Deciphering the Role of B Cells in Multiple Sclerosis—Towards Specific Targeting of Pathogenic Function

Abstract
:1. Introduction
2. The Role of B Cells in Multiple Sclerosis (MS)
2.1. Pathogenic Contribution of B Cell-Derived Antibodies
2.2. Antigen-Activated B Cells Contribute as Potent Antigen-Presenting Cells
2.3. The Affected CNS Itself Provides a B Cell Fostering Milieu in MS
2.4. B Cells as Source of Pro- and Anti-Inflammatory Cytokines
3. B Cell-Directed Therapeutic Interventions
3.1. Plasmapheresis—Second Line Therapy for MS Relapses
3.2. Approved Therapies Partially or Indirectly Effecting B Cells
3.3. Strategies Directly Targeting B Cells—Emerging Therapies
3.3.1. Anti-CD20 Antibodies Directly Target B Cells
3.3.2. Anti-CD19 (MEDI-551)—A Broader B cell-Depleting Approach.
3.3.3. Lessons from the Atacicept Trial
4. Conclusions and Outlook
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lehmann-Horn, K.; Kronsbein, H.C.; Weber, M.S. Targeting B cells in the treatment of multiple sclerosis: Recent advances and remaining challenges. Ther. Adv. Neurol. Disord. 2013, 6, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Kabat, E.A.; Freedman, D.A.; Murray, J.P.; Knaub, V. A study of the crystalline albumin, γ globulin and total protein in the cerebrospinal fluid of 100 cases of multiple sclerosis and in other diseases. Am. J. Med. Sci. 1950, 219, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Siritho, S.; Freedman, M.S. The prognostic significance of cerebrospinal fluid in multiple sclerosis. J. Neurol. Sci. 2009, 279, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Link, H.; Huang, Y.-M. Oligoclonal bands in multiple sclerosis cerebrospinal fluid: An update on methodology and clinical usefulness. J. Neuroimmunol. 2006, 180, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Mentele, R.; Malotka, J.; Kellermann, J.; Kumpfel, T.; Wekerle, H.; Lottspeich, F.; Hohlfeld, R.; Dornmair, K. Matching of oligoclonal immunoglobulin transcriptomes and proteomes of cerebrospinal fluid in multiple sclerosis. Nat. Med. 2008, 14, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Beltrán, E.; Obermeier, B.; Moser, M.; Coret, F.; Simó-Castelló, M.; Boscá, I.; Pérez-Miralles, F.; Villar, L.M.; Senel, M.; Tumani, H.; et al. Intrathecal somatic hypermutation of IgM in multiple sclerosis and neuroinflammation. Brain 2014, 137, 2703–2714. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Duquette, P.; Zhang, Y.; Talbot, P.; Poole, R.; Antel, J. Clonal expansion and somatic hypermutation of V(H) genes of B cells from cerebrospinal fluid in multiple sclerosis. J. Clin. Investig. 1998, 102, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.S.; Hemmer, B.; Cepok, S. The role of antibodies in multiple sclerosis. Biochim. Biophys. Acta 2011, 1812, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Reiber, H.; Ungefehr, S.; Jacobi, C. The intrathecal, polyspecific and oligoclonal immune response in multiple sclerosis. Mult. Scler. J. 1998, 4, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Genain, C.P.; Cannella, B.; Hauser, S.L.; Raine, C.S. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat. Med. 1999, 5, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Blauth, K.; Soltys, J.; Matschulat, A.; Reiter, C.R.; Ritchie, A.; Baird, N.L.; Bennett, J.L.; Owens, G.P. Antibodies produced by clonally expanded plasma cells in multiple sclerosis cerebrospinal fluid cause demyelination of spinal cord explants. Acta Neuropathol. 2015, 130, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Elliott, C.; Lindner, M.; Arthur, A.; Brennan, K.; Jarius, S.; Hussey, J.; Chan, A.; Stroet, A.; Olsson, T.; Willison, H.; et al. Functional identification of pathogenic autoantibody responses in patients with multiple sclerosis. Brain 2012, 135, 1819–1833. [Google Scholar] [CrossRef] [PubMed]
