
Osteogenic Differentiation in Healthy and Pathological Conditions




Abstract
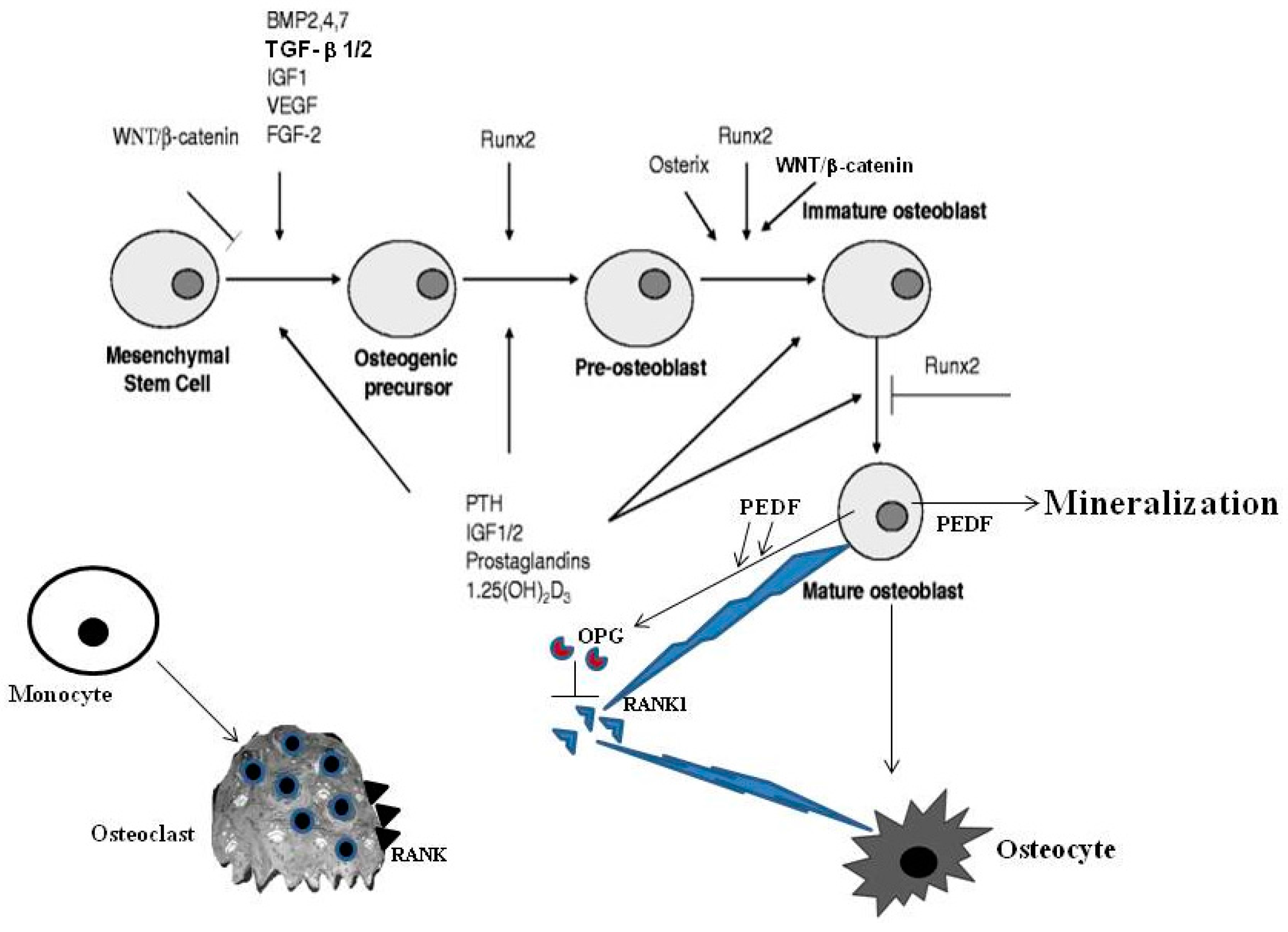
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Bone Remodeling
3. Molecular Pathways
4. MSC and Systemic Disorders Affecting Bone
5. MSCs and Heritable Bone Diseases
6. MSCs and Cancer
7. Clinical Applications of MSCs in Bone Regeneration and Repair
8. Concluding Remarks
Author Contributions
Conflicts of Interest
References
- Berendsen, A.D.; Olsen, B.R. Bone development. Bone 2015, 80, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Atkins, G.J.; Findlay, D.M. Osteocyte regulation of bone mineral: A little give and take. Osteoporos. Int. 2012, 23, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.P.; Paulson, C.; Shao, J.Z.; Zhang, X.; Wu, M.; Chen, W. Wnt and the Wnt signaling pathway in bone development and disease. Front. Biosci. 2014, 19, 379–407. [Google Scholar] [CrossRef]
- Li, M.; Li, Y.; Deng, W.; Zhang, Z.; Deng, Z.; Hu, Y.; Xia, W.; Xu, L. Chinese bone turnover marker study: Reference ranges for C-terminal telopeptide of type I collagen and procollagen I N-terminal peptide by age and gender. PLoS ONE 2014, 9, e103841. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Song, N.; Tombran-Tink, J.; Niyibizi, C. Pigment epithelium-derived factor enhances differentiation and mineral deposition of human mesenchymal stem cells. Stem Cells 2013, 31, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Bogan, R.; Riddle, R.C.; Li, Z.; Kumar, S.; Nandal, A.; Faugere, M.C.; Boskey, A.; Crawford, S.E.; Clemens, T.L. A mouse model for human osteogenesis imperfecta type VI. J. Bone Miner. Res. 2013, 28, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- Valenti, M.T.; Carbonare, L.D.; Mottes, M. Hypophosphatasia and Mesenchymal. Int. J. Stem. Cell Res. Ther. 2016, 3, 20. [Google Scholar]
- Cao, X.; Chen, D. The BMP signaling and in vivo bone formation. Gene 2005, 357, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Day, T.F.; Guo, X.; Garrett-Beal, L.; Yang, Y. Wnt/β-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev. Cell 2005, 8, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.O.; Insogna, K.L. Where Wnts went: The exploding field of Lrp5 and Lrp6 signaling in bone. J. Bone Miner. Res. 2009, 24, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Honma, M.; Ikebuchi, Y.; Kariya, Y.; Suzuki, H. Regulatory mechanisms of RANKL presentation to osteoclast precursors. Curr. Osteoporos. Rep. 2014, 12, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Takada, I.; Mihara, M.; Suzawa, M.; Ohtake, F.; Kobayashi, S.; Igarashi, M.; Youn, M.Y.; Takeyama, K.; Nakamura, T.; Mezaki, Y.; et al. A histone lysine methyltransferase activated by non-canonical Wnt signalling suppresses PPAR-γ transactivation. Nat. Cell Biol. 2007, 9, 1273–1285. [Google Scholar] [CrossRef] [PubMed]
