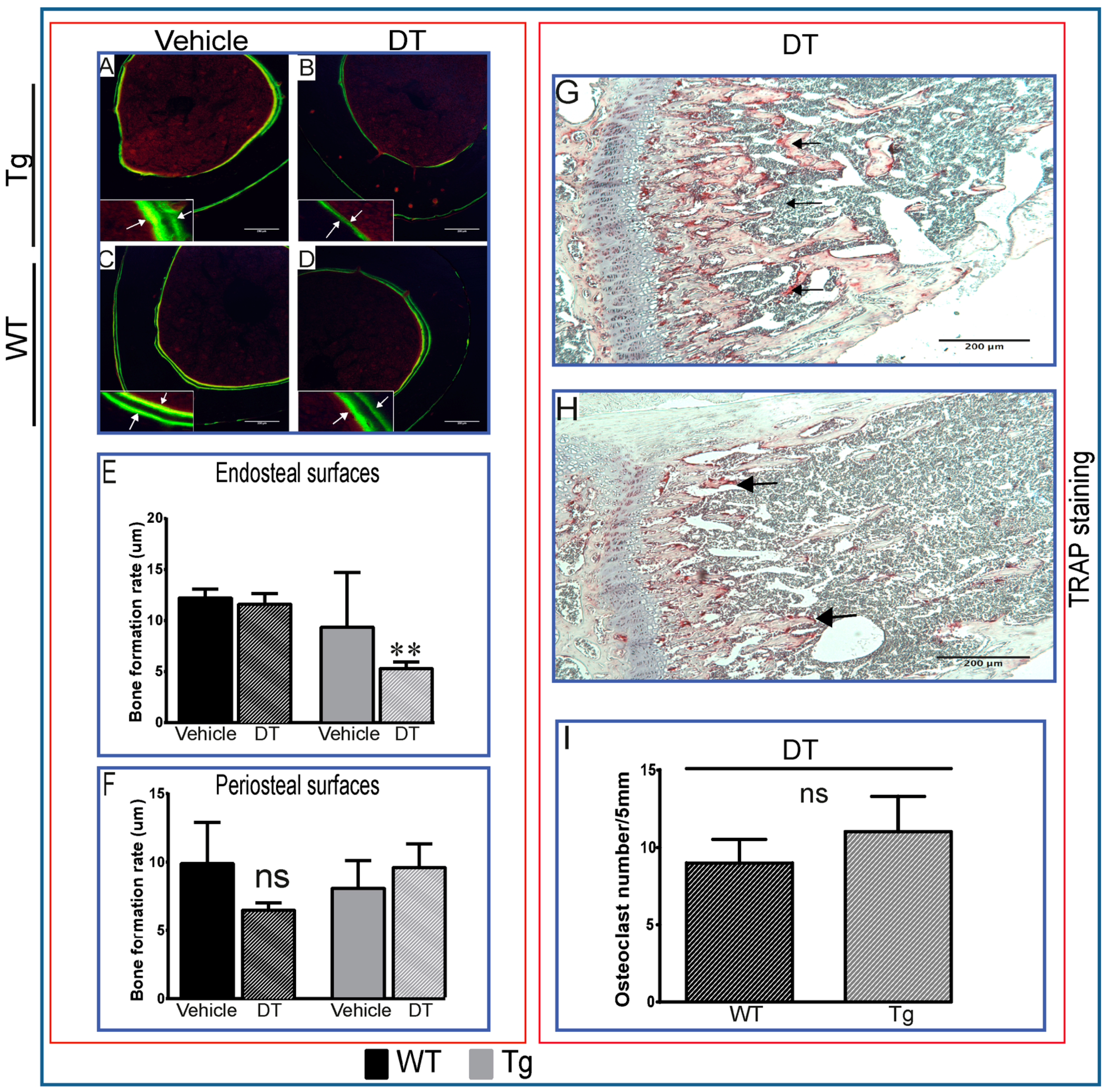{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. DT Treatment Leads to Changes in Bone Formation Despite Inefficient Ablation of Osteocytes
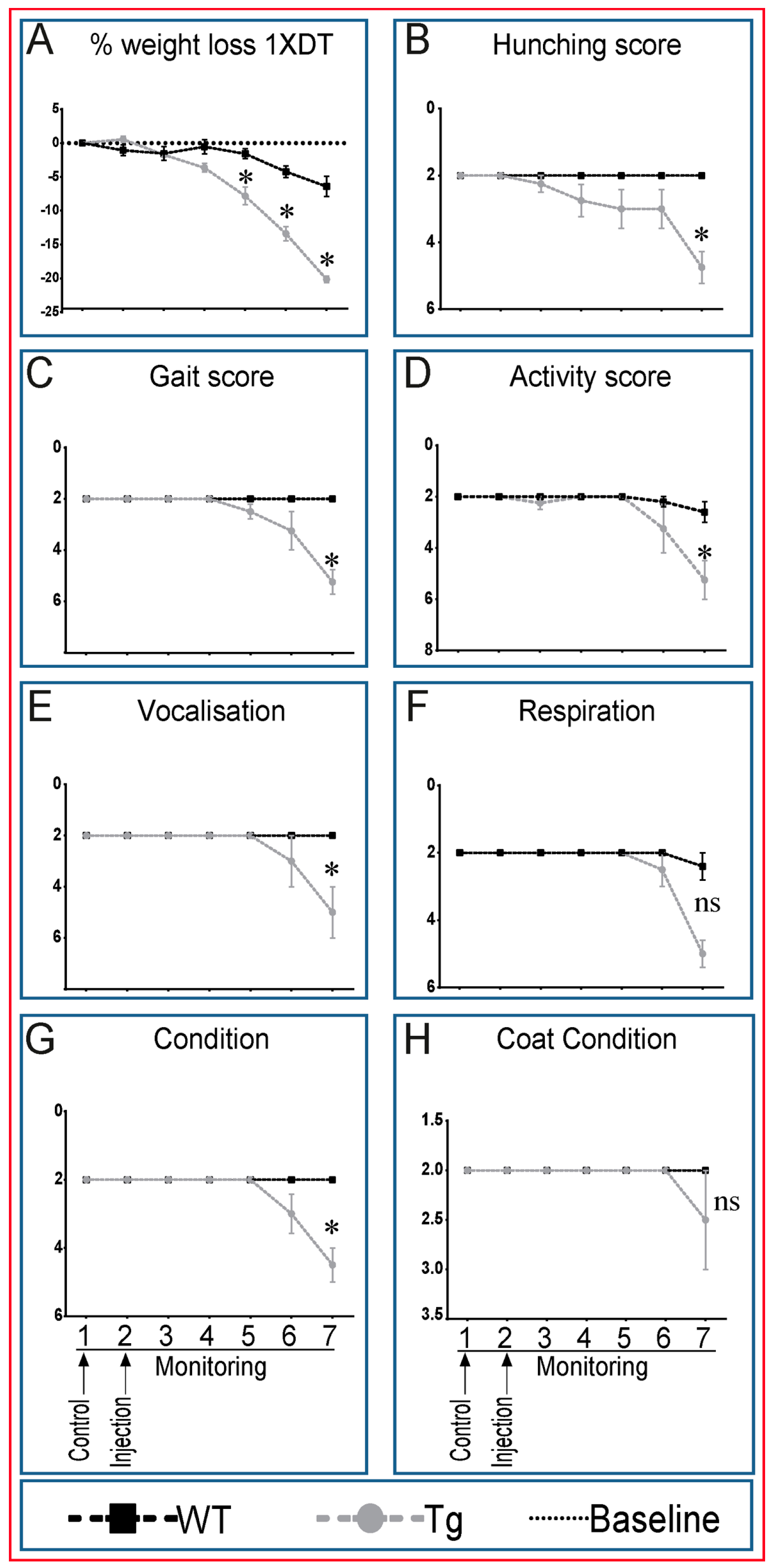
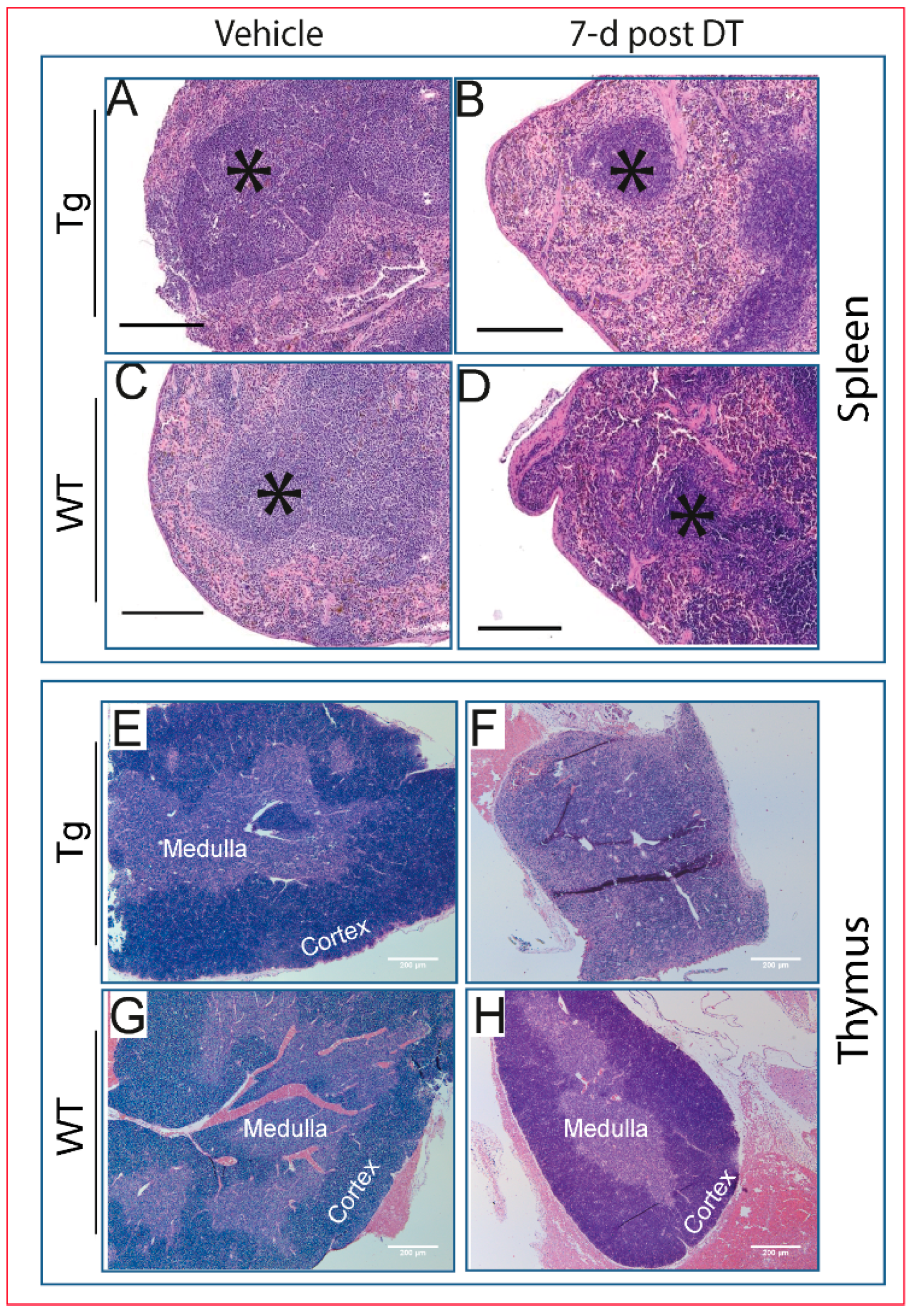
2.2. Transgenic Mice Exhibit Generalized Adverse Health Status with DT Treatment
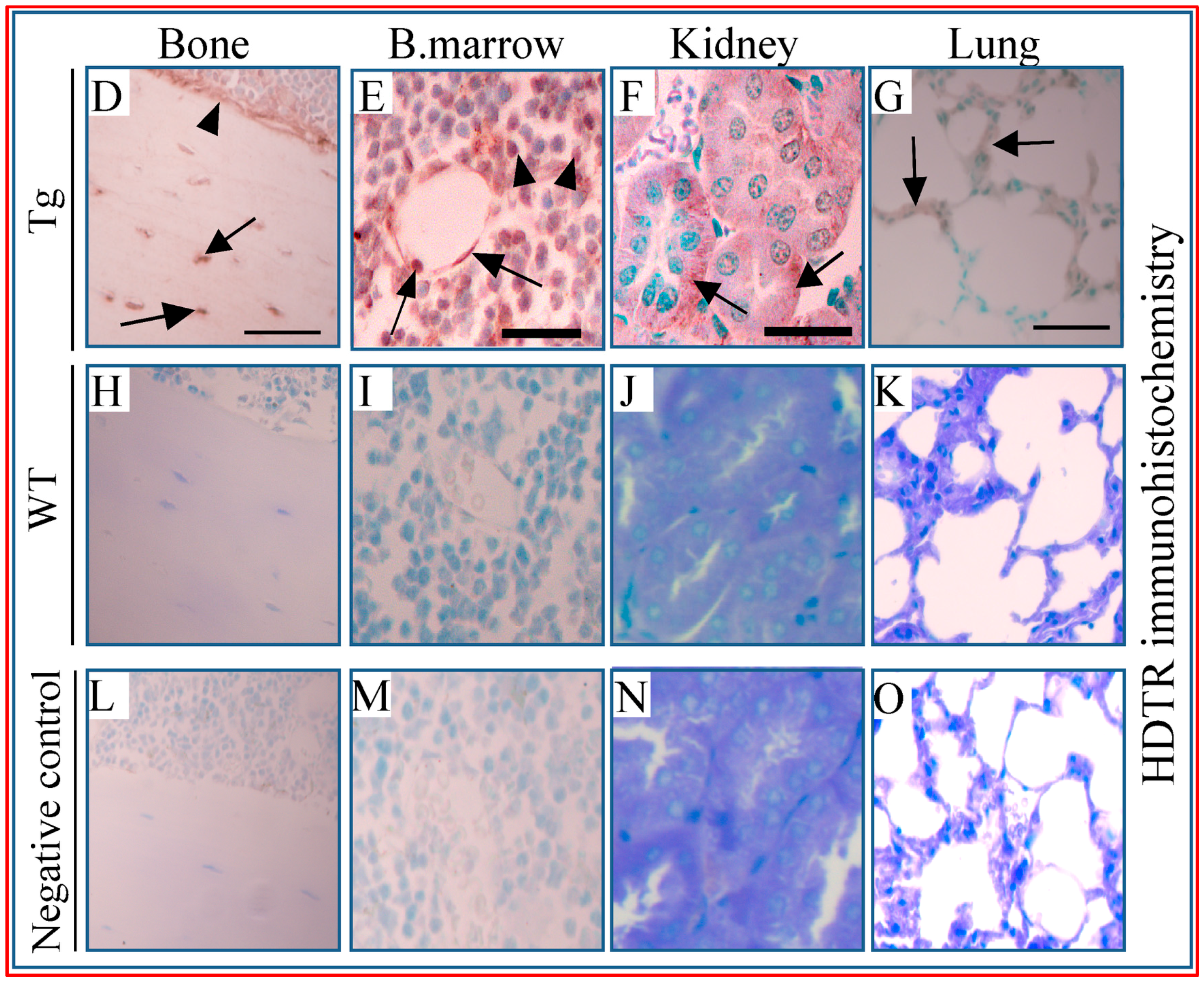
2.3. Multiple Tissues Show Misexpression of Dmp1-Driven HDTR mRNA and Broad Expression of Dmp1
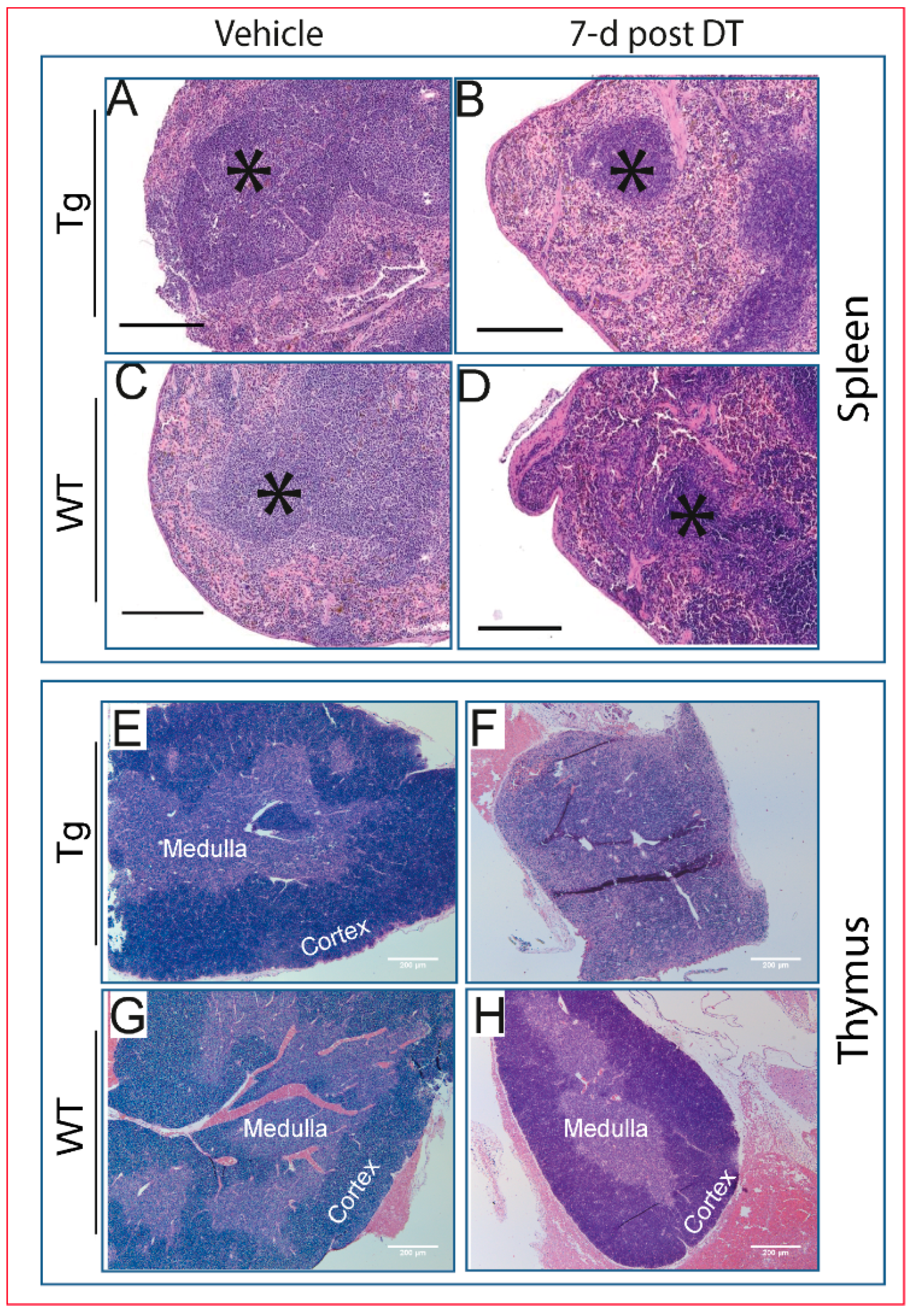
2.4. Pathological Changes Are Evident in Multiple Tissues Following DT Injection
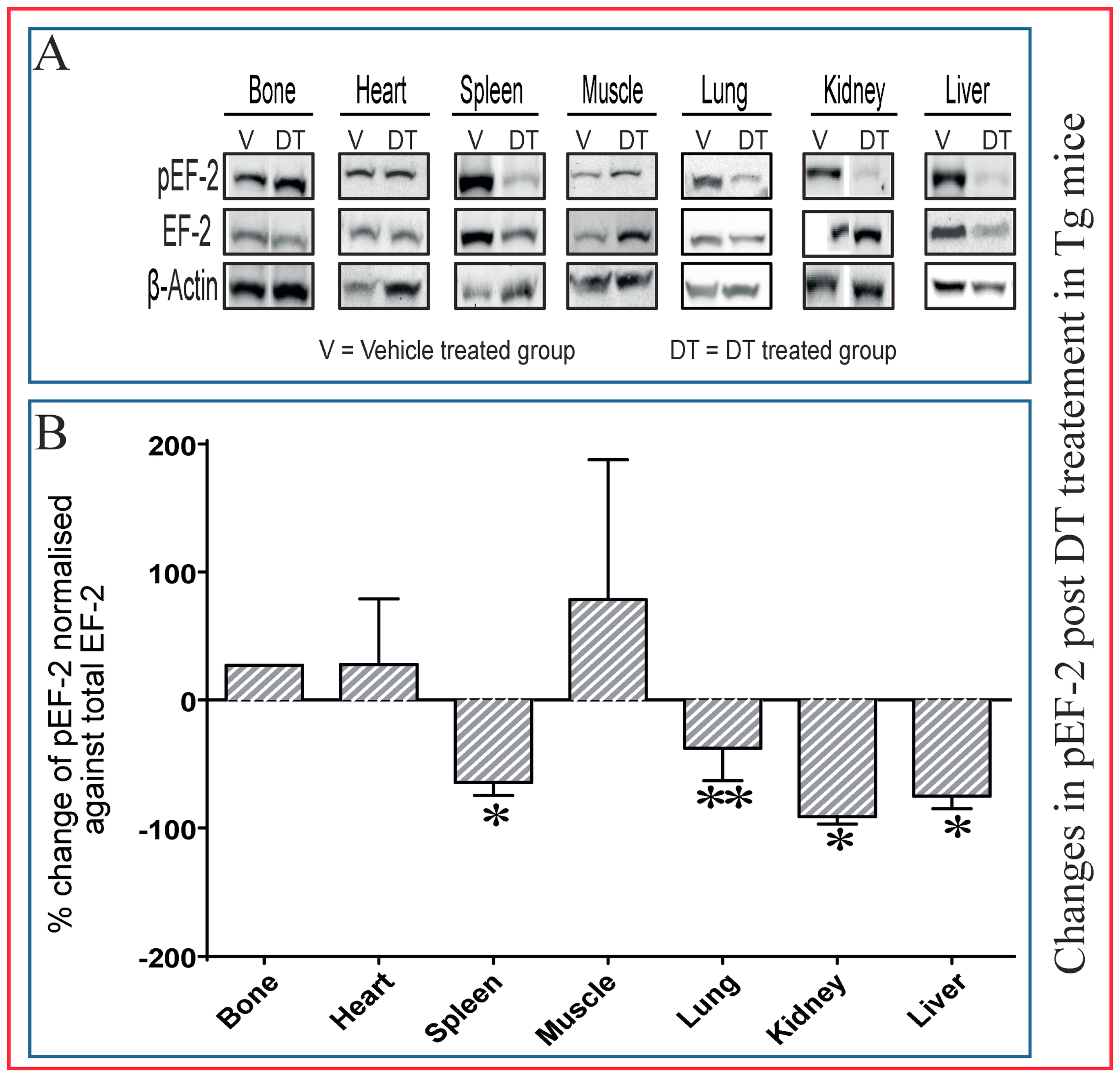
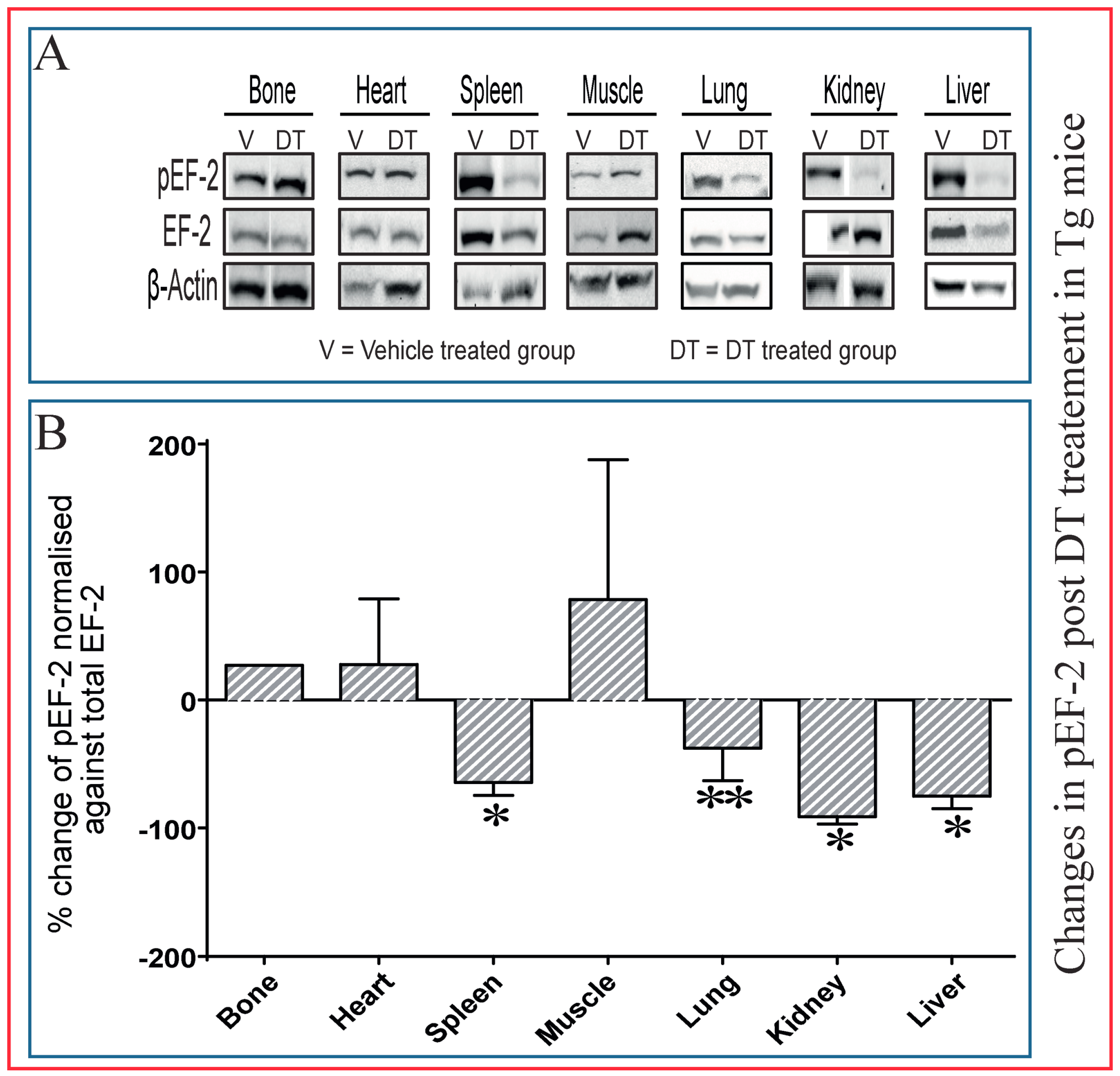
2.5. Dephosphorylation of EF-2 Reveals Direct DT Toxicity in Multiple Tissues in Tg Mice
3. Discussion
4. Materials and Methods
4.1. Animal Model
4.2. Diphtheria Toxin Administration
4.3. Calcium and Phosphorus Measurements
4.4. Health Evaluation
4.5. Histological Analysis
4.6. TUNEL Staining
4.7. TRAP Staining
4.8. Total RNA Isolation and PCR
4.9. Quantitative PCR
4.10. Western Blotting
4.11. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Manolagas, S.C. Birth and death of bone cells: Basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 2000, 21, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Klein-Nulend, J.; van der Plas, A.; Semeins, C.M.; Ajubi, N.E.; Frangos, J.A.; Nijweide, P.J.; Burger, E.H. Sensitivity of osteocytes to biomechanical stress in vitro. FASEB J. 1995, 9, 441–445. [Google Scholar] [PubMed]
- Weinbaum, S.; Cowin, S.C.; Zeng, Y. A model for the excitation of osteocytes by mechanical loading-induced bone fluid shear stresses. J. Biomech. 1994, 27, 339–360. [Google Scholar] [CrossRef]
- Rubin, C.T.; McLeod, K.J.; Bain, S.D. Functional strains and cortical bone adaptation: Epigenetic assurance of skeletal integrity. J. Biomech. 1990, 23, 43–54. [Google Scholar] [CrossRef]
- Pitsillides, A.A.; Rawlinson, S.C.F.; Suswillo, R.F.L.; Bourrin, S.; Zaman, G.; Lanyon, L.E. Mechanical strain-induced no production by bone cells: A possible role in adaptive bone (re)modeling? FASEB J. 1995, 9, 1614–1622. [Google Scholar] [PubMed]