- Brennan, K.M.; Galban-Horcajo, F.; Rinaldi, S.; O’Leary, C.P.; Goodyear, C.S.; Kalna, G.; Arthur, A.; Elliot, C.; Barnett, S.; Linington, C.; et al. Lipid arrays identify myelin-derived lipids and lipid complexes as prominent targets for oligoclonal band antibodies in multiple sclerosis. J. Neuroimmunol. 2011, 238, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Lalive, P.H.; Menge, T.; Delarasse, C.; Della Gaspera, B.; Pham-Dinh, D.; Villoslada, P.; von Büdingen, H.C.; Genain, C.P. Antibodies to native myelin oligodendrocyte glycoprotein are serologic markers of early inflammation in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2006, 103, 2280–2285. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.G.; Catz, I. Relative frequency of autoantibodies to myelin basic protein and proteolipid protein in optic neuritis and multiple sclerosis cerebrospinal fluid. J. Neurol. Sci. 1994, 121, 66–73. [Google Scholar] [CrossRef]
- Mathey, E.K.; Derfuss, T.; Storch, M.K.; Williams, K.R.; Hales, K.; Woolley, D.R.; Al-Hayani, A.; Davies, S.N.; Rasband, M.N.; Olsson, T.; et al. Neurofascin as a novel target for autoantibody-mediated axonal injury. J. Exp. Med. 2007, 204, 2363–2372. [Google Scholar] [CrossRef] [PubMed]
- Derfuss, T.; Parikh, K.; Velhin, S.; Braun, M.; Mathey, E.; Krumbholz, M.; Kümpfel, T.; Moldenhauer, A.; Rader, C.; Sonderegger, P.; et al. Contactin-2/TAG-1-directed autoimmunity is identified in multiple sclerosis patients and mediates gray matter pathology in animals. Proc. Natl. Acad. Sci. USA 2009, 106, 8302–8307. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Kalman, B. Autoreactive IgG to intracellular proteins in sera of MS patients. J. Neuroimmunol. 1999, 99, 72–81. [Google Scholar] [CrossRef]
- Lalive, P.H.; Molnarfi, N.; Benkhoucha, M.; Weber, M.S.; Santiago-Raber, M.-L. Antibody response in MOG35–55 induced EAE. J. Neuroimmunol. 2011, 240, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Kinzel, S.; Lehmann-Horn, K.; Torke, S.; Hausler, D.; Winkler, A.; Stadelmann, C.; Payne, N.; Feldmann, L.; Saiz, A.; Reindl, M.; et al. Myelin-reactive antibodies initiate T cell-mediated CNS autoimmune disease by opsonization of endogenous antigen. Acta Neuropathol. 2016, 132, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Cserr, H.F.; Knopf, P.M. Cervical lymphatics, the blood-brain barrier and the immunoreactivity of the brain: A new view. Immunol. Today 1992, 13, 507–512. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337. [Google Scholar] [CrossRef] [PubMed]
- Duddy, M.; Niino, M.; Adatia, F.; Hebert, S.; Freedman, M.; Atkins, H.; Kim, H.J.; Bar-Or, A. Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J. Immunol. 2007, 178, 6092–6099. [Google Scholar] [CrossRef] [PubMed]
- Mathias, A.; Perriard, G.; Canales, M.; Soneson, C.; Delorenzi, M.; Schluep, M.; Du Pasquier, R.A. Increased ex vivo antigen presentation profile of B cells in multiple sclerosis. Mult. Scler. J. 2017, 23, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Aung, L.L.; Balashov, K.E. Decreased Dicer expression is linked to increased expression of co-stimulatory molecule CD80 on B cells in multiple sclerosis. Mult. Scler. J. 2015, 21, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Genc, K.; Dona, D.L.; Reder, A.T. Increased CD80+ B cells in active multiple sclerosis and reversal by interferon β-1b therapy. J. Clin. Investig. 1997, 99, 2664–2671. [Google Scholar] [CrossRef] [PubMed]
- Harp, C.T.; Ireland, S.; Davis, L.S.; Remington, G.; Cassidy, B.; Cravens, P.D.; Stuve, O.; Lovett-Racke, A.E.; Eagar, T.N.; Greenberg, B.M.; et al. Memory B cells from a subset of treatment-naive relapsing-remitting multiple sclerosis patients elicit CD4+ T-cell proliferation and IFN-γ production in response to myelin basic protein and myelin oligodendrocyte glycoprotein. Eur. J. Immunol. 2010, 40, 2942–2956. [Google Scholar] [CrossRef] [PubMed]
- Lanzavecchia, A. Antigen-specific interaction between T and B cells. Nature 1985, 314, 537–539. [Google Scholar] [CrossRef] [PubMed]