- Dalle Carbonare, L.; Innamorati, G.; Valenti, M.T. Transcription factor Runx2 and its application to bone tissue engineering. Stem Cell Rev. 2012, 8, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Islam, R.; Yoon, W.J.; Ryoo, H.M. Pin1, the master orchestrator of bone cell differentiation. J. Cell. Physiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.K.; Young, D.W.; Montecino, M.; van Wijnen, A.J.; Stein, J.L.; Lian, J.B.; Stein, G.S. Bookmarking the genome: Maintenance of epigenetic information. J. Biol. Chem. 2011, 286, 18355–18361. [Google Scholar] [CrossRef] [PubMed]
- Jing, D.; Hao, J.; Shen, Y.; Tang, G.; Li, M.L.; Huang, S.H.; Zhao, Z.H. The role of microRNAs in bone remodeling. Int. J. Oral. Sci. 2015, 7, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Sugatani, T.; Hildreth, B.E., 3rd; Toribio, R.E.; Malluche, H.H.; Hruska, K.A. Expression of DGCR8-dependent micrornas is indispensable for osteoclastic development and bone-resorbing activity. J. Cell. Biochem. 2014, 115, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Qin, A.P.; Liao, B.; Shao, H.G.; Guo, L.J.; Xie, G.Q.; Yang, L.; Jiang, T.J. A novel microRNA regulates osteoclast differentiation via targeting protein inhibitor of activated STAT3 (PIAS3). Bone 2014, 67, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.F.; Yang, G.H.; Pan, X.H.; Zhang, S.J.; Zhao, C.; Qiu, B.S.; Gu, H.F.; Hong, J.F.; Cao, L.; Chen, Y.; et al. Altered microrna expression profile in exosomes during osteogenic differentiation of human bone marrow-derived mesenchymal stem cells. PLoS ONE 2014, 9, e114627. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Luan, J.; Li, H.; Zhou, X.; Han, J. Exosomes derived from mineralizing osteoblasts promote ST2 cell osteogenic differentiation by alteration of microRNA expression. FEBS Lett. 2016, 590, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Rosen, C.J.; Bouxsein, M.L. Mechanisms of disease: Is osteoporosis the obesity of bone? Nat. Clin. Pract. Rheumatol. 2006, 2, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Valenti, M.T.; Garbin, U.; Pasini, A.; Zanatta, M.; Stranieri, C.; Manfro, S.; Zucal, C.; Dalle Carbonare, L. Role of ox-PAPCs in the differentiation of mesenchymal stem cells (MSCs) and Runx2 and PPARγ2 expression in MSCs-like of osteoporotic patients. PLoS ONE 2011, 6, e20363. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, N.; Huang, X.; Xu, J.; Fernandes, J.C.; Dai, K.; Zhang, X. Dexamethasone shifts bone marrow stromal cells from osteoblasts to adipocytes by C/EBPα promoter methylation. Cell Death Dis. 2013, 4, e832. [Google Scholar] [CrossRef] [PubMed]
- Stenderup, K.; Justesen, J.; Clausen, C.; Kassem, M. Aging is associated with decreased maximal life span and accelerated senescence of bone marrow stromal cells. Bone 2003, 33, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Dalle Carbonare, L.; Matte, A.; Valenti, M.T.; Siciliano, A.; Mori, A.; Schweiger, V.; Zampieri, G.; Perbellini, L.; De Franceschi, L. Hypoxia-reperfusion affects osteogenic lineage and promotes sickle cell bone disease. Blood 2015, 126, 2320–2328. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.X.; O’Neill, K.D.; Allen, M.R.; Newman, C.L.; Moe, S.M. Low bone turnover in chronic kidney disease is associated with decreased VEGF-A expression and osteoblast differentiation. Am. J. Nephrol. 2015, 41, 464–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, Y.; Hayashi, Y.; Schlieve, C.R.; Ikeya, M.; Kim, H.; Nguyen, T.D.; Sami, S.; Baba, S.; Barruet, E.; Nasu, A.; et al. Induced pluripotent stem cells from patients with human fibrodysplasia ossificans progressiva show increased mineralization and cartilage formation. Orphanet J. Rare Dis. 2013, 8, 190. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Zuo, Y.; Chen, Y.; Song, L.; Zhu, Q.; Yu, J.; Shan, C.; Cai, Z.; Hao, J.; Kaplan, F.S.; et al. ACVR1-Fc suppresses BMP signaling and chondro-osseous differentiation in an in vitro model of fibrodysplasia ossificans progressiva. Bone 2016, 92, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.T.; Gao, Y.H.; Inada, M.; et al. Targeted disruption of CBFA1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef]
- Ding, B.; Li, C.; Xuan, K.; Liu, N.; Tang, L.; Liu, Y.; Guo, W.; Liu, W.; Jin, Y. The effect of the cleidocranial dysplasia-related novel 1116_1119insC mutation in the RUNX2 gene on the biological function of mesenchymal cells. Eur. J. Med. Genet. 2013, 56, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Forlino, A.; Marini, J.C. Osteogenesis imperfecta. Lancet 2016, 387, 1657–1671. [Google Scholar] [CrossRef]
- Glorieux, F.H.; Ward, L.M.; Rauch, F.; Lalic, L.; Roughley, P.J.; Travers, R. Osteogenesis imperfecta type VI: A form of brittle bone disease with a mineralization defect. J. Bone Miner. Res. 2002, 17, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.L. Hypophosphatasia: An overview of the disease and its treatment. Osteoporos. Int. 2015, 26, 2743. [Google Scholar] [CrossRef] [PubMed]
- Taketani, T.; Oyama, C.; Mihara, A.; Tanabe, Y.; Abe, M.; Hirade, T.; Yamamoto, S.; Bo, R.; Kanai, R.; Tadenuma, T.; et al. Ex vivo expanded allogeneic mesenchymal stem cells with bone marrow transplantation improved osteogenesis in infants with severe hypophosphatasia. Cell Transplant. 2015, 24, 1931–1943. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-de-Souza, P.; Comito, G.; Pons-Segura, C.; Taddei, M.L.; Gori, V.; Becherucci, V.; Bambi, F.; Margheri, F.; Laurenzana, A.; Del Rosso, M.; et al. Mesenchymal stem cells are recruited and activated into carcinoma-associated fibroblasts by prostate cancer microenvironment-derived TGF-β1. Stem Cells 2016. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-mesenchymal transition in cancer: Parallels between normal development and tumor progression. J. Mammary Gland Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Vergara, D.; Merlot, B.; Lucot, J.P.; Collinet, P.; Vinatier, D.; Fournier, I.; Salzet, M. Epithelial-mesenchymal transition in ovarian cancer. Cancer Lett. 2010, 291, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Valenti, M.T.; Serafini, P.; Innamorati, G.; Gili, A.; Cheri, S.; Bassi, C.; Dalle Carbonare, L. Runx2 expression: A mesenchymal stem marker for cancer. Oncol. Lett. 2016, 12, 4167–4172. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Yuan, J.; Li, K. EMT transcription factors: Implication in osteosarcoma. Med. Oncol. 2013, 30, 697. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Shin, Y.W.; Yang, K.H.; Kim, S.B.; Yoo, M.J.; Han, S.K.; Im, S.A.; Won, Y.D.; Sung, Y.B.; Jeon, T.S.; et al. A multi-center, randomized, clinical study to compare the effect and safety of autologous cultured osteoblast (OssronTM) injection to treat fractures. BMC Musculoskelet. Disord. 2009, 10, 20. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Cui, D.; Wang, B.; Tian, F.; Guo, L.; Yang, L.; Liu, B.; Yu, X. Treatment of early stage osteonecrosis of the femoral head with autologous implantation of bone marrow-derived and cultured mesenchymal stem cells. Bone 2012, 50, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.A.; Giannoudis, P.V.; Kouroupis, D. Bone repair with skeletal stem cells: Rationale, progress to date and clinical application. Ther. Adv. Musculoskelet. Dis. 2016, 8, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, E.M.; Gordon, P.L.; Koo, W.K.; Marx, J.C.; Neel, M.D.; McNall, R.Y.; Muul, L.; Hofmann, T. Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: Implications for cell therapy of bone. Proc. Natl. Acad. Sci. USA 2002, 99, 8932–8937. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.K.; Gotherstrom, C. Prenatal transplantation of mesenchymal stem cells to treat osteogenesis imperfecta. Front. Pharmacol. 2014, 5, 223. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valenti, M.T.; Dalle Carbonare, L.; Mottes, M. Osteogenic Differentiation in Healthy and Pathological Conditions. Int. J. Mol. Sci. 2017, 18, 41. https://doi.org/10.3390/ijms18010041
Valenti MT, Dalle Carbonare L, Mottes M. Osteogenic Differentiation in Healthy and Pathological Conditions. International Journal of Molecular Sciences. 2017; 18(1):41. https://doi.org/10.3390/ijms18010041
Chicago/Turabian StyleValenti, Maria Teresa, Luca Dalle Carbonare, and Monica Mottes. 2017. "Osteogenic Differentiation in Healthy and Pathological Conditions" International Journal of Molecular Sciences 18, no. 1: 41. https://doi.org/10.3390/ijms18010041
APA StyleValenti, M. T., Dalle Carbonare, L., & Mottes, M. (2017). Osteogenic Differentiation in Healthy and Pathological Conditions. International Journal of Molecular Sciences, 18(1), 41. https://doi.org/10.3390/ijms18010041