- Bonewald, L.F. Mechanosensation and transduction in osteocytes. BoneKEy Osteovision 2006, 3, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Javaheri, B.; Stern, A.R.; Lara, N.; Dallas, M.; Zhao, H.; Liu, Y.; Bonewald, L.F.; Johnson, M.L. Deletion of a single β-catenin allele in osteocytes abolishes the bone anabolic response to loading. J. Bone Miner. Res. 2014, 29, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Klein-Nulend, J.; Bakker, A.D.; Bacabac, R.G.; Vatsa, A.; Weinbaum, S. Mechanosensation and transduction in osteocytes. Bone 2013, 54, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Kingsmill, V.J.; Boyde, A. Mineralisation density of human mandibular bone: Quantitative backscattered electron image analysis. J. Anat. 1998, 192, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Quarles, L.D. Skeletal secretion of FGF-23 regulates phosphate and vitamin D metabolism. Nat. Rev. Endocrinol. 2012, 8, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Poole, K.E.; van Bezooijen, R.L.; Loveridge, N.; Hamersma, H.; Papapoulos, S.E.; Löwik, C.W.; Reeve, J. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005, 19, 1842–1844. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, N.; Chong, W.H.; Gafni, R.I.; Collins, M.T. Fibroblast growth factor 23: State of the field and future directions. Trends Endocrinol. Metab. 2012, 23, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Kogianni, G.; Mann, V.; Noble, B.S. Apoptotic bodies convey activity capable of initiating osteoclastogenesis and localized bone destruction. J. Bone Miner. Res. 2008, 23, 915–927. [Google Scholar] [CrossRef] [PubMed]
- Verborgt, O.; Tatton, N.A.; Majeska, R.J.; Schaffler, M.B. Spatial distribution of BAX and BCL-2 in osteocytes after bone fatigue: Complementary roles in bone remodeling regulation? J. Bone Miner. Res. 2002, 17, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.H.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011, 17, 1235–1262. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, S.; Ishii, K.; Amizuka, N.; Li, M.; Kobayashi, T.; Kohno, K.; Ito, M.; Takeshita, S.; Ikeda, K. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007, 5, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Asada, N.; Katayama, Y.; Sato, M.; Minagawa, K.; Wakahashi, K.; Kawano, H.; Kawano, Y.; Sada, A.; Ikeda, K.; Matsui, T.; et al. Matrix-embedded osteocytes regulate mobilization of hematopoietic stem/progenitor cells. Cell Stem Cell 2013, 12, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Asada, N.; Kawano, Y.; Wakahashi, K.; Minagawa, K.; Kawano, H.; Sada, A.; Ikeda, K.; Matsui, T.; Katayama, Y. Osteocytes regulate primary lymphoid organs and fat metabolism. Cell Metab. 2013, 18, 749–758. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Plotkin, L.I.; Galli, C.; Goellner, J.J.; Gortazar, A.R.; Allen, M.R.; Robling, A.G.; Bouxsein, M.; Schipani, E.; Turner, C.H.; et al. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE 2008, 3, e2942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, Y.; Allen, M.R.; Condon, K.; Lezcano, V.; Ronda, A.C.; Galli, C.; Olivos, N.; Passeri, G.; O’Brien, C.A.; Bivi, N.; et al. PTH receptor signaling in osteocytes governs periosteal bone formation and intracortical remodeling. J. Bone Miner. Res. 2011, 26, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xie, Y.; Zhang, S.; Dusevich, V.; Bonewald, L.; Feng, J. Dmp1-targeted Cre expression in odontoblasts and osteocytes. J. Dent. Res. 2007, 86, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Powell, W.F., Jr.; Barry, K.J.; Tulum, I.; Kobayashi, T.; Harris, S.E.; Bringhurst, F.R.; Pajevic, P.D. Targeted ablation of the PTH/PTHrP receptor in osteocytes impairs bone structure and homeostatic calcemic responses. J. Endocrinol. 2011, 209, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Burkholder, T.; Foltz, C.; Karlsson, E.; Linton, C.G.; Smith, J.M. Health evaluation of experimental laboratory mice. Curr. Protoc. Mouse Biol. 2012, 2, 145–165. [Google Scholar] [PubMed]
- Foltz, C.J.; Ullman-Cullere, M. Guidelines for assessing the health and condition of mice. Lab. Anim. 1999, 28, 28–32. [Google Scholar]
- Al-Jazzar, A. The Role of Osteocytes in the Regulation of Bone Marrow Mesenchymal Stem Cells; Royal Veterinary College: London, UK, December 2016. [Google Scholar]
- Saito, M.; Iwawaki, T.; Taya, C.; Yonekawa, H.; Noda, M.; Inui, Y.; Mekada, E.; Kimata, Y.; Tsuru, A.; Kohno, K. Diphtheria toxin receptor-mediated conditional and targeted cell ablation in transgenic mice. Nat. Biotechnol. 2001, 19, 746–750. [Google Scholar] [CrossRef] [PubMed]
- Collier, R.J.; Kandel, J. Structure and activity of diptheria toxin. J. Biol. Chem. 1971, 246, 1496–1503. [Google Scholar] [PubMed]
- Gill, D.M.; Pappenheimer, A.M., Jr. Structure-activity relationships in diphtheria toxin. J. Biol. Chem. 1971, 246, 1492–1495. [Google Scholar] [PubMed]
- Murphy, J.R. Mechanism of diphtheria toxin catalytic domain delivery to the eukaryotic cell cytosol and the cellular factors that directly participate in the process. Toxins 2011, 3, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.; Bennett, M.J.; Fujii, G.; Curmi, P.M.; Kantardjieff, K.A.; Collier, R.J.; Eisenberg, D. The crystal structure of diphtheria toxin. Nature 1992, 357, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Kochi, S.K.; Collier, R.J. DNA fragmentation and cytolysis in u937 cells treated with diphtheria-toxin or other inhibitors of protein-synthesis. Exp. Cell Res. 1993, 208, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, P.F.; Saelinger, C.B. Inhibition of protein synthesis after intravenous or intramuscular challenge with diphtheria toxin. Infect. Immun. 1972, 6, 418–421. [Google Scholar] [PubMed]
- Li, C.C.; Xie, X.H.; Wang, X.F.; Sun, Y.; Liu, P.H.; Chen, L.; Qin, C.L. Differential expression and localization of dentin matrix protein 1 (Dmp1) fragments in mouse submandibular glands. J. Mol. Histol. 2013, 44, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Terasawa, M.; Shimokawa, R.; Terashima, T.; Ohya, K.; Takagi, Y.; Shimokawa, H. Expression of dentin matrix protein 1 (Dmp1) in nonmineralized tissues. J. Bone Miner. Metab. 2004, 22, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Ogbureke, K.U.E.; Fisher, L.W. Renal expression of sibling proteins and their partner matrix metalloproteinases (MMPs). Kidney Int. 2005, 68, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Ogbureke, K.U.E.; Fisher, L.W. Expression of siblings and their partner MMPs in salivary glands. J. Dent. Res. 2004, 83, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Dragomir, A.C.; Kocabayoglu, P.; Rahman, A.H.; Chow, A.; Hashimoto, D.; Leboeuf, M.; Kraus, T.; Moran, T.; Carrasco-Avino, G.; et al. Central role of conventional dendritic cells in regulation of bone marrow release and survival of neutrophils. J. Immunol. 2014, 192, 3374–3382. [Google Scholar] [CrossRef] [PubMed]
- Tittel, A.P.; Heuser, C.; Ohliger, C.; Llanto, C.; Yona, S.; Hammerling, G.J.; Engel, D.R.; Garbi, N.; Kurts, C. Functionally relevant neutrophilia in CD11c diphtheria toxin receptor transgenic mice. Nat. Methods 2012, 9, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Van Blijswijk, J.; Schraml, B.U.; Reis Sousa, C. Advantages and limitations of mouse models to deplete dendritic cells. Eur. J. Immunol. 2013, 43, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Unutmaz, D.; Wong, P.; Sano, G.-I.; De los Santos, K.; Sparwasser, T.; Wu, S.; Vuthoori, S.; Ko, K.; Zavala, F. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity 2002, 17, 211–220. [Google Scholar] [CrossRef]