- Pollinger, B.; Krishnamoorthy, G.; Berer, K.; Lassmann, H.; Bosl, M.R.; Dunn, R.; Domingues, H.S.; Holz, A.; Kurschus, F.C.; Wekerle, H. Spontaneous relapsing-remitting EAE in the SJL/J mouse: MOG-reactive transgenic T cells recruit endogenous MOG-specific B cells. J. Exp. Med. 2009, 206, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, G.; Lassmann, H.; Wekerle, H.; Holz, A. Spontaneous opticospinal encephalomyelitis in a double-transgenic mouse model of autoimmune T cell/B cell cooperation. J. Clin. Investig. 2006, 116, 2385–2392. [Google Scholar] [CrossRef] [PubMed]
- Molnarfi, N.; Schulze-Topphoff, U.; Weber, M.S.; Patarroyo, J.C.; Prod’homme, T.; Varrin-Doyer, M.; Shetty, A.; Linington, C.; Slavin, A.J.; Hidalgo, J.; et al. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J. Exp. Med. 2013, 210, 2921–2937. [Google Scholar] [CrossRef] [PubMed]
- Cepok, S.; Jacobsen, M.; Schock, S.; Omer, B.; Jaekel, S.; Boddeker, I.; Oertel, W.H.; Sommer, N.; Hemmer, B. Patterns of cerebrospinal fluid pathology correlate with disease progression in multiple sclerosis. Brain 2001, 124, 2169–2176. [Google Scholar] [CrossRef] [PubMed]
- Lucchinetti, C.; Bruck, W.; Parisi, J.; Scheithauer, B.; Rodriguez, M.; Lassmann, H. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann. Neurol. 2000, 47, 707–717. [Google Scholar] [CrossRef]
- Krumbholz, M.; Theil, D.; Cepok, S.; Hemmer, B.; Kivisäkk, P.; Ransohoff, R.M.; Hofbauer, M.; Farina, C.; Derfuss, T.; Hartle, C.; et al. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain 2006, 129, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Rentzos, M.; Cambouri, C.; Rombos, A.; Nikolaou, C.; Anagnostouli, M.; Tsoutsou, A.; Dimitrakopoulos, A.; Triantafyllou, N.; Vassilopoulos, D. IL-15 is elevated in serum and cerebrospinal fluid of patients with multiple sclerosis. J. Neurol. Sci. 2006, 241, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Kowarik, M.C.; Cepok, S.; Sellner, J.; Grummel, V.; Weber, M.S.; Korn, T.; Berthele, A.; Hemmer, B. CXCL13 is the major determinant for B cell recruitment to the CSF during neuroinflammation. J. Neuroinflamm. 2012, 9, 93. [Google Scholar] [CrossRef] [PubMed]
- Krumbholz, M.; Theil, D.; Derfuss, T.; Rosenwald, A.; Schrader, F.; Monoranu, C.M.; Kalled, S.L.; Hess, D.M.; Serafini, B.; Aloisi, F.; et al. BAFF is produced by astrocytes and up-regulated in multiple sclerosis lesions and primary central nervous system lymphoma. J. Exp. Med. 2005, 201, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; van Horssen, J.; Mahad, D. Progressive multiple sclerosis: Pathology and pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Serafini, B.; Rosicarelli, B.; Magliozzi, R.; Stigliano, E.; Aloisi, F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004, 14, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Howell, O.W.; Reeves, C.A.; Nicholas, R.; Carassiti, D.; Radotra, B.; Gentleman, S.M.; Serafini, B.; Aloisi, F.; Roncaroli, F.; Magliozzi, R.; et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011, 134, 2755–2771. [Google Scholar] [CrossRef] [PubMed]
- Lovato, L.; Willis, S.N.; Rodig, S.J.; Caron, T.; Almendinger, S.E.; Howell, O.W.; Reynolds, R.; O’Connor, K.C.; Hafler, D.A. Related B cell clones populate the meninges and parenchyma of patients with multiple sclerosis. Brain 2011, 134, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Palanichamy, A.; Apeltsin, L.; Kuo, T.C.; Sirota, M.; Wang, S.; Pitts, S.J.; Sundar, P.D.; Telman, D.; Zhao, L.Z.; Derstine, M.; et al. Immunoglobulin class-switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci. Transl. Med. 2014, 6, 248ra106. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.N.H.; Yaari, G.; Vander Heiden, J.A.; Church, G.; Donahue, W.F.; Hintzen, R.Q.; Huttner, A.J.; Laman, J.D.; Nagra, R.M.; Nylander, A.; et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci. Transl. Med. 2014, 6, 248ra107. [Google Scholar] [CrossRef] [PubMed]
