Proteome Stability as a Key Factor of Genome Integrity


Abstract
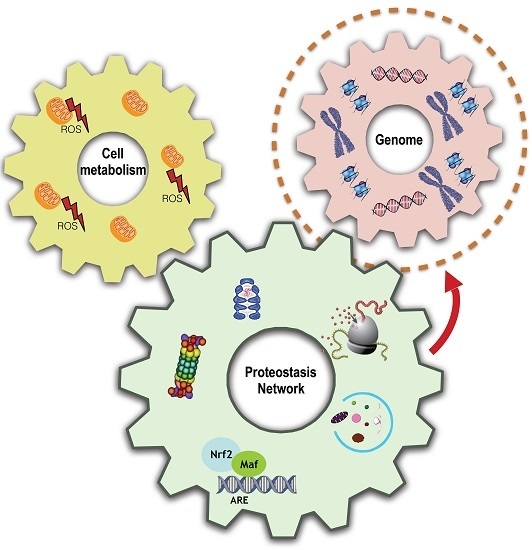
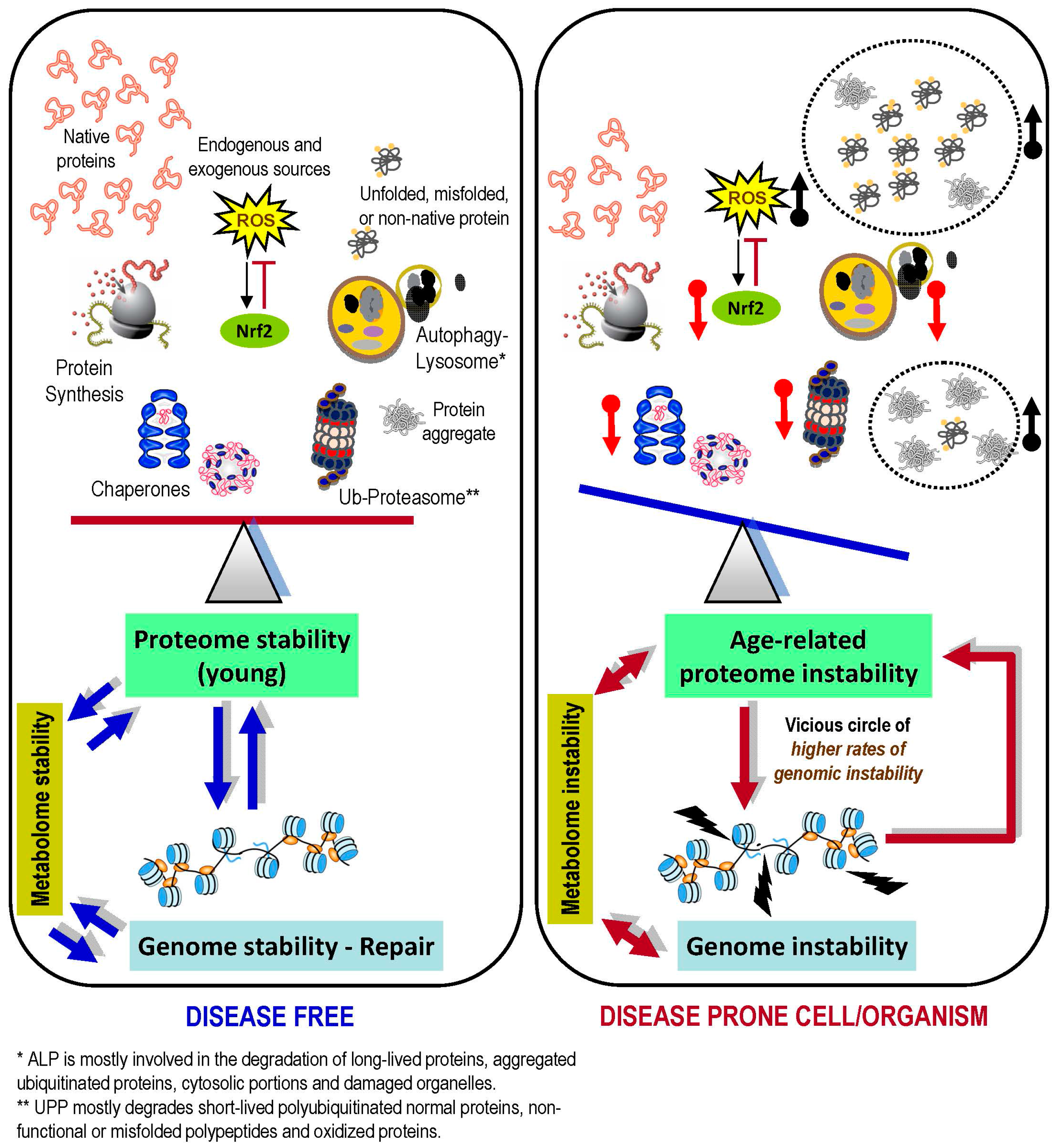
:
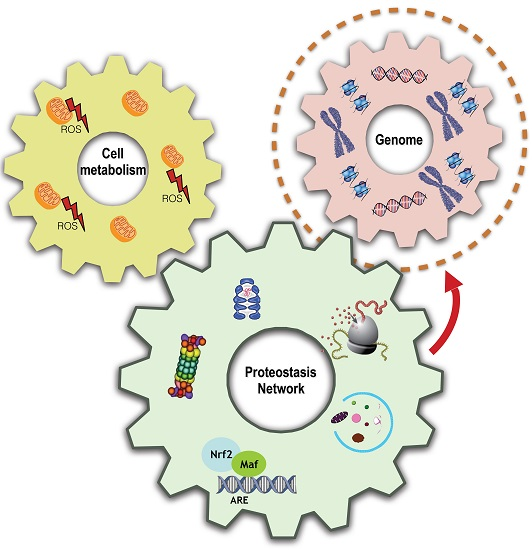{kind=link}
{kind=link}
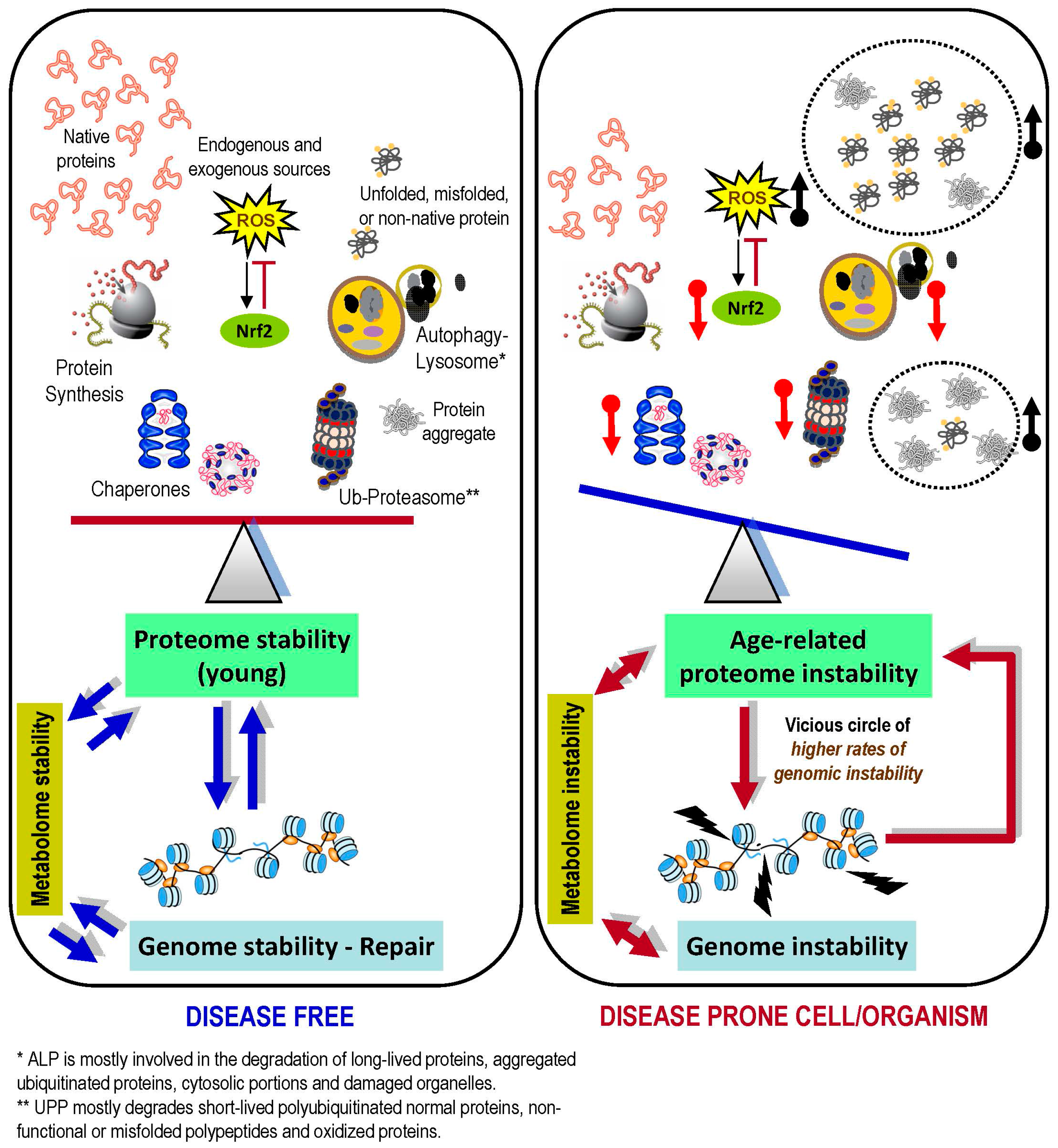{kind=link}
1. Introduction
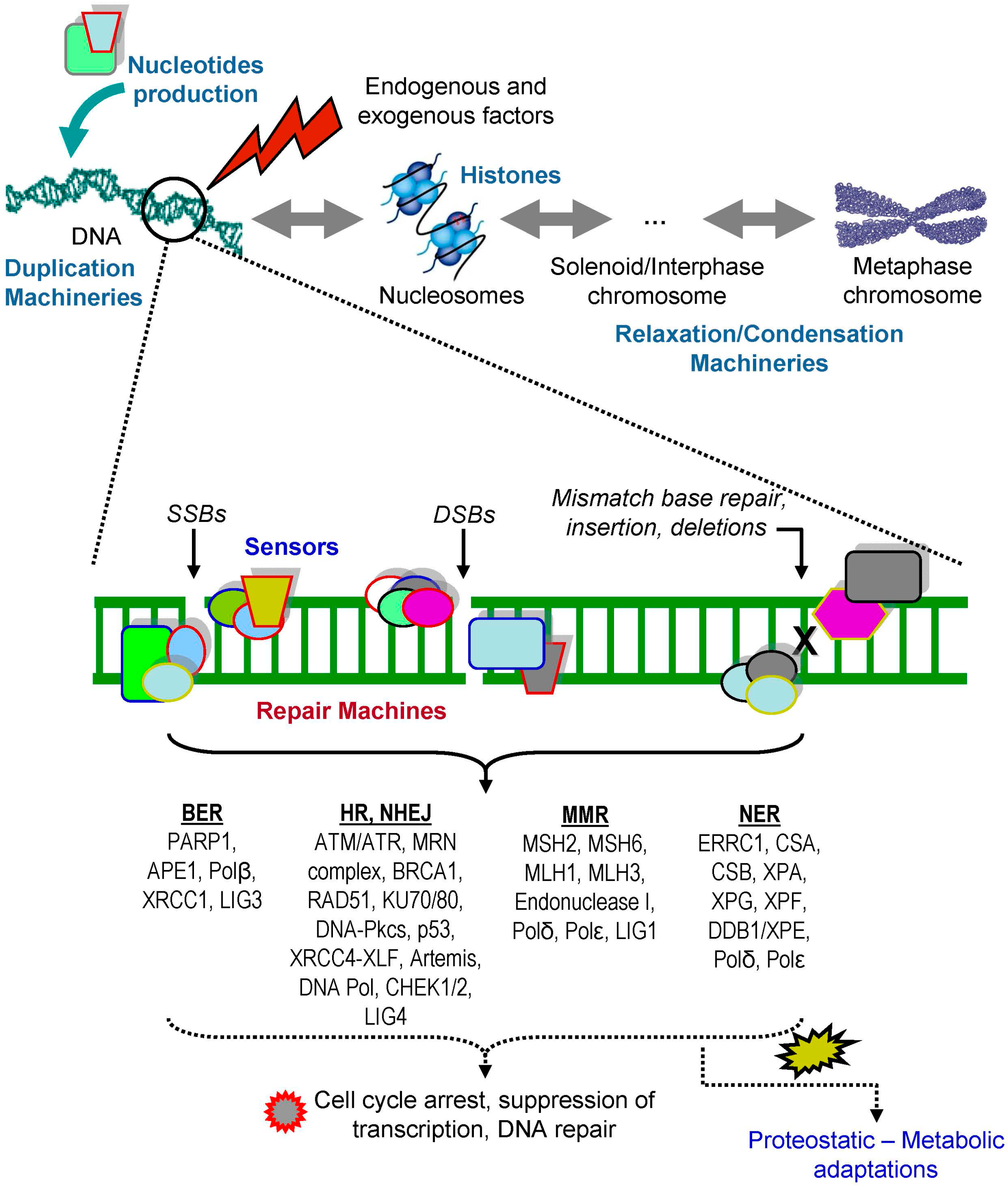
2. DNA Damage Responses
3. The Proteostasis Network
4. Endoplasmic Reticulum Unfolded Protein Response (UPRER) in Genome Instability
5. Oxidative Stress in Genome Integrity
6. Impact of Molecular Chaperones Function on Genome Stability
7. Ubiquitin-Proteasome Pathway (UPP) and Genome Integrity
8. The Ubiquitin-Proteasome liaison in DNA Damaged Responses (DDR)
8.1. Ubiquitination and Sumoylation
8.2. Proteasomal Degradation of DDR Factors
9. Autophagy-Lysosome Pathway (ALP) in Genome Integrity
10. Concluding Remarks
Acknowledgments
Conflicts of Interests
Abbreviations
| 53BP1 | p53-binding protein 1 |
| ALP | autophagy-lysosome pathway |
| AMPK | adenosine monophosphate-activated protein kinase |
| AP | apurinic/apyrimidinic sites |
| ATF | activating transcription factor |
| ATM | ataxia telangiectasia mutated |
| ATR | ataxia telangiectasia mutated and Rad3 related |
| ATRIP | ataxia telangiectasia mutated and Rad3 related interacting protein |
| BER | base-excision repair |
| BRCA1 | breast cancer type 1 susceptibility protein |
| CDK | cyclin-dependent kinase |
| CHEK1 | checkpoint kinase-1 |
| CHEK2 | checkpoint kinase-2 |
| DDR | DNA damage response |
| DNA-PKcs | DNA-dependent protein kinase catalytic subunit |
| DSB | double strand break |
| ER | endoplasmic reticulum |
| ERAD | endoplasmic reticulum-associated degradation |
| FA | Fanconi anemia complex |
| FIP200 | 200-kDa FAK-family-interacting protein |
| FLNA | filamin A |
| FoxO | forkhead box O |
| GPX1 | glutathione peroxidase 1 |
| GSTD1 | glutathione S-transferase 1 |
| HDACs | histone deacetylases |
| HR | homologous recombination |
| HIF-1 | hypoxia-inducible factor 1 |
| Hsf1 | heat shock factor 1 |
| Hsp | heat shock protein |
| IRE1 | inositol-requiring enzyme 1 |
| IR | ionizing radiation |
| Keap1 | kelch-like ECH-associated protein 1 |
| KLHL15 | kelch-like protein 15 |
| LAD | lamin-associated domain |
| maf | v-Maf avian musculoaponeurotic fibrosarcoma oncogene homolog |
| MDC1 | mediator of DNA damage checkpoint 1 |
| MDM2 | mouse double minute 2 homolog |
| MMR | mismatch repair |
| MRN | MRE11/RAD50/NBS1 complex |
| NQO1 | NA(D)PH quonine dehydrogenase 1 |
| NER | nucleotide excision repair |
| NHEJ | non-homologous end joining |
| Nrf2 | nuclear factor erythroid 2-related factor 2 |
| PERK | protein kinase R-like endoplasmic reticulum kinase |
| PIAS | protein inhibitor of activated STAT |
| PICHROS | protein-induced chromatin stress |
| PIKK | phosphatidylinositol 3-kinase-related kinase |
| PKR | protein kinase R |
| PN | proteostasis network |
| PTM | post-translational modification |
| RB1CC1 | RB1-inducible coiled-coil 1 |
| RNF168 | ring finger protein 168 |
| ROS | reactive oxygen species |
| SIRT1 | sirtuin 1 |
| sHSP | small heat-shock protein |
| SOD | superoxide dismutase |
| SSB | single strand brake |
| TIGAR | TP53-inducible glycolysis and apoptosis regulator |
| TOR | target of rapamycin |
| UBD | ubiquitin binding domain |
| UBL | ubiquitin-like protein |
| ULK-1 | unc-51 like autophagy activating kinase 1 |
| UPP | ubiquitin-proteasome pathway |
| UPR | unfolded protein response |
| UPRER | endoplasmic reticulum unfolded protein response |
| UV | ultra violet |
| VCP | valosin-containing protein |
| XBP1 | X box-binding protein |
References
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Tsakiri, E.N.; Trougakos, I.P. The amazing ubiquitin-proteasome system: Structural components and implication in aging. Int. Rev. Cell Mol. Biol. 2015, 314, 171–237. [Google Scholar] [PubMed]
- Vilchez, D.; Saez, I.; Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 2014, 5, 5659. [Google Scholar] [CrossRef] [PubMed]
- Akerfelt, M.; Morimoto, R.I.; Sistonen, L. Heat shock factors: Integrators of cell stress, development and lifespan. Nat. Rev. Mol. Cell. Biol. 2010, 11, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.; Grune, T. PARP-mediated proteasome activation: A co-ordination of DNA repair and protein degradation? Bioessays 2002, 24, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I.; Cuervo, A.M. Proteostasis and the aging proteome in health and disease. J. Gerontol. Ser. A 2014, 69, S33–S38. [Google Scholar] [CrossRef] [PubMed]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F.M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Seto, E. HATs and HDACs: From structure, function and regulation to novel strategies for therapy and prevention. Oncogene 2007, 26, 5310–5318. [Google Scholar] [CrossRef] [PubMed]
- Tarrant, M.K.; Cole, P.A. The chemical biology of protein phosphorylation. Annu. Rev. Biochem. 2009, 78, 797–825. [Google Scholar] [CrossRef] [PubMed]
- Welchman, R.L.; Gordon, C.; Mayer, R.J. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat. Rev. Mol. Cell Biol. 2005, 6, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Zharkov, D.O. Base excision DNA repair. Cell. Mol. Life Sci. 2008, 65, 1544–1565. [Google Scholar] [CrossRef] [PubMed]
- Kolodner, R.D.; Marsischky, G.T. Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 1999, 9, 89–96. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Erie, D.A. DNA mismatch repair. Annu. Rev. Biochem. 2005, 74, 681–710. [Google Scholar] [CrossRef] [PubMed]