- Probst, H.; Tschannen, K.; Odermatt, B.; Schwendener, R.; Zinkernagel, R.; van Den Broek, M. Histological analysis of CD11c-dtr/GFP mice after in vivo depletion of dendritic cells. Clin. Exp. Immunol. 2005, 141, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Henique, C.; Tharaux, P.L. Targeting signaling pathways in glomerular diseases. Curr. Opin. Nephrol. Hypertens. 2012, 21, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.B.; Sugimoto, K.; Dhawan, P.; Harris, R.C. Juxtacrine activation of EGFR regulates claudin expression and increases transepithelial resistance. Am. J. Physiol. Cell Physiol. 2007, 293, C1660–C1668. [Google Scholar] [CrossRef] [PubMed]
- Lorch, G.; Gilmore, J.L.; Koltz, P.F.; Gonterman, R.M.; Laughner, R.; Lewis, D.A.; Konger, R.L.; Nadella, K.S.; Toribio, R.E.; Rosol, T.J.; et al. Inhibition of epidermal growth factor receptor signalling reduces hypercalcaemia induced by human lung squamous-cell carcinoma in athymic mice. Br. J. Cancer 2007, 97, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Macleod, R.J.; Chattopadhyay, N.; Tfelt-Hansen, J.; Kifor, O.; Butters, R.R.; Brown, E.M. Calcium-sensing receptor activation stimulates parathyroid hormone-related protein secretion in prostate cancer cells: Role of epidermal growth factor receptor transactivation. Bone 2004, 35, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. Nih image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.Q.; Huang, H.; Lu, Y.; Ye, L.; Xie, Y.; Tsutsui, T.W.; Kunieda, T.; Castranio, T.; Scott, G.; Bonewald, L.B.; et al. The dentin matrix protein 1 (Dmp1) is specifically expressed in mineralized, but not soft, tissues during development. J. Dent. Res. 2003, 82, 776–780. [Google Scholar] [CrossRef] [PubMed]
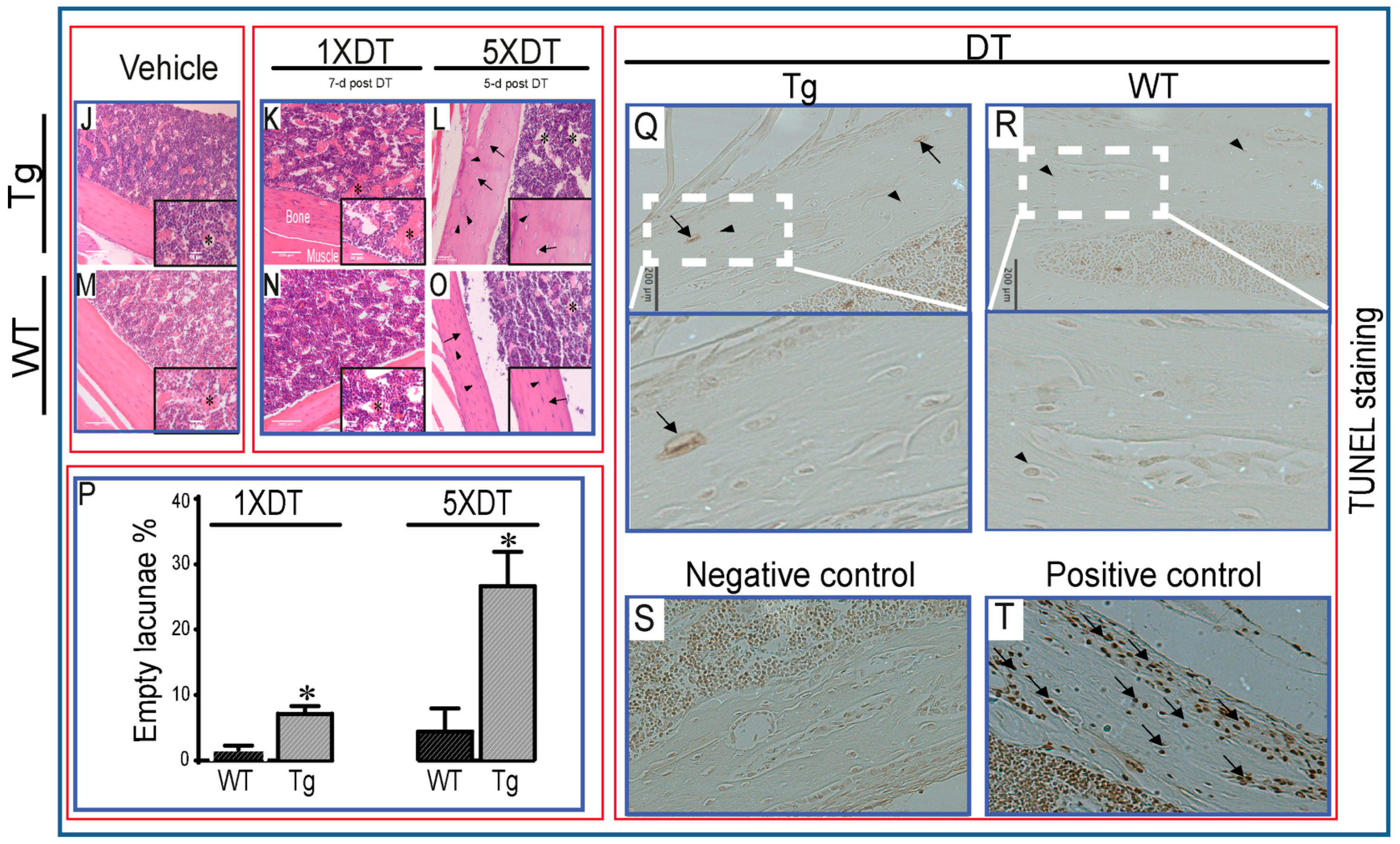
 ), viable osteocyte; shown by arrow (
), viable osteocyte; shown by arrow (  ), empty lacuna) was only observed in Tg mice treated with DT for five consecutive days (M) and only low levels (<10%) in WT and Tg mice after single DT treatment (P). Statistical comparisons: * p < 0.05 WT and Tg. Assessment of DT-induced apoptosis by TUNEL staining after single DT treatment in cortical bone of Tg (Q) and WT (R) revealed a very low number of apoptosis-positive osteocytes (arrow); arrowheads indicate negative cells. White-dotted boxes show a magnification of similar regions to better visualise the presence or lack of apoptotic cells. Negative (S) and positive (T) controls demonstrate a lack of staining in negative and many stained cells in positive control. Scale bar, 200 µm in the full overview images and 50 µm in the insets. Graphs represent the means ± SEM; ns = not significant.