- Lehmann-Horn, K.; Wang, S.Z.; Sagan, S.A.; Zamvil, S.S.; von Budingen, H.C. B cell repertoire expansion occurs in meningeal ectopic lymphoid tissue. JCI Insight 2016, 1, e87234. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Mitsdoerffer, M.; Croxford, A.L.; Awasthi, A.; Dardalhon, V.A.; Galileos, G.; Vollmar, P.; Stritesky, G.L.; Kaplan, M.H.; Waisman, A.; et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2008, 105, 18460–18465. [Google Scholar] [CrossRef] [PubMed]
- Barr, T.A.; Shen, P.; Brown, S.; Lampropoulou, V.; Roch, T.; Lawrie, S.; Fan, B.; O’Connor, R.A.; Anderton, S.M.; Bar-Or, A.; et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6–producing B cells. J. Exp. Med. 2012, 209, 1001–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, Y.; Li, R.; Rezk, A.; Misirliyan, H.; Moore, C.; Farooqi, N.; Solis, M.; Goiry, L.G.; de Faria Junior, O.; Dang, V.D.; et al. A Novel MicroRNA-132-Surtuin-1 Axis Underlies Aberrant B-cell Cytokine Regulation in Patients with Relapsing-Remitting Multiple Sclerosis. PLoS ONE 2014, 9, e105421. [Google Scholar] [CrossRef] [PubMed]
- Bracarda, S.; Porta, C.; Sisani, M.; Marrocolo, F.; Paglino, C.; Hamzaj, A.; Buono, S.D.; Sternberg, C.N. Comparing comparators: A look at control arms in kidney cancer studies over the years. Br. J. Cancer 2015, 112, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Fillatreau, S.; Sweenie, C.H.; McGeachy, M.J.; Gray, D.; Anderton, S.M. B cells regulate autoimmunity by provision of IL-10. Nat. Immunol. 2002, 3, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Roch, T.; Lampropoulou, V.; O’Connor, R.A.; Stervbo, U.; Hilgenberg, E.; Ries, S.; Dang, V.D.; Jaimes, Y.; Daridon, C.; et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 2014, 507, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Bjarnadottir, K.; Benkhoucha, M.; Merkler, D.; Weber, M.S.; Payne, N.L.; Bernard, C.C.; Molnarfi, N.; Lalive, P.H. B cell-derived transforming growth factor-β1 expression limits the induction phase of autoimmune neuroinflammation. Sci. Rep. 2016, 6, 34594. [Google Scholar] [CrossRef] [PubMed]
- Iwata, Y.; Matsushita, T.; Horikawa, M.; Dilillo, D.J.; Yanaba, K.; Venturi, G.M.; Szabolcs, P.M.; Bernstein, S.H.; Magro, C.M.; Williams, A.D.; et al. Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood 2011, 117, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Lehmann-Horn, K.; Schleich, E.; Hertzenberg, D.; Hapfelmeier, A.; Kumpfel, T.; von Bubnoff, N.; Hohlfeld, R.; Berthele, A.; Hemmer, B.; Weber, M.S. Anti-CD20 B-cell depletion enhances monocyte reactivity in neuroimmunological disorders. J. Neuroinflamm. 2011, 8, 146. [Google Scholar] [CrossRef] [PubMed]
- Duddy, M.E.; Alter, A.; Bar-Or, A. Distinct profiles of human B cell effector cytokines: A role in immune regulation? J. Immunol. 2004, 172, 3422–3427. [Google Scholar] [CrossRef] [PubMed]
- Weinshenker, B.G.; O’Brien, P.C.; Petterson, T.M.; Noseworthy, J.H.; Lucchinetti, C.F.; Dodick, D.W.; Pineda, A.A.; Stevens, L.N.; Rodriguez, M. A randomized trial of plasma exchange in acute central nervous system inflammatory demyelinating disease. Ann. Neurol. 1999, 46, 878–886. [Google Scholar] [CrossRef]
- Ehler, J.; Koball, S.; Sauer, M.; Hickstein, H.; Mitzner, S.; Benecke, R.; Zettl, U.K. Therapeutic plasma exchange in glucocorticosteroid-unresponsive patients with Clinically Isolated Syndrome. Ther. Apher.Dial. 2014, 18, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Ehler, J.; Koball, S.; Sauer, M.; Mitzner, S.; Hickstein, H.; Benecke, R.; Zettl, U.K. Response to Therapeutic Plasma Exchange as a Rescue Treatment in Clinically Isolated Syndromes and Acute Worsening of Multiple Sclerosis: A Retrospective Analysis of 90 Patients. PLoS ONE 2015, 10, e0134583. [Google Scholar] [CrossRef] [PubMed]