- Plotz, G.; Raedle, J.; Brieger, A.; Trojan, J.; Zeuzem, S. hMutSα forms an ATP-dependent complex with hMutLα and hMutLβ on DNA. Nucleic Acids Res. 2002, 30, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Li, G.M.; Modrich, P. Restoration of mismatch repair to nuclear extracts of H6 colorectal tumor cells by a heterodimer of human MutL homologs. Proc. Natl. Acad. Sci. USA 1995, 92, 1950–1954. [Google Scholar] [CrossRef] [PubMed]
- Li, G.M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Hanawalt, A.; Spivak, G. Transcription-coupled DNA repair: Two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008, 9, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Spivak, G. Nucleotide excision repair in humans. DNA Repair 2015, 36, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Sugasawa, K. Molecular mechanisms of DNA damage recognition for mammalian nucleotide excision repair. DNA Repair 2016, 44, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Zha, S.; Guo, C.; Boboila, C.; Oksenych, V.; Cheng, H.L.; Zhang, Y.; Wesemann, D.R.; Yuen, G.; Patel, H.; Goff, P.H.; et al. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature 2011, 469, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Hiom, K. Coping with DNA double strand breaks. DNA Repair 2010, 9, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- McVey, M.; Lee, S.E. MMEJ repair of double-strand breaks (director’s cut): Deleted sequences and alternative endings. Trends Genet. 2008, 24, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.V.; Graham, M.E.; Jakob, B.; Tobias, F.; Kijas, A.W.; Tanuji, M.; Chen, P.; Robinson, P.J.; Taucher-Scholz, G.; Suzuki, K.; et al. Autophosphorylation and ATM activation: Additional sites add to the complexity. J. Biol. Chem. 2011, 286, 9107–9119. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, D.; Xu, X.; Zhong, X.; Ahmed, F.; Zhong, J.; Liao, J.; Dykxhoorn, D.M.; Weinstock, D.M.; Pfeifer, G.P.; Lieberman, J. A PP4-phosphatase complex dephosphorylates γ-H2AX generated during DNA replication. Mol. Cell 2008, 31, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, E.; Takeishi, Y.; Ueda, S.; Tsurimoto, T. Interaction between Rad9-Hus1-Rad1 and TopBP1 activates ATR-ATRIP and promotes TopBP1 recruitment to sites of UV-damage. DNA Repair 2014, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sideridou, M.; Zakopoulou, R.; Evangelou, K.; Liontos, M.; Kotsinas, A.; Rampakakis, E.; Gagos, S.; Kahata, K.; Grabusic, K.; Gkouskou, K.; et al. Cdc6 expression represses E-cadherin transcription and activates adjacent replication origins. J. Cell Biol. 2011, 195, 1123–1140. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., III; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Bennetzen, M.V.; Larsen, D.H.; Bunkenborg, J.; Bartek, J.; Lukas, J.; Andersen, J.S. Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Mol. Cell. Proteom. 2010, 9, 1314–1323. [Google Scholar] [CrossRef] [PubMed]
- Stucki, M.; Clapperton, J.A.; Mohammad, D.; Yaffe, M.B.; Smerdon, S.J.; Jackson, S.P. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005, 123, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- Stewart, G.S.; Wang, B.; Bignell, C.R.; Taylor, A.M.; Elledge, S.J. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 2003, 421, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. DNA damage checkpoints: From initiation to recovery or adaptation. Curr. Opin. Cell Biol. 2007, 19, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.K.; Lovly, C.M.; Piwnica-Worms, H. Regulation of the Chk2 protein kinase by oligomerization-mediated cis- and trans-phosphorylation. Mol. Cancer Res. 2003, 1, 598–609. [Google Scholar] [PubMed]
- Velimezi, G.; Liontos, M.; Vougas, K.; Roumeliotis, T.; Bartkova, J.; Sideridou, M.; Dereli-Oz, A.; Kocylowski, M.; Pateras, I.S.; Evangelou, K.; et al. Functional interplay between the DNA-damage-response kinase ATM and ARF tumour suppressor protein in human cancer. Nat. Cell Biol. 2013, 15, 967–977. [Google Scholar] [CrossRef] [PubMed]
- Olsson, A.; Manzl, C.; Strasser, A.; Villunger, A. How important are post-translational modifications in p53 for selectivity in target-gene transcription and tumour suppression? Cell Death Differ. 2007, 14, 1561–1575. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, K.; Yamaguchi-Shinozaki, K.; Seki, M. Regulatory network of gene expression in the drought and cold stress responses. Curr. Opin. Plant Biol. 2003, 6, 410–417. [Google Scholar] [CrossRef]
- Maya, R.; Balass, M.; Kim, S.T.; Shkedy, D.; Leal, J.F.; Shifman, O.; Moas, M.; Buschmann, T.; Ronai, Z.; Shiloh, Y.; et al. ATM-dependent phosphorylation of Mdm2 on serine 395: Role in p53 activation by DNA damage. Genes Dev. 2001, 15, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Papamichos-Chronakis, M.; Peterson, C.L. Chromatin and the genome integrity network. Nat. Rev. Genet. 2013, 14, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Trougakos, I.P.; Sesti, F.; Tsakiri, E.; Gorgoulis, V.G. Non-enzymatic post-translational protein modifications and proteostasis network deregulation in carcinogenesis. J. Proteom. 2013, 92, 274–298. [Google Scholar] [CrossRef] [PubMed]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef] [PubMed]
- Fribley, A.; Zhang, K.; Kaufman, R.J. Regulation of apoptosis by the unfolded protein response. Methods Mol. Biol. 2009, 559, 191–204. [Google Scholar] [PubMed]