), viable osteocyte; shown by arrow ( ), empty lacuna) was only observed in Tg mice treated with DT for five consecutive days (M) and only low levels (<10%) in WT and Tg mice after single DT treatment (P). Statistical comparisons: * p < 0.05 WT and Tg. Assessment of DT-induced apoptosis by TUNEL staining after single DT treatment in cortical bone of Tg (Q) and WT (R) revealed a very low number of apoptosis-positive osteocytes (arrow); arrowheads indicate negative cells. White-dotted boxes show a magnification of similar regions to better visualise the presence or lack of apoptotic cells. Negative (S) and positive (T) controls demonstrate a lack of staining in negative and many stained cells in positive control. Scale bar, 200 µm in the full overview images and 50 µm in the insets. Graphs represent the means ± SEM; ns = not significant.
), empty lacuna) was only observed in Tg mice treated with DT for five consecutive days (M) and only low levels (<10%) in WT and Tg mice after single DT treatment (P). Statistical comparisons: * p < 0.05 WT and Tg. Assessment of DT-induced apoptosis by TUNEL staining after single DT treatment in cortical bone of Tg (Q) and WT (R) revealed a very low number of apoptosis-positive osteocytes (arrow); arrowheads indicate negative cells. White-dotted boxes show a magnification of similar regions to better visualise the presence or lack of apoptotic cells. Negative (S) and positive (T) controls demonstrate a lack of staining in negative and many stained cells in positive control. Scale bar, 200 µm in the full overview images and 50 µm in the insets. Graphs represent the means ± SEM; ns = not significant.
), viable osteocyte; shown by arrow ( ), empty lacuna) was only observed in Tg mice treated with DT for five consecutive days (M) and only low levels (<10%) in WT and Tg mice after single DT treatment (P). Statistical comparisons: * p < 0.05 WT and Tg. Assessment of DT-induced apoptosis by TUNEL staining after single DT treatment in cortical bone of Tg (Q) and WT (R) revealed a very low number of apoptosis-positive osteocytes (arrow); arrowheads indicate negative cells. White-dotted boxes show a magnification of similar regions to better visualise the presence or lack of apoptotic cells. Negative (S) and positive (T) controls demonstrate a lack of staining in negative and many stained cells in positive control. Scale bar, 200 µm in the full overview images and 50 µm in the insets. Graphs represent the means ± SEM; ns = not significant.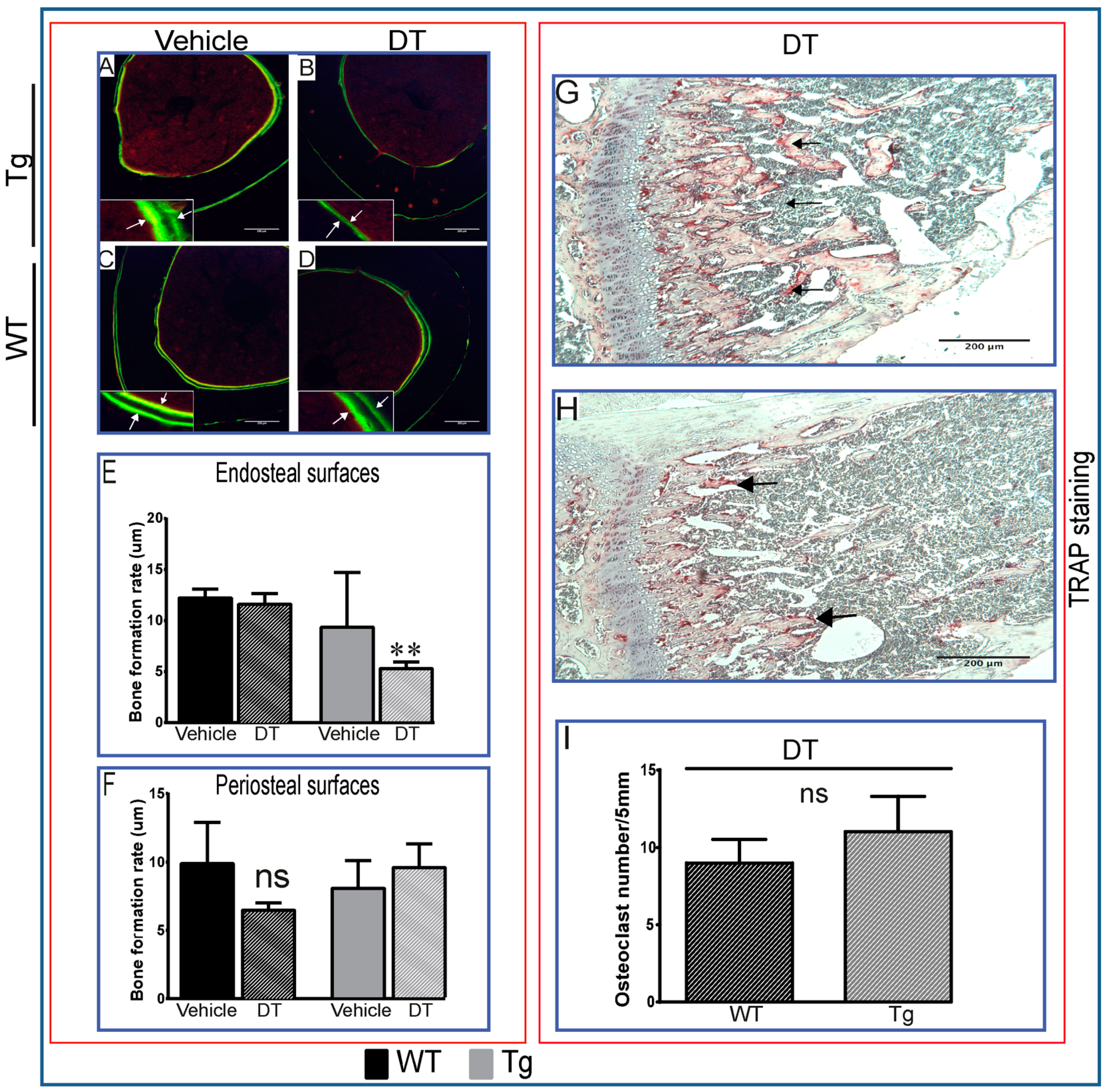


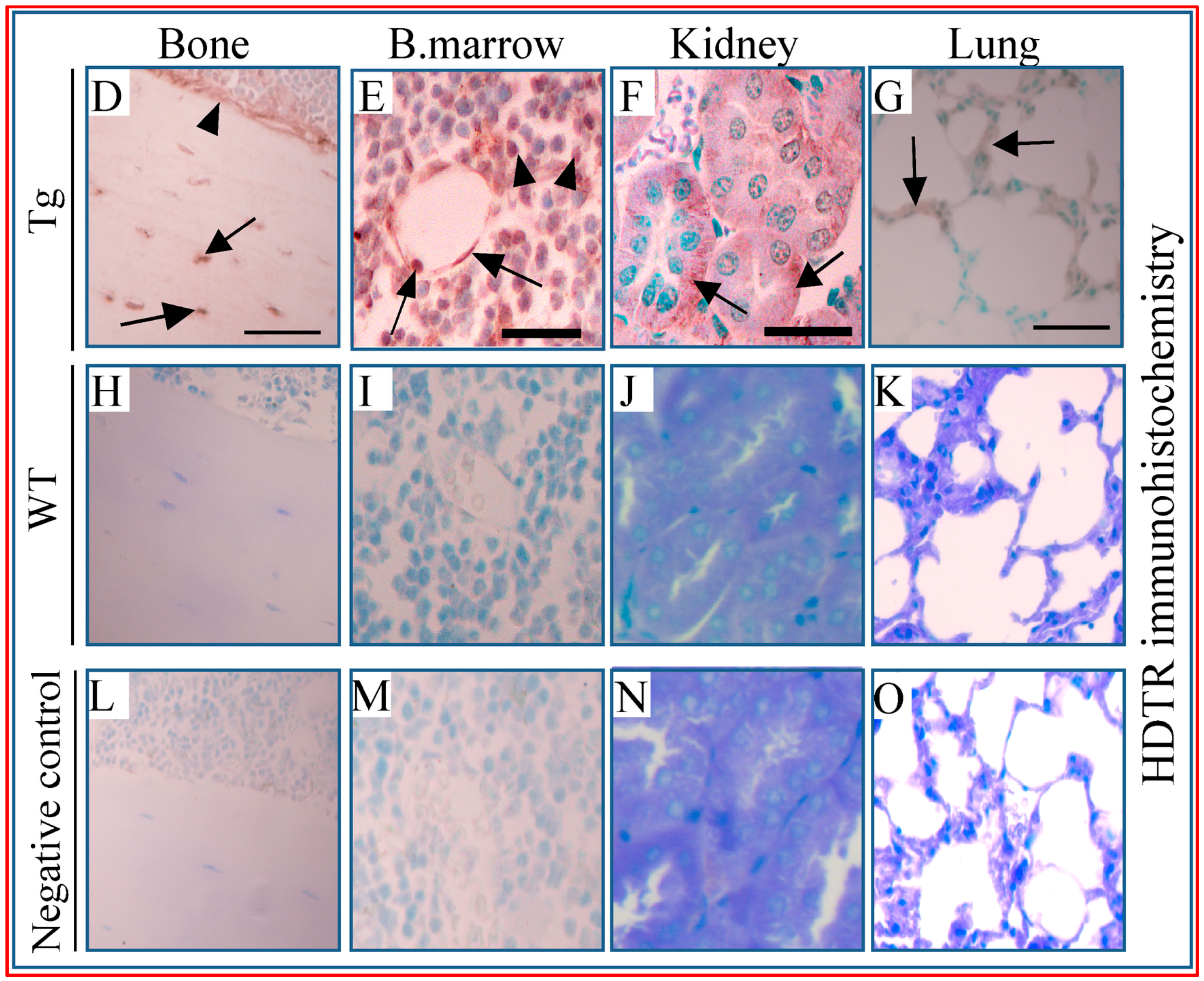
 ) as wells as osteoblasts (D, arrowhead;
) as wells as osteoblasts (D, arrowhead;  ), bone marrow cells (E, arrowhead) and around vessels (E, arrow), kidney tubules (F, arrow), and lung alveolar wall (G, arrow). Respective tissues show negative staining in WT (H–K). (L–O) are PBS negative controls. Scale bar, 200 µm.