- Ruprecht, K.; Klinker, E.; Dintelmann, T.; Rieckmann, P.; Gold, R. Plasma exchange for severe optic neuritis: Treatment of 10 patients. Neurology 2004, 63, 1081–1083. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, R.; Gueguen, A.; Parquet, N.; Saheb, S.; Driss, F.; Mesnil, M.; Vignal, C.; Aboab, J.; Depaz, R.; Gout, O. Plasma exchange response in 34 patients with severe optic neuritis. J. Neurol. 2016, 263, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Zettl, U.K.; Hartung, H.P.; Pahnke, A.; Brueck, W.; Benecke, R.; Pahnke, J. Lesion pathology predicts response to plasma exchange in secondary progressive MS. Neurology 2006, 67, 1515–1516. [Google Scholar] [CrossRef] [PubMed]
- Keegan, M.; Konig, F.; McClelland, R.; Bruck, W.; Morales, Y.; Bitsch, A.; Panitch, H.; Lassmann, H.; Weinshenker, B.; Rodriguez, M.; et al. Relation between humoral pathological changes in multiple sclerosis and response to therapeutic plasma exchange. Lancet 2005, 366, 579–582. [Google Scholar] [CrossRef]
- Seifert, C.L.; Wegner, C.; Sprenger, T.; Weber, M.S.; Bruck, W.; Hemmer, B.; Sellner, J. Favorable response to plasma exchange in tumefactive CNS demyelination with delayed B-cell response. Mult. Scler. J. 2011, 18, 1045–1049. [Google Scholar] [CrossRef] [PubMed]
- Severa, M.; Rizzo, F.; Giacomini, E.; Salvetti, M.; Coccia, E.M. IFN-β and multiple sclerosis: Cross-talking of immune cells and integration of immunoregulatory networks. Cytokine Growth Factor Rev. 2015, 26, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Galboiz, Y.; Shapiro, S.; Lahat, N.; Rawashdeh, H.; Miller, A. Matrix metalloproteinases and their tissue inhibitors as markers of disease subtype and response to interferon-β therapy in relapsing and secondary-progressive multiple sclerosis patients. Ann. Neurol. 2001, 50, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Niino, M.; Hirotani, M.; Miyazaki, Y.; Sasaki, H. Memory and naive B-cell subsets in patients with multiple sclerosis. Neurosci. Lett. 2009, 464, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, F.; Giacomini, E.; Mechelli, R.; Buscarinu, M.C.; Salvetti, M.; Severa, M.; Coccia, E.M. Interferon-β therapy specifically reduces pathogenic memory B cells in multiple sclerosis patients by inducing a FAS-mediated apoptosis. Immunol. Cell Biol. 2016, 94, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Schubert, R.D.; Hu, Y.; Kumar, G.; Szeto, S.; Abraham, P.; Winderl, J.; Guthridge, J.M.; Pardo, G.; Dunn, J.; Steinman, L.; et al. IFN-β treatment requires B cells for efficacy in neuroautoimmunity. J. Immunol. 2015, 194, 2110–2116. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.S.; Starck, M.; Wagenpfeil, S.; Meinl, E.; Hohlfeld, R.; Farina, C. Multiple sclerosis: Glatiramer acetate inhibits monocyte reactivity in vitro and in vivo. Brain 2004, 127, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Vieira, P.L.; Heystek, H.C.; Wormmeester, J.; Wierenga, E.A.; Kapsenberg, M.L. Glatiramer acetate (copolymer-1, copaxone) promotes Th2 cell development and increased IL-10 production through modulation of dendritic cells. J. Immunol. 2003, 170, 4483–4488. [Google Scholar] [CrossRef] [PubMed]
- Lalive, P.H.; Neuhaus, O.; Benkhoucha, M.; Burger, D.; Hohlfeld, R.; Zamvil, S.S.; Weber, M.S. Glatiramer acetate in the treatment of multiple sclerosis: Emerging concepts regarding its mechanism of action. CNS Drugs 2011, 25, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.S.; Prod’homme, T.; Youssef, S.; Dunn, S.E.; Rundle, C.D.; Lee, L.; Patarroyo, J.C.; Stuve, O.; Sobel, R.A.; Steinman, L.; et al. Type II monocytes modulate T cell-mediated central nervous system autoimmune disease. Nat. Med. 2007, 13, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Duda, P.W.; Schmied, M.C.; Cook, S.L.; Krieger, J.I.; Hafler, D.A. Glatiramer acetate (Copaxone) induces degenerate, Th2-polarized immune responses in patients with multiple sclerosis. J. Clin. Investig. 2000, 105, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, O.; Farina, C.; Yassouridis, A.; Wiendl, H.; Then Bergh, F.; Dose, T.; Wekerle, H.; Hohlfeld, R. Multiple sclerosis: Comparison of copolymer-1- reactive T cell lines from treated and untreated subjects reveals cytokine shift from T helper 1 to T helper 2 cells. Proc. Natl. Acad. Sci. USA 2000, 97, 7452–7457. [Google Scholar] [CrossRef] [PubMed]