- Nakka, V.P.; Prakash-Babu, P.; Vemuganti, R. Crosstalk between endoplasmic reticulum stress, oxidative stress, and autophagy: Potential therapeutic targets for acute CNS injuries. Mol. Neurobiol. 2016, 53, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Niforou, K.; Cheimonidou, C.; Trougakos, I.P. Molecular chaperones and proteostasis regulation during redox imbalance. Redox Biol. 2014, 2, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Hartl, F.U. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J. Degradation of oxidized proteins by the 20S proteasome. Biochimie 2001, 83, 301–310. [Google Scholar] [CrossRef]
- Grune, T.; Merker, K.; Sandig, G.; Davies, K.J. Selective degradation of oxidatively modified protein substrates by the proteasome. Biochem. Biophys. Res. Commun. 2003, 305, 709–718. [Google Scholar] [CrossRef]
- Saeki, Y.; Tanaka, K. Assembly and function of the proteasome. Methods Mol. Biol. 2012, 832, 315–337. [Google Scholar] [PubMed]
- Wang, H.; Song, P.; Du, L.; Tian, W.; Yue, W.; Liu, M.; Li, D.; Wang, B.; Zhu, Y.; Cao, C.; et al. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: Implication of dysregulated mitochondrial dynamics in Parkinson disease. Biol. Chem. 2011, 286, 11649–11658. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, N.; Stiller, S.B.; Pfanner, N. Activation and degradation of mitofusins: Two pathways regulate mitochondrial fusion by reversible ubiquitylation. Mol. Cell 2013, 49, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Gumeni, S.; Trougakos, I.P. Cross talk of proteostasis and mitostasis in cellular homeodynamics, ageing, and disease. Oxid. Med. Cell. Longev. 2016, 2016, 4587691. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Nedić, O.; Rattan, S.I.; Grune, T.; Trougakos, I.P. Molecular effects of advanced glycation end products on cell signalling pathways, ageing and pathophysiology. Free Radic. Res. 2013, 47, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M. Chaperone-mediated autophagy: Selectivity pays off. Trends Endocrinol. Metab. 2010, 21, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.B.; Schumacher, B. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Murshid, A.; Prince, T. The shock of aging: Molecular chaperones and the heat shock response in longevity and aging—A mini-review. Gerontology 2009, 55, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Van der Horst, A.; Burgering, B.M. Stressing the role of FoxO proteins in lifespan and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Sykiotis, G.P.; Bohmann, D. Stress-activated cap‘n’collar transcription factors in aging and human disease. Sci. Signal. 2010, 3, re3. [Google Scholar] [CrossRef] [PubMed]
- Tsakiri, E.N.; Sykiotis, G.P.; Papassideri, I.S.; Terpos, E.; Dimopoulos, M.A.; Gorgoulis, V.G.; Bohmann, D.; Trougakos, I.P. Proteasome dysfunction in Drosophila signals to an Nrf2-dependent regulatory circuit aiming to restore proteostasis and prevent premature aging. Aging Cell 2013, 12, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I.; Driessen, A.J.; Hegde, R.S.; Langer, T. The life of proteins: The good, the mostly good and the ugly. Nat. Struct. Mol. Biol. 2011, 18, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. Repression of the heat shock response is a programmed event at the onset of reproduction. Mol. Cell 2015, 59, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Kaufman, R.J. That which does not kill me makes me stronger: Adapting to chronic ER stress. Trends Biochem. Sci. 2007, 32, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.C.; Weissman, A.M. The unfolded protein response, degradation from endoplasmic reticulum and cancer. Genes Cancer 2010, 1, 764–778. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, N. Cellular stress/the unfolded protein response: Relevance to sleep and sleep disorders. Sleep Med. Rev. 2009, 13, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Yamamori, T.; Meike, S.; Nagane, M.; Yasui, H.; Inanami, O. ER stress suppresses DNA double-strand break repair and sensitizes tumor cells to ionizing radiation by stimulating proteasomal degradation of Rad51. FEBS Lett. 2013, 587, 3348–3353. [Google Scholar] [CrossRef] [PubMed]
- Bindra, R.S.; Schaffer, P.J.; Meng, A.; Woo, J.; Måseide, K.; Roth, M.E.; Lizardi, P.; Hedley, D.W.; Bristow, R.G.; Glazer, P.M. Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Mol. Cell. Biol. 2004, 24, 8504–8518. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, K.; Li, Z. Unfolded protein response in cancer: The physician’s perspective. J. Hematol. Oncol. 2011, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Oommen, D.; Prise, K.M. Down-regulation of PERK enhances resistance to ionizing radiation. Biochem. Biophys. Res. Commun. 2013, 441, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Raven, J.F.; Baltzis, D.; Wang, S.; Mounir, Z.; Papadakis, A.I.; Gao, H.Q.; Koromilas, A.E. PKR and PKR-like endoplasmic reticulum kinase induce the proteasome-dependent degradation of cyclin D1 via a mechanism requiring eukaryotic initiation factor 2α phosphorylation. J. Biol. Chem. 2008, 283, 3097–3108. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Wang, C.; Li, Z.; Sakamaki, T.; Pestell, R.G. Minireview: Cyclin D1: Normal and abnormal functions. Endocrinology 2004, 145, 5439–5447. [Google Scholar] [CrossRef] [PubMed]
- Shimura, T.; Kakuda, S.; Ochiai, Y.; Nakagawa, H.; Kuwahara, Y.; Takai, Y.; Kobayashi, J.; Komatsu, K.; Fukumoto, M. Acquired radioresistance of human tumor cells by DNA-PK/AKT/GSK3β-mediated cyclin D1 overexpression. Oncogene 2010, 29, 4826–4837. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S. The glucose-regulated proteins: Stress induction and clinical applications. Trends Biochem. Sci. 2001, 26, 504–510. [Google Scholar] [CrossRef]