) as wells as osteoblasts (D, arrowhead; ), bone marrow cells (E, arrowhead) and around vessels (E, arrow), kidney tubules (F, arrow), and lung alveolar wall (G, arrow). Respective tissues show negative staining in WT (H–K). (L–O) are PBS negative controls. Scale bar, 200 µm.
), bone marrow cells (E, arrowhead) and around vessels (E, arrow), kidney tubules (F, arrow), and lung alveolar wall (G, arrow). Respective tissues show negative staining in WT (H–K). (L–O) are PBS negative controls. Scale bar, 200 µm.
) as wells as osteoblasts (D, arrowhead; ), bone marrow cells (E, arrowhead) and around vessels (E, arrow), kidney tubules (F, arrow), and lung alveolar wall (G, arrow). Respective tissues show negative staining in WT (H–K). (L–O) are PBS negative controls. Scale bar, 200 µm.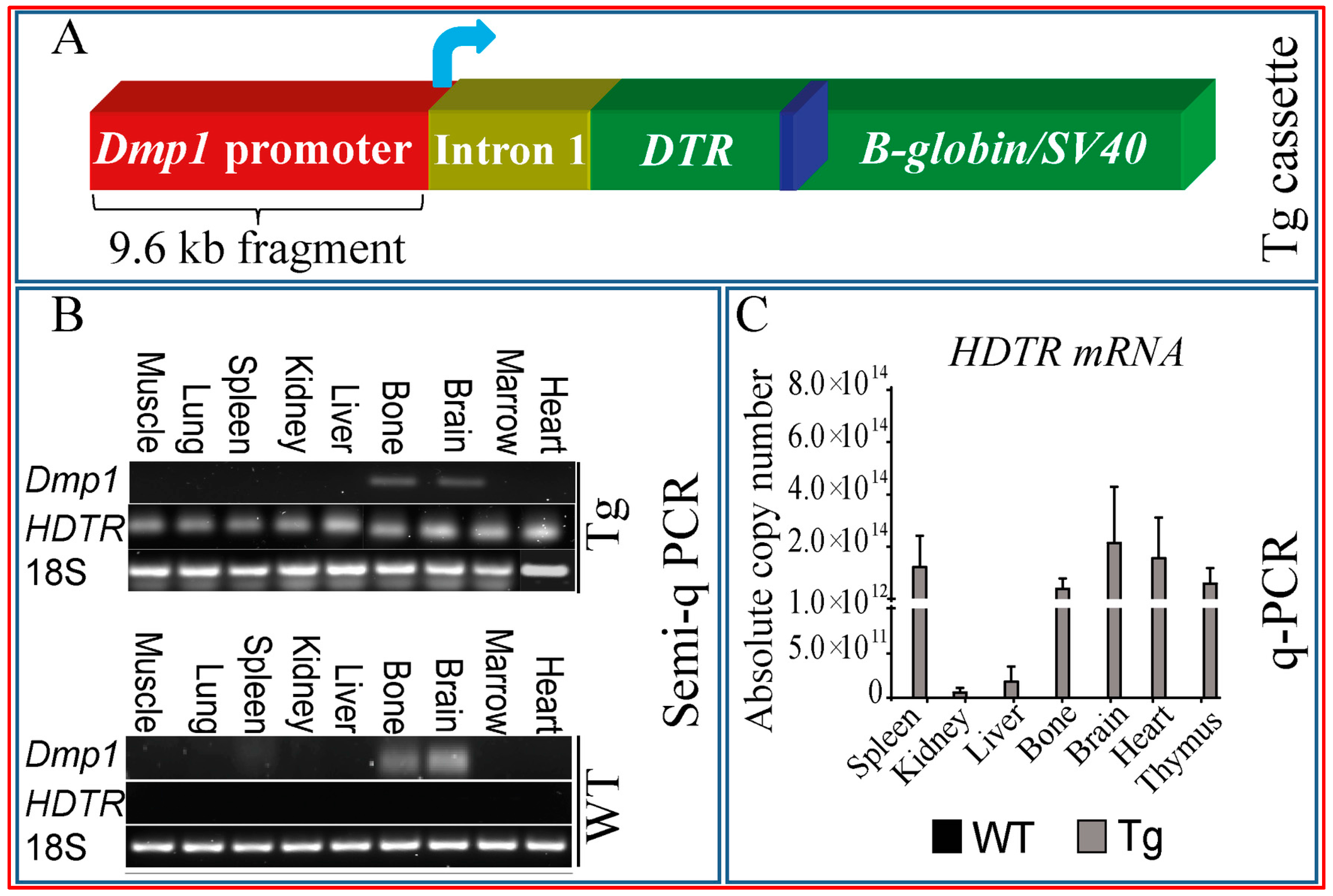
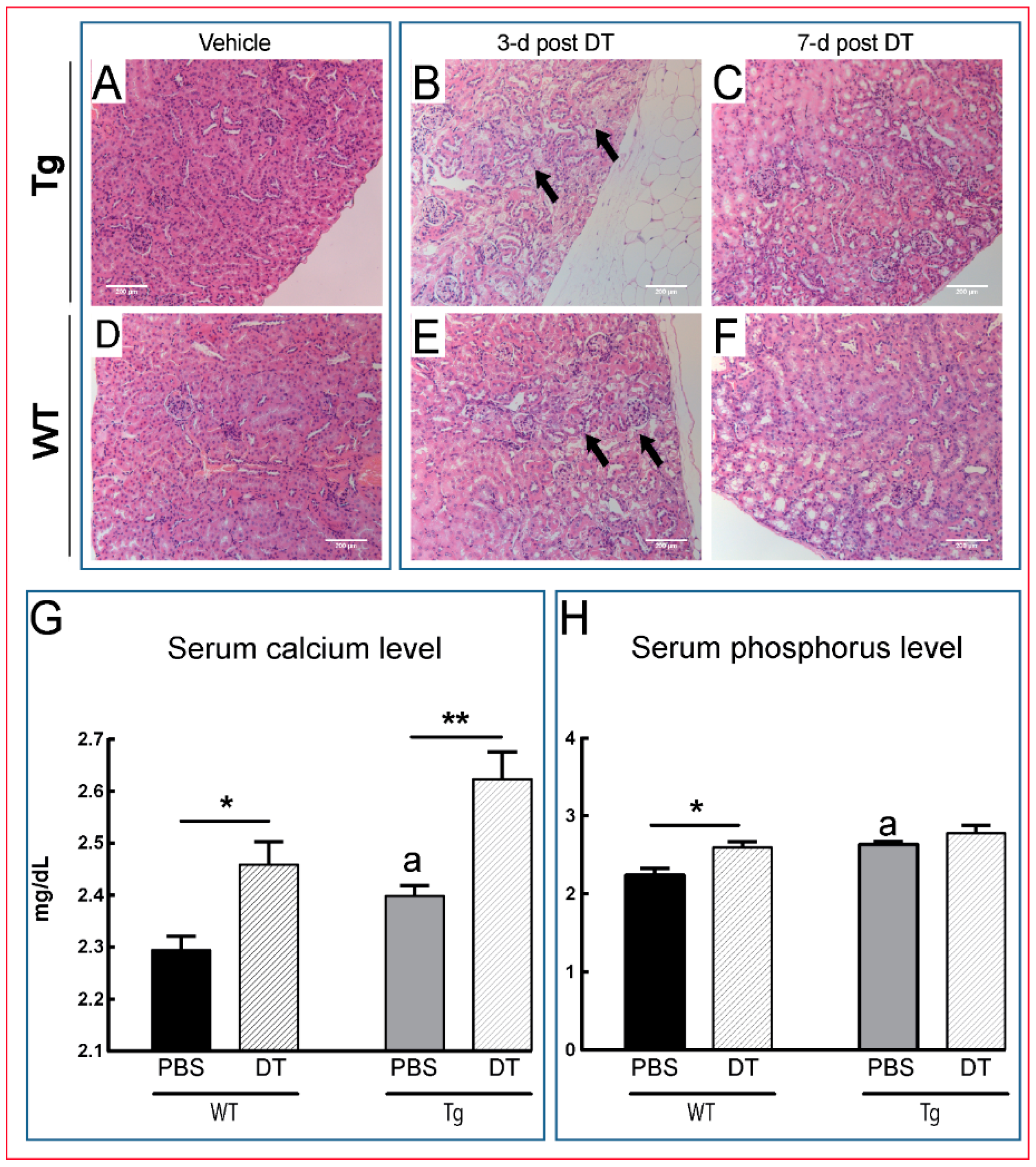
 ) as early as three days after DT treatment (B,E), before any overt osteocyte death (Figure 1J–P). Kidney shows an apparent recovery in these pathological changes seven days post DT (C,F) with increased serum calcium and phosphorous levels in treated Tg mice seven days post DT (G,H). Scale bar 200 µm. Graphs represent means ± SEM. Statistical comparisons: * p < 0.05 and ** p < 0.01 vehicle and DT treated within each genotype; (a) denotes significant differences between vehicle-treated WT and Tg mice (p < 0.05).