- Ireland, S.J.; Guzman, A.A.; O’Brien, D.E.; Hughes, S.; Greenberg, B.; Flores, A.; Graves, D.; Remington, G.; Frohman, E.M.; Davis, L.S.; et al. The effect of glatiramer acetate therapy on functional properties of B cells from patients with relapsing-remitting multiple sclerosis. JAMA Neurol. 2014, 71, 1421–1428. [Google Scholar] [CrossRef] [PubMed]
- Ireland, S.J.; Blazek, M.; Harp, C.T.; Greenberg, B.; Frohman, E.M.; Davis, L.S.; Monson, N.L. Antibody-independent B cell effector functions in relapsing remitting multiple sclerosis: Clues to increased inflammatory and reduced regulatory B cell capacity. Autoimmunity 2012, 45, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Begum-Haque, S.; Sharma, A.; Christy, M.; Lentini, T.; Ochoa-Reparaz, J.; Fayed, I.F.; Mielcarz, D.; Haque, A.; Kasper, L.H. Increased expression of B cell-associated regulatory cytokines by glatiramer acetate in mice with experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2010, 219, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Begum-Haque, S.; Christy, M.; Ochoa-Reparaz, J.; Nowak, E.C.; Mielcarz, D.; Haque, A.; Kasper, L.H. Augmentation of regulatory B cell activity in experimental allergic encephalomyelitis by glatiramer acetate. J. Neuroimmunol. 2011, 232, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Sellner, J.; Koczi, W.; Harrer, A.; Oppermann, K.; Obregon-Castrillo, E.; Pilz, G.; Wipfler, P.; Afazel, S.; Haschke-Becher, E.; Trinka, E.; et al. Glatiramer acetate attenuates the pro-migratory profile of adhesion molecules on various immune cell subsets in multiple sclerosis. Clin. Exp. Immunol. 2013, 173, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Bielekova, B.; Richert, N.; Herman, M.L.; Ohayon, J.; Waldmann, T.A.; McFarland, H.; Martin, R.; Blevins, G. Intrathecal effects of daclizumab treatment of multiple sclerosis. Neurology 2011, 77, 1877–1886. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Winokur, P.; Blake, A.; Wu, T.; Romm, E.; Bielekova, B. Daclizumab reverses intrathecal immune cell abnormalities in multiple sclerosis. Ann. Clin. Transl. Neurol. 2015, 2, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Winokur, P.; Blake, A.; Wu, T.; Manischewitz, J.; King, L.R.; Romm, E.; Golding, H.; Bielekova, B. Patients with MS under daclizumab therapy mount normal immune responses to influenza vaccination. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e196. [Google Scholar] [CrossRef] [PubMed]
- Kowarik, M.C.; Pellkofer, H.L.; Cepok, S.; Korn, T.; Kumpfel, T.; Buck, D.; Hohlfeld, R.; Berthele, A.; Hemmer, B. Differential effects of fingolimod (FTY720) on immune cells in the CSF and blood of patients with MS. Neurology 2011, 76, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Blumenfeld, S.; Staun-Ram, E.; Miller, A. Fingolimod therapy modulates circulating B cell composition, increases B regulatory subsets and production of IL-10 and TGFβ in patients with Multiple Sclerosis. J. Autoimmun. 2016, 70, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Grutzke, B.; Hucke, S.; Gross, C.C.; Herold, M.V.; Posevitz-Fejfar, A.; Wildemann, B.T.; Kieseier, B.C.; Dehmel, T.; Wiendl, H.; Klotz, L. Fingolimod treatment promotes regulatory phenotype and function of B cells. Ann. Clin. Transl. Neurol. 2015, 2, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Bail, K.; Notz, Q.; Rovituso, D.M.; Schampel, A.; Wunsch, M.; Koeniger, T.; Schropp, V.; Bharti, R.; Scholz, C.J.; Foerstner, K.U.; et al. Differential effects of FTY720 on the B cell compartment in a mouse model of multiple sclerosis. J. Neuroinflamm. 2017, 14, 148. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.D.; Martin, K.A.; Calabresi, P.A.; Bhargava, P. Dimethyl fumarate alters B-cell memory and cytokine production in MS patients. Ann. Clin. Transl. Neurol. 2017, 4, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Longbrake, E.E.; Cantoni, C.; Chahin, S.; Cignarella, F.; Cross, A.H.; Piccio, L. Dimethyl fumarate induces changes in B- and T-lymphocyte function independent of the effects on absolute lymphocyte count. Mult. Scler. J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lundy, S.K.; Wu, Q.; Wang, Q.; Dowling, C.A.; Taitano, S.H.; Mao, G.; Mao-Draayer, Y. Dimethyl fumarate treatment of relapsing-remitting multiple sclerosis influences B-cell subsets. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e211. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Rezk, A.; Ghadiri, M.; Luessi, F.; Zipp, F.; Li, H.; Giacomini, P.S.; Antel, J.; Bar-Or, A. Dimethyl Fumarate Treatment Mediates an Anti-Inflammatory Shift in B Cell Subsets of Patients with Multiple Sclerosis. J. Immunol. 2017, 198, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Krumbholz, M.; Meinl, I.; Kumpfel, T.; Hohlfeld, R.; Meinl, E. Natalizumab disproportionately increases circulating pre-B and B cells in multiple sclerosis. Neurology 2008, 71, 1350–1354. [Google Scholar] [CrossRef] [PubMed]
- Stuve, O.; Youssef, S.; Weber, M.S.; Nessler, S.; von Budingen, H.C.; Hemmer, B.; Prod’homme, T.; Sobel, R.A.; Steinman, L.; Zamvil, S.S. Immunomodulatory synergy by combination of atorvastatin and glatiramer acetate in treatment of CNS autoimmunity. J. Clin. Investig. 2006, 116, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Warnke, C.; Stettner, M.; Lehmensiek, V.; Dehmel, T.; Mausberg, A.K.; von Geldern, G.; Gold, R.; Kumpfel, T.; Hohlfeld, R.; Maurer, M.; et al. Natalizumab exerts a suppressive effect on surrogates of B cell function in blood and CSF. Mult. Scler. J. 2015, 21, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Mellergard, J.; Edstrom, M.; Jenmalm, M.C.; Dahle, C.; Vrethem, M.; Ernerudh, J. Increased B cell and cytotoxic NK cell proportions and increased T cell responsiveness in blood of natalizumab-treated multiple sclerosis patients. PLoS ONE 2013, 8, e81685. [Google Scholar] [CrossRef] [PubMed]
- Planas, R.; Jelcic, I.; Schippling, S.; Martin, R.; Sospedra, M. Natalizumab treatment perturbs memory- and marginal zone-like B-cell homing in secondary lymphoid organs in multiple sclerosis. Eur. J. Immunol. 2012, 42, 790–798. [Google Scholar] [CrossRef] [PubMed]
- von Glehn, F.; Farias, A.S.; de Oliveira, A.C.; Damasceno, A.; Longhini, A.L.; Oliveira, E.C.; Damasceno, B.P.; Santos, L.M.; Brandao, C.O. Disappearance of cerebrospinal fluid oligoclonal bands after natalizumab treatment of multiple sclerosis patients. Mult. Scler. J. 2012, 18, 1038–1041. [Google Scholar] [CrossRef] [PubMed]
- Lehmann-Horn, K.; Sagan, S.A.; Bernard, C.C.; Sobel, R.A.; Zamvil, S.S. B-cell very late antigen-4 deficiency reduces leukocyte recruitment and susceptibility to central nervous system autoimmunity. Ann. Neurol. 2015, 77, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Lehmann-Horn, K.; Sagan, S.A.; Winger, R.C.; Spencer, C.M.; Bernard, C.C.; Sobel, R.A.; Zamvil, S.S. CNS accumulation of regulatory B cells is VLA-4-dependent. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e212. [Google Scholar] [CrossRef] [PubMed]
- Coles, A.J.; Twyman, C.L.; Arnold, D.L.; Cohen, J.A.; Confavreux, C.; Fox, E.J.; Hartung, H.P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: A randomised controlled phase 3 trial. Lancet 2012, 380, 1829–1839. [Google Scholar] [CrossRef]
- Cohen, J.A.; Coles, A.J.; Arnold, D.L.; Confavreux, C.; Fox, E.J.; Hartung, H.P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; Fisher, E.; et al. Alemtuzumab versus interferon β 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: A randomised controlled phase 3 trial. Lancet 2012, 380, 1819–1828. [Google Scholar] [CrossRef]
- Baker, D.; Herrod, S.S.; Alvarez-Gonzalez, C.; Giovannoni, G.; Schmierer, K. Interpreting Lymphocyte Reconstitution Data From the Pivotal Phase 3 Trials of Alemtuzumab. JAMA Neurol. 2017, 74, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Waubant, E.; Arnold, D.L.; Vollmer, T.; Antel, J.; Fox, R.J.; Bar-Or, A.; Panzara, M.; Sarkar, N.; Agarwal, S.; et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 2008, 358, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Li, D.; Calabresi, P.A.; O’Connor, P.; Bar-Or, A.; Barkhof, F.; Yin, M.; Leppert, D.; Glanzman, R.; Tinbergen, J.; et al. Ocrelizumab in relapsing-remitting multiple sclerosis: A phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011, 378, 1779–1787. [Google Scholar] [CrossRef]
- Sorensen, P.S.; Lisby, S.; Grove, R.; Derosier, F.; Shackelford, S.; Havrdova, E.; Drulovic, J.; Filippi, M. Safety and efficacy of ofatumumab in relapsing-remitting multiple sclerosis: A phase 2 study. Neurology 2014, 82, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus Interferon β-1a in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Hawker, K.; O’Connor, P.; Freedman, M.S.; Calabresi, P.A.; Antel, J.; Simon, J.; Hauser, S.; Waubant, E.; Vollmer, T.; Panitch, H.; et al. Rituximab in patients with primary progressive multiple sclerosis: Results of a randomized double-blind placebo-controlled multicenter trial. Ann. Neurol. 2009, 66, 460–471. [Google Scholar] [CrossRef] [PubMed]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Martin Mdel, P.; Cravens, P.D.; Winger, R.; Kieseier, B.C.; Cepok, S.; Eagar, T.N.; Zamvil, S.S.; Weber, M.S.; Frohman, E.M.; Kleinschmidt-Demasters, B.K.; et al. Depletion of B lymphocytes from cerebral perivascular spaces by rituximab. Arch. Neurol. 2009, 66, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Monson, N.L.; Cravens, P.D.; Frohman, E.M.; Hawker, K.; Racke, M.K. Effect of rituximab on the peripheral blood and cerebrospinal fluid B cells in patients with primary progressive multiple sclerosis. Arch. Neurol. 2005, 62, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Cross, A.H.; Stark, J.L.; Lauber, J.; Ramsbottom, M.J.; Lyons, J.A. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J. Neuroimmunol. 2006, 180, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.L.; Combs, D.; Rosenberg, J.; Levy, A.; McDermott, M.; Damon, L.; Ignoffo, R.; Aldape, K.; Shen, A.; Lee, D.; et al. Rituximab therapy for CNS lymphomas: Targeting the leptomeningeal compartment. Blood 2003, 101, 466–468. [Google Scholar] [CrossRef] [PubMed]
- Lehmann-Horn, K.; Kinzel, S.; Feldmann, L.; Radelfahr, F.; Hemmer, B.; Traffehn, S.; Bernard, C.C.; Stadelmann, C.; Bruck, W.; Weber, M.S. Intrathecal anti-CD20 efficiently depletes meningeal B cells in CNS autoimmunity. Ann. Clin. Transl. Neurol. 2014, 1, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Studer, V.; Rossi, S.; Motta, C.; Buttari, F.; Centonze, D. Peripheral B cell depletion and central proinflammatory cytokine reduction following repeated intrathecal administration of rituximab in progressive Multiple Sclerosis. J. Neuroimmunol. 2014, 276, 229–231. [Google Scholar] [CrossRef] [PubMed]
- Topping, J.; Dobson, R.; Lapin, S.; Maslyanskiy, A.; Kropshofer, H.; Leppert, D.; Giovannoni, G.; Evdoshenko, E. The effects of intrathecal rituximab on biomarkers in multiple sclerosis. Mult. Scler. Relat. Disord. 2016, 6, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Svenningsson, A.; Bergman, J.; Dring, A.; Vagberg, M.; Birgander, R.; Lindqvist, T.; Gilthorpe, J.; Bergenheim, T. Rapid depletion of B lymphocytes by ultra-low-dose rituximab delivered intrathecally. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e79. [Google Scholar] [CrossRef] [PubMed]
- Costello, F.; Stuve, O.; Weber, M.S.; Zamvil, S.S.; Frohman, E. Combination therapies for multiple sclerosis: Scientific rationale, clinical trials, and clinical practice. Curr. Opin. Neurol. 2007, 20, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Komori, M.; Lin, Y.C.; Cortese