- Little, E.; Ramakrishnan, M.; Roy, B.; Gazit, G.; Lee, A.S. The glucose-regulated proteins (GRP78 and GRP94): Functions, gene regulation, and applications. Crit. Rev. Eukaryot. Gene Expr. 1994, 4, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.V.; Castro-Obregon, S.; Frankowski, H.; Schuler, M.; Stoka, V.; del Rio, G.; Bredesen, D.E.; Ellerby, H.M. Coupling endoplasmic reticulum stress to the cell death program. An Apaf-1-independent intrinsic pathway. J. Biol. Chem. 2002, 277, 21836–21842. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.K.; Mao, C.; Baumeister, P.; Austin, R.C.; Kaufman, R.J.; Lee, A.S. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: Role of ATP binding site in suppression of caspase-7 activation. J. Biol. Chem. 2003, 278, 20915–20924. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, P.; Luo, S.; Skarnes, W.C.; Sui, G.; Seto, E.; Shi, Y.; Lee, A.S. Endoplasmic reticulum stress induction of the Grp78/BiP promoter: Activating mechanisms mediated by YY1 and its interactive chromatin modifiers. Mol. Cell. Biol. 2005, 25, 4529–4540. [Google Scholar] [CrossRef] [PubMed]
- Boisvert, F.M.; Dery, U.; Masson, J.Y.; Richard, S. Arginine methylation of MRE11 by PRMT1 is required for DNA damage checkpoint control. Genes Dev. 2005, 19, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Groenendyk, J.; Agellon, L.B.; Michalak, M. Coping with endoplasmic reticulum stress in the cardiovascular system. Annu. Rev. Physiol. 2013, 75, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Kasper, L.H.; Boussouar, F.; Boyd, K.; Xu, W.; Biesen, M.; Rehg, J.; Baudino, T.A.; Cleveland, J.L.; Brindle, P.K. Two transactivation mechanisms cooperate for the bulk of HIF-1-responsive gene expression. EMBO J. 2005, 24, 3846–3858. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.E.; Lee, H.G.; ChoI, H.; Chung, D.H.; Yoon, S.H.; Yang, Y.M.; Lee, J.W.; Choi, S.; Park, J.W.; Ye, S.K.; et al. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J. 2005, 19, 1296–1298. [Google Scholar] [CrossRef] [PubMed]
- Islam, K.N.; Mendelson, C.R. Permissive effects of oxygen on cyclic AMP and interleukin1 stimulation of surfactant protein A gene expression are mediated by epigenetic mechanisms. Mol. Cell. Biol. 2006, 26, 2901–2912. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.B.; Barton, M.C. Hypoxia-induced and stress-specific changes in chromatin structure and function. Mutat. Res. 2007, 618, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Bristow, R.G.; Hill, R.P. Hypoxia and metabolism: Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.Y.; Dhingra, K.; Hittelman, W.N.; Liehr, J.G.; deAndrade, M.; Li, D.H. Lipid peroxidation-induced putative malondialdehyde–DNA adducts in human breast tissues. Cancer Epidemiol. Biomark. Prev. 1996, 5, 705–710. [Google Scholar]
- Boonstra, J.; Post, J.A. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene 2004, 337, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative stress, inflammation, and cancer: How are they linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/β-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.D.; Hannink, M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol. Cell. Biol. 2003, 23, 8137–8151. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Wakabayashi, N.; Greenlaw, J.L.; Yamamoto, M.; Kensler, T.W. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signalling pathway. Mol. Cell. Biol. 2003, 23, 8786–8794. [Google Scholar] [CrossRef] [PubMed]
- Tsakiri, E.N.; Sykiotis, G.P.; Papassideri, I.S.; Gorgoulis, V.G.; Bohmann, D.; Trougakos, I.P. Differential regulation of proteasome functionality in reproductive vs. somatic tissues of Drosophila during aging or oxidative stress. FASEB J. 2013, 27, 2407–2420. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C. DNA damage and repair. Nature 2003, 421, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, J.S. The DNA Damage Response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Gutowski, M.; Kowalczyk, S. A study of free radical chemistry: Their role and pathophysiological significance. Acta Biochim. Pol. 2013, 60, 1–16. [Google Scholar] [PubMed]
- Wiseman, H.; Halliwell, B. Damage to DNA by reactive oxygen and nitrogen species: Role in inflammatory disease and progression to cancer. Biochem. J. 1996, 313, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Gamez, J.A.; Rodriguez-Vargas, J.M.; Quiles-Perez, R.; Aguilar-Quesada, R.; Martin-Oliva, D.; de Murcia, G.; de Murcia, J.M.; Almendros, A.; de Almodóvar, M.R.; Oliver, F.J. PARP-1 is involved in autophagy induced by DNA damage. Autophagy 2009, 5, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Vargas, J.M.; Ruiz-Magaña, M.J.; Ruiz-Ruiz, C.; Majuelos-Melguizo, J.; Peralta-Leal, A.; Rodríguez, M.I.; Muñoz-Gámez, J.A.; de Almodóvar, M.R.; Siles, E.; Rivas, A.L.; et al. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res. 2012, 22, 1181–1198. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Ogrunc, M.; Di Micco, R.; Liontos, M.; Bombardelli, L.; Mione, M.; Fumagalli, M.; Gorgoulis, V.G.; d’Adda di Fagagna, F. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 2014, 21, 998–1012. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Kenneth, H.Y.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. Noncanonical Mechanism of Nrf2 Activation by Autophagy Deficiency: Direct Interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [PubMed]
- Chio, I.I.; Jafarnejad, S.M.; Ponz-Sarvise, M.; Park, Y.; Rivera, K.; Palm, W.; Wilson, J.; Sangar, V.; Hao, Y.; Öhlund, D.; et al. NRF2 promotes tumor maintenance by modulating mRNA translation in pancreatic cancer. Cell 2016, 166, 963–976. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; Ludwig, R.L.; Vousden, K.H. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 20491–20496. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.B.; Pandita, R.K.; Eskiocak, U.; Ly, P.; Kaisani, A.; Kumar, R.; Cornelius, C.; Wright, W.E.; Pandita, T.K.; Shay, J.W. Targeting of Nrf2 induces DNA damage signalling and protects colonic epithelial cells from ionizing radiation. Proc. Natl. Acad. Sci. USA 2012, 109, E2949–E2955. [Google Scholar] [CrossRef] [PubMed]
- Jódar, L.; Mercken, E.M.; Ariza, J.; Younts, C.; González-Reyes, J.A.; Alcaín, F.J.; Burón, I.; de Cabo, R.; Villalba, J.M. Genetic deletion of Nrf2 promotes immortalization and decreases life span of murine embryonic fibroblasts. J. Gerontol. Ser. A 2011, 66, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Faraonio, R.; Vergara, P.; Di Marzo, D.; Pierantoni, M.G.; Napolitano, M.; Russo, T.; Cimino, F. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J. Biol. Chem. 2006, 281, 39776–39784. [Google Scholar] [CrossRef] [PubMed]
- Walerych, D.; Lisek, K.; Sommaggio, R.; Piazza, S.; Ciani, Y.; Dalla, E.; Rajkowska, K.; Gaweda-Walerych, K.; Ingallina, E.; Tonelli, C.; et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat. Cell Biol. 2016, 18, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Galanos, P.; Vougas, K.; Walter, D.; Polyzos, A.; Maya-Mendoza, A.; Haagensen, E.J.; Kokkalis, A.; Roumelioti, F.M.; Gagos, S.; Tzetis, M.; et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat. Cell Biol. 2016, 18, 777–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriegenburg, F.; Jakopec, V.; Poulsen, E.G.; Nielsen, S.V.; Roguev, A.; Krogan, N.; Gordon, C.; Fleig, U.; Hartmann-Petersen, R. A chaperone-assisted degradation pathway targets kinetochore proteins to ensure genome stability. PLoS Genet. 2014, 10, e1004140. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.L.; Chiang, Y.R.; Narberhaus, F.; Baron, C.; Lai, E.M. The small heat-shock protein HspL is a VirB8 chaperone promoting type IV secretion-mediated DNA transfer. J. Biol. Chem. 2010, 285, 19757–19766. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Xi, J.H.; Wawrousek, E.F.; Fleming, T.P.; Andley, U.P. Hyperproliferation and p53 status of lens epithelial cells derived from a B-crystallin knockout mice. J. Biol. Chem. 2003, 278, 36876–36886. [Google Scholar] [CrossRef] [PubMed]
- Hunt, C.R.; Dix, D.J.; Sharma, G.G.; Pandita, R.K.; Gupta, A.; Funk, M.; Pandita, T.K. Genomic instability and enhanced radiosensitivity in Hsp70.1- and Hsp70.3-deficient mice. Mol. Cell. Biol. 2004, 24, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Dorard, C.; de Thonel, A.; Collura, A.; Marisa, L.; Svrcek, M.; Lagrange, A.; Jego, G.; Wanherdrick, K.; Joly, A.L.; Buhard, O.; et al. Expression of a mutant HSP110 sensitizes colorectal cancer cells to chemotherapy and improves disease prognosis. Nat. Med. 2011, 17, 1283–1289. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhu, Y.; Zhou, W.; Molinier, J.; Dong, A.; Shen, W.H. NAP1 family histone chaperones are required for somatic homologous recombination in Arabidopsis. Plant Cell 2012, 24, 1437–1447. [Google Scholar] [CrossRef] [PubMed]
- Nystrom, T.; Yang, J.; Molin, M. Peroxiredoxins, gerontogenes linking aging to genome instability and cancer. Genes Dev. 2012, 26, 2001–2008. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, R.A.; Ellis, R.J. Chaperones: Needed for both the good times and the bad times. Philos. Trans. R. Soc. Lond. B 2013, 368, 20130091. [Google Scholar] [CrossRef] [PubMed]
- Vaz, B.; Halder, S.; Ramadan, K. Role of p97/VCP (Cdc48) in genome stability. Front. Genet. 2013, 4, 60. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, K.; Bruderer, R.; Spiga, F.M.; Popp, O.; Baur, T.; Gotta, M.; Meyer, H.H. Cdc48/p97promotes reformation of the nucleus by extracting the kinase Aurora B from chromatin. Nature 2007, 450, 1258–1262. [Google Scholar] [CrossRef] [PubMed]
- Franz, A.; Orth, M.; Pirson, P.A.; Sonneville, R.; Blow, J.J.; Gartner, A.; Stemmann, O.; Hoppe, T. CDC48/p97 coordinates CDT1 degradation with GINS chromatin dissociation to ensure faithful DNA replication. Mol. Cell 2011, 44, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Meerang, M.; Ritz, D.; Paliwal, S.; Garajova, Z.; Bosshard, M.; Mailand, N.; Janscak, P.; Hübscher, U.; Meyer, H.; Ramadan, K. The ubiquitin-selective segregase VCP/p97 orchestrates the response to DNA double strand breaks. Nat. Cell Biol. 2011, 13, 1376–1382. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.; Havens, C.G.; Walter, J.C.; Harper, J.W. A genome wide screen identifies p97 as an essential regulator of DNA damage-dependent CDT1destruction. Mol. Cell 2011, 44, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Oania, R.; Fang, R.; Smith, G.T.; Deshaies, R.J. Cdc48/p97 mediates UV-dependent turnover of RNAPolII. Mol. Cell 2011, 41, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Mouysset, J.; Deichsel, A.; Moser, S.; Hoege, C.; Hyman, A.A.; Gartner, A.; Hoppe, T. Cell cycle progression requires the CDC48-UFD1/NPL-4 complex for efficient DNA replication. Proc. Natl. Acad. Sci. USA 2008, 105, 12879–12884. [Google Scholar] [CrossRef] [PubMed]
- Deichsel, A.; Mouysset, J.; Hoppe, T. The ubiquitin-selective chaperone CDC48/p97, a new player in DNA replication. Cell Cycle 2009, 8, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Mittelman, D.; Sykoudis, K.; Hersh, M.; Lin, Y.; Wilson, J.H. Hsp90 modulates CAG repeat instability in human cells. Cell Stress Chaperones 2010, 15, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Yu, D.; Hirayama, R.; Ninomiya, Y.; Sekine, E.; Kubota, N.; Ando, K.; Okayasu, R. Inhibition of homologous recombination repair in irradiated tumor cells pretreated with Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Biochem. Biophys. Res. Commun. 2006, 351, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Enders, G.H. Expanded roles for Chk1 in genome maintenance. J. Biol. Chem. 2008, 283, 17749–17752. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, K.B.; Li, R. A prescription for “stress” the role of Hsp90 in genome stability and cellular adaptation. Trends Cell Biol. 2012, 22, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Buszczak, M.; Signer, R.A.; Morrison, S.J. Cellular differences in protein synthesis regulate tissue homeostasis. Cell 2014, 159, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef] [PubMed]
- Hochstrasser, M. Origin and function of ubiquitin-like proteins. Nature 2009, 458, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Wiśniewski, J.R.; Hein, M.Y.; Cox, J.; Mann, M. A “proteomic ruler” for protein copy number and concentration estimation without spike-in standards. Mol. Cell. Proteom. 2014, 13, 3497–3506. [Google Scholar] [CrossRef] [PubMed]
- Peth, A.; Nathan, J.A.; Goldberg, A.L. The ATP costs and time required to degrade ubiquitinated proteins by the 26 S proteasome. J. Biol. Chem. 2013, 288, 29215–29222. [Google Scholar] [CrossRef] [PubMed]
- Gendron, J.M.; Webb, K.; Yang, B.; Rising, L.; Zuzow, N.; Bennett, E.J. Using the ubiquitin-modified proteome to monitor distinct and spatially restricted protein homeostasis dysfunction. Mol. Cell. Proteom. 2016, 15, 2576–2593. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.W.; Bennett, E.J. Proteome complexity and the forces that drive proteome imbalance. Nature 2016, 537, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.T.; Bonneau, A.R.; Takacs, C.M.; Bazzini, A.A.; DiVito, K.R.; Fleming, E.S.; Giraldez, A.J. Nanog, Pou5f1 and SoxB1 activate zygotic gene expression during the maternal-to-zygotic transition. Nature 2013, 503, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, M.; Rooney, M.S.; Mertins, P.; Przybylski, D.; Chevrier, N.; Satija, R.; Rodriguez, E.H.; Fields, A.P.; Schwartz, S.; Raychowdhury, R.; et al. Dynamic profiling of the protein life cycle in response to pathogens. Science 2015, 347, 1259038. [Google Scholar] [CrossRef] [PubMed]
- Walter, D.; Hoffmann, S.; Komseli, E.S.; Rappsilber, J.; Gorgoulis, V.; Sørensen, C.S. SCF(Cyclin F)-dependent degradation of CDC6 suppresses DNA re-replication. Nat. Commun. 2016, 7, 10530. [Google Scholar] [CrossRef] [PubMed]
- Roseaulin, L.C.; Noguchi, C.; Noguchi, E. Proteasome-dependent degradation of replisome components regulates faithful DNA replication. Cell Cycle 2013, 12, 2564–2569. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, O.; Reinheckel, T.; Sitte, N.; Hass, R.; Grune, T.; Davies, K.J. Poly-ADP ribose polymerase activates nuclear proteasome to degrade oxidatively damaged histones. Proc. Natl. Acad. Sci. USA 1999, 96, 6223–6228. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.W.; Lamond, A.I.; Mann, M.; Andersen, J.S. Analysis of nucleolar protein dynamics reveals the nuclear degradation of ribosomal proteins. Curr. Biol. 2007, 17, 749–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, A.; Nagashima, Y.; Matsumoto, L.; Suzuki, T.; Yamanaka, T.; Date, H.; Deoka, K.; Nukina, N.; Tsuji, S. Intranuclear degradation of polyglutamine aggregates by the ubiquitin-proteasome system. J. Biol. Chem. 2009, 284, 9796–9803. [Google Scholar] [CrossRef] [PubMed]
- McBride, W.H.; Iwamoto, K.S.; Syljuasen, R.; Pervan, M.; Pajonk, F. The role of theubiquitin/proteasome system in cellular responses to radiation. Oncogene 2003, 22, 5755–5773. [Google Scholar] [CrossRef] [PubMed]
- Yew, P.R. Ubiquitin-mediated proteolysis of vertebrate G1- and S-phase regulators. J. Cell. Physiol. 2001, 187, 1–10. [Google Scholar] [CrossRef]
- Deshaies, R.J. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol. 2014, 12, 94. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.A.; Cleveland, D.W. Does aneuploidy cause cancer? Curr. Opin. Cell Biol. 2006, 18, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.; Cox, J.; Mann, M. Proteomic changes resulting from gene copy number variations in cancer cells. PLoS Genet. 2010, 6, e1001090. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.L.; Walton, Z.E.; Altman, B.J.; Stine, Z.E.; Dang, C.V. MYC and metabolism on the path to cancer. Semin. Cell Dev. Biol. 2015, 43, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Ruggero, D. Translational control in cancer etiology. Cold Spring Harb. Perspect. Biol. 2013, 5, a012336. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Bekker-Jensen, S.; Mailand, N. Assembly and function of DNA double-strand breakrepair foci in mammalian cells. DNA Repair 2010, 9, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Komander, D.; Rape, M. The ubiquitin code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef] [PubMed]
- Bergink, S.; Jentsch, S. Principles of ubiquitin and SUMO modifications in DNA repair. Nature 2009, 458, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Panier, S.; Durocher, D. Regulatory ubiquitylation in response to DNA double-strand breaks. DNA Repair 2009, 8, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Mailand, N.; Bekker-Jensen, S.; Faustrup, H.; Melander, F.; Bartek, J.; Lukas, C.; Lukas, J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 2007, 131, 887–900. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Lukas, C.; Bartek, J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 2011, 13, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Schwertman, P.; Bekker-Jensen, S.; Mailand, N. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat. Rev. Mol. Cell Biol. 2016, 17, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Galanty, Y.; Belotserkovskaya, R.; Coates, J.; Polo, S.; Miller, K.M.; Jackson, S.P. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature 2009, 462, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Leverson, J.D.; Hunter, T. Conserved function of RNF4 family proteins in eukaryotes: Targeting a ubiquitin ligase to SUMOylated proteins. EMBO J. 2007, 26, 4102–4112. [Google Scholar] [CrossRef] [PubMed]
- Kusumoto, R.; Masutani, C.; Sugasawa, K.; Iwai, S.; Araki, M.; Uchida, A.; Mizukoshi, T.; Hanaoka, F. Diversity of the damage recognition step in the global genomic nucleotide excision repair In Vitro. Mutat. Res. 2001, 485, 219–227. [Google Scholar] [CrossRef]
- Ulrich, H.D. Regulating post-translational modifications of the eukaryotic replication clamp PCNA. DNA Repair 2009, 8, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, C.; DeMartino, G.N. Intracellular localization of proteasomes. Int. J. Biochem. Cell Biol. 2003, 35, 579–589. [Google Scholar] [CrossRef]
- Kouranti, I.; Peyroche, A. Protein degradation in DNA damage response. Semin. Cell Dev. Biol. 2012, 23, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Ma, Z.; Willers, H.; Akhtar, K.; Scott, S.P.; Zhang, J.; Powell, S.; Zhang, J. Disassembly of MDC1 foci is controlled by ubiquitin-proteasome-dependent degradation. J. Biol. Chem. 2008, 283, 31608–31616. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Sato, K.; Wu, W. The BRCA1 ubiquitin ligase and homologous recombination repair. FEBS Lett. 2011, 585, 2836–2844. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, L.P.; Himmels, S.F.; Trenner, A.; Walker, C.; von Aesch, C.; Eggenschwiler, A.; Murina, O.; Enchev, R.I.; Peter, M.; Freire, R.; et al. Cullin3-KLHL15 ubiquitin ligase mediates CtIP protein turnover to fine-tune DNA-end resection. Nat. Commun. 2016, 7, 12628. [Google Scholar] [CrossRef] [PubMed]
- Menendez, D.; Inga, A.; Resnick, M.A. The expanding universe of p53 targets. Nat. Rev. Cancer 2009, 9, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. p53 regulation by ubiquitin. FEBS Lett. 2011, 585, 2803–2809. [Google Scholar] [CrossRef] [PubMed]
- Manfredi, J.J. The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 2010, 24, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Khoo, K.H.; Verma, C.S.; Lane, D.P. Drugging the p53 pathway: Understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 2014, 13, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, K.; Meerang, M. Degradation-linked ubiquitin signal and proteasome are integral components of DNA double strand break repair: New perspectives for anti-cancer therapy. FEBS Lett. 2011, 585, 2868–2875. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Pigeon, H.; Laurent, G.; Humbert, O.; Salles, B.; Lautier, D. Degadration of mismatch repair hMutSα heterodimer by the ubiquitin-proteasome pathway. FEBS Lett. 2004, 562, 40–44. [Google Scholar] [CrossRef]
- El-Shemerly, M.; Janscak, P.; Hess, D.; Jiricny, J.; Ferrari, S. Degradation of human exonuclease 1b upon DNA synthesis inhibition. Cancer Res. 2005, 65, 3604–3609. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.L.; Tait, P.S.; Finch, D.; Dianova, I.I.; Edelmann, M.J.; Khoronenkova, S.V.; Kessler, B.M.; Sharma, R.A.; McKenna, W.G.; Dianov, G.L. Ubiquitin ligase ARF-BP1/Mule modulates base excision repair. EMBO J. 2009, 28, 3207–3215. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.L.; Tait, P.S.; Finch, D.; Dianova, I.I.; Allinson, S.L.; Dianov, G.L. CHIP-mediated degradation and DNA damage-dependent stabilization regulate base excision repair proteins. Mol. Cell 2008, 29, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.L.; Dianova, I.I.; Finch, D.; Tait, P.S.; Ström, C.E.; Helleday, T.; Dianov, G.L. XRCC1 phosphorylation by CK2 is required for its stability and efficient DNA repair. DNA Repair 2010, 9, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Simbulan-Rosenthal, C.M.; Smulson, M.E.; Chock, P.B.; Yang, D.C. Polyubiquitylation of PARP-1 through ubiquitin K48 is modulated by activated DNA, NAD+, and dipeptides. J. Cell Biochem. 2008, 104, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Messner, S.; Schuermann, D.; Altmeyer, M.; Kassner, I.; Schmidt, D.; Schär, P.; Müller, S.; Hottiger, M.O. Sumoylation of poly(ADP-ribose) polymerase 1 inhibits its acetylation and restrains transcriptional coactivator function. FASEB J. 2009, 23, 3978–3989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurihara, Y.; Kanki, T.; Aoki, Y.; Hirota, Y.; Saigusa, T.; Uchiumi, T.; Kang, D. Mitophagy plays an essential role in reducing mitochondrial production of reactive oxygen species and mutation of mitochondrial DNA by maintaining mitochondrial quantity and quality in yeast. J. Biol. Chem. 2012, 287, 3265–3272. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress-responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I. Autophagy basics. Microbiol. Immunol. 2011, 55, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Eliopoulos, A.G.; Havaki, S.; Gorgoulis, V.G. DNA damage response and autophagy: A meaningful partnership. Front. Genet. 2016, 7, 204. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Karantza-Wadsworth, V.; Patel, S.; Kravchuk, O.; Chen, G.; Mathew, R.; Jin, S.; White, E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007, 21, 1621–1635. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Narzt, M.S.; Nagelreiter, I.M.; Hohensinner, P.; Terlecki-Zaniewicz, L.; Tschachler, E. Autophagy deficient keratinocytes display increased DNA damage, senescence and aberrant lipid composition after oxidative stress In Vitro and In Vivo. Redox Biol. 2017, 11, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Jin, S. Autophagy, mitochondrial quality control, and oncogenesis. Autophagy 2006, 2, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; White, E. Role of autophagy in cancer: Management of metabolic stress. Autophagy 2007, 3, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G.; Bovina, C.; D’Aurelio, M.; Fato, R.; Formiggini, G.; Genova, M.L.; Giuliano, G.; Pich, M.M.; Paolucci, U.G.O.; Castelli, G.P.; et al. Role of mitochondria in oxidative stress and aging. Ann. N. Y. Acad. Sci. 2002, 959, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karp, C.M.; Beaudoin, B.; Vuong, N.; Chen, G.; Chen, H.Y.; Bray, K.; Reddy, A.; Bhanot, G.; Gelinas, C.; et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009, 137, 1062–1075. [Google Scholar] [CrossRef] [PubMed]
- Rello-Varona, S.; Lissa, D.; Shen, S.; Niso-Santano, M.; Senovilla, L.; Mariño, G.; Vitale, I.; Jemaá, M.; Harper, F.; Pierron, G.; et al. Autophagic removal of micronuclei. Cell Cycle 2012, 11, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.P.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 2013, 202, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Evangelou, K.; Lougiakis, N.; Rizou, S.V.; Kotsinas, A.; Kletsas, D.; Muñoz-Espín, D.; Kastrinakis, N.G.; Pouli, N.; Marakos, P.; Townsend, P.; et al. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell 2017, 16, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Xu, C.; Donahue, G.; Shimi, T.; Pan, J.A.; Zhu, J.; Ivanov, A.; Capell, B.C.; Drake, A.M.; Shah, P.P.; et al. Autophagy mediates degradation of nuclear lamina. Nature 2015, 527, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Kongara, S.; Beaudoin, B.; Karp, C.M.; Bray, K.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007, 21, 1367–1381. [Google Scholar] [CrossRef] [PubMed]
- Robert, T.; Vanoli, F.; Chiolo, I.; Shubassi, G.; Bernstein, K.A.; Rothstein, R.; Botrugno, O.A.; Parazzoli, D.; Oldani, A.; Minucci, S.; et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011, 471, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Yuan, N.; Wang, Z.; Cao, Y.; Fang, Y.; Li, X.; Xu, F.; Song, L.; Wang, J.; Zhang, H.; et al. Autophagy confers DNA damage repair pathways to protect the hematopoietic system from nuclear radiation injury. Sci. Rep. 2015, 5, 12362. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.Y.; Xu, N.; O’Prey, J.; Lao, L.Y.; Joshi, S.; Long, J.S.; O’Prey, M.; Croft, D.R.; Beaumatin, F.; Baudot, A.D.; et al. Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc. Natl. Acad. Sci. USA 2015, 112, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.; Guan, J.L. Suppression of autophagy by FIP200 deletion impairs DNA damage repair and increases cell death upon treatments with anti-cancer agents. Mol. Cancer Res. 2011, 9, 1232–1241. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, N.; Zhang, L.; Li, R.; Fu, W.; Ma, K.; Li, X.; Wang, L.; Wang, J.; Zhang, H.; et al. Autophagy regulates chromatin ubiquitination in DNA damage response through elimination of SQSTM1/p62. Mol. Cell 2016, 63, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.; Carroll, B.; Sarallah, R.; Correia-Melo, C.; Ogrodnik, M.; Nelson, G.; Otten, E.G.; Manni, D.; Antrobus, R.; Morgan, B.A.; et al. SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy 2016, 12, 1917–1930. [Google Scholar] [CrossRef] [PubMed]
- Tsakiri, E.N.; Iliaki, K.K.; Höhn, A.; Grimm, S.; Papassideri, I.S.; Grune, T.; Trougakos, I.P. Diet-derived advanced glycation end products or lipofuscin disrupts proteostasis and reduces life span in Drosophila melanogaster. Free Radic. Biol. Med. 2013, 65, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Moreau, P.; Palumbo, A.; Joshua, D.; Pour, L.; Hájek, R.; Facon, T.; Ludwig, H.; Oriol, A.; Goldschmidt, H.; et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): A randomised, phase 3, open-label, multicentre study. Lancet Oncol. 2016, 17, 27–38. [Google Scholar] [CrossRef]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gumeni, S.; Evangelakou, Z.; Gorgoulis, V.G.; Trougakos, I.P. Proteome Stability as a Key Factor of Genome Integrity. Int. J. Mol. Sci. 2017, 18, 2036. https://doi.org/10.3390/ijms18102036
Gumeni S, Evangelakou Z, Gorgoulis VG, Trougakos IP. Proteome Stability as a Key Factor of Genome Integrity. International Journal of Molecular Sciences. 2017; 18(10):2036. https://doi.org/10.3390/ijms18102036
Chicago/Turabian StyleGumeni, Sentiljana, Zoi Evangelakou, Vassilis G. Gorgoulis, and Ioannis P. Trougakos. 2017. "Proteome Stability as a Key Factor of Genome Integrity" International Journal of Molecular Sciences 18, no. 10: 2036. https://doi.org/10.3390/ijms18102036