) as early as three days after DT treatment (B,E), before any overt osteocyte death (Figure 1J–P). Kidney shows an apparent recovery in these pathological changes seven days post DT (C,F) with increased serum calcium and phosphorous levels in treated Tg mice seven days post DT (G,H). Scale bar 200 µm. Graphs represent means ± SEM. Statistical comparisons: * p < 0.05 and ** p < 0.01 vehicle and DT treated within each genotype; (a) denotes significant differences between vehicle-treated WT and Tg mice (p < 0.05).
) as early as three days after DT treatment (B,E), before any overt osteocyte death (Figure 1J–P). Kidney shows an apparent recovery in these pathological changes seven days post DT (C,F) with increased serum calcium and phosphorous levels in treated Tg mice seven days post DT (G,H). Scale bar 200 µm. Graphs represent means ± SEM. Statistical comparisons: * p < 0.05 and ** p < 0.01 vehicle and DT treated within each genotype; (a) denotes significant differences between vehicle-treated WT and Tg mice (p < 0.05).
) as early as three days after DT treatment (B,E), before any overt osteocyte death (Figure 1J–P). Kidney shows an apparent recovery in these pathological changes seven days post DT (C,F) with increased serum calcium and phosphorous levels in treated Tg mice seven days post DT (G,H). Scale bar 200 µm. Graphs represent means ± SEM. Statistical comparisons: * p < 0.05 and ** p < 0.01 vehicle and DT treated within each genotype; (a) denotes significant differences between vehicle-treated WT and Tg mice (p < 0.05).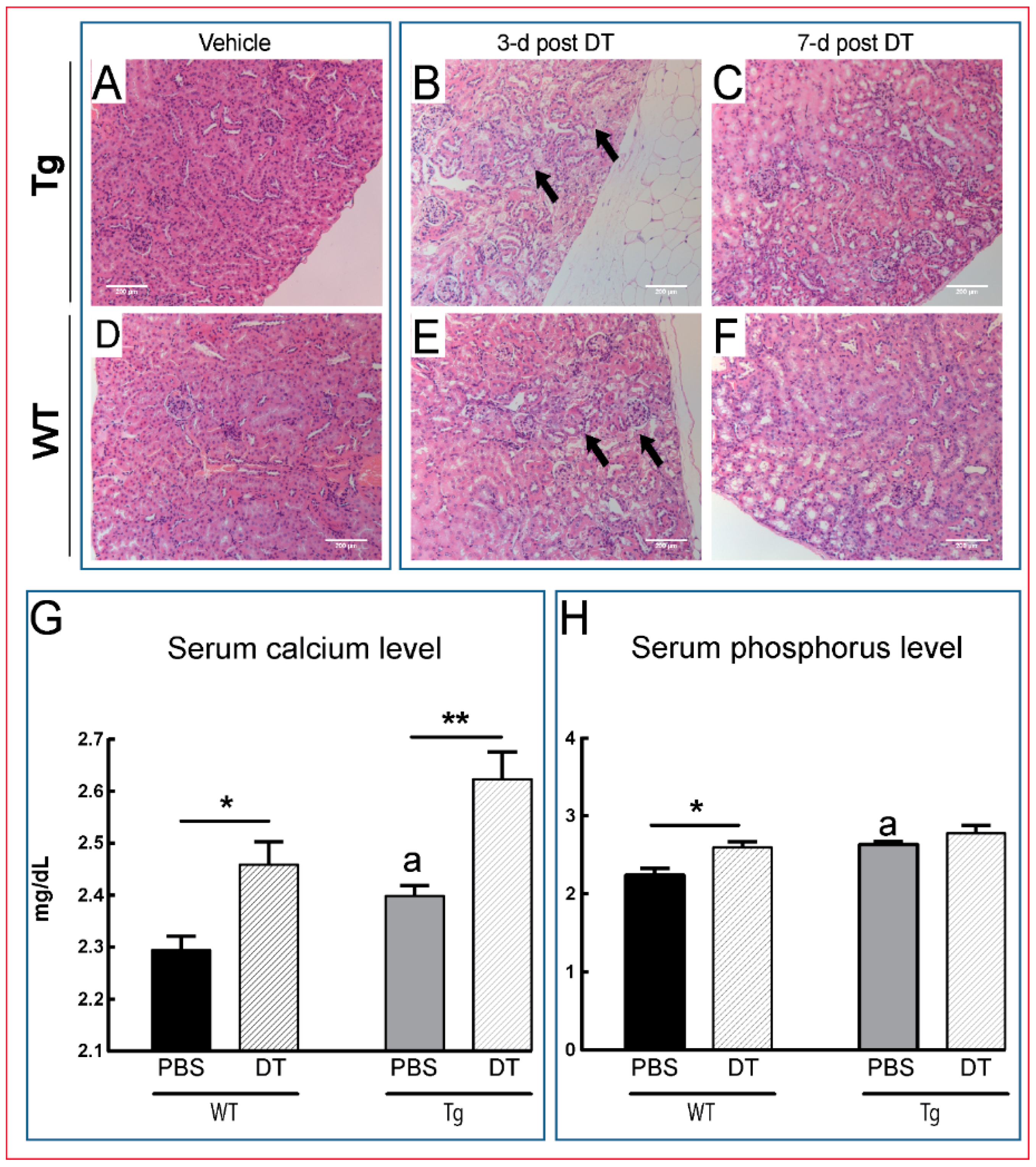
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Jazzar, A.; Javaheri, B.; Prideaux, M.; Boyde, A.; Scudamore, C.L.; Cherifi, C.; Hay, E.; Hopkinson, M.; Boyd, M.; Cohen-Solal, M.; et al. Dmp1 Promoter-Driven Diphtheria Toxin Receptor Transgene Expression Directs Unforeseen Effects in Multiple Tissues. Int. J. Mol. Sci. 2017, 18, 29. https://doi.org/10.3390/ijms18010029
Al-Jazzar A, Javaheri B, Prideaux M, Boyde A, Scudamore CL, Cherifi C, Hay E, Hopkinson M, Boyd M, Cohen-Solal M, et al. Dmp1 Promoter-Driven Diphtheria Toxin Receptor Transgene Expression Directs Unforeseen Effects in Multiple Tissues. International Journal of Molecular Sciences. 2017; 18(1):29. https://doi.org/10.3390/ijms18010029
Chicago/Turabian StyleAl-Jazzar, Ahmed, Behzad Javaheri, Matt Prideaux, Alan Boyde, Cheryl L. Scudamore, Chahrazad Cherifi, Eric Hay, Mark Hopkinson, Michael Boyd, Martine Cohen-Solal, and et al. 2017. "Dmp1 Promoter-Driven Diphtheria Toxin Receptor Transgene Expression Directs Unforeseen Effects in Multiple Tissues" International Journal of Molecular Sciences 18, no. 1: 29. https://doi.org/10.3390/ijms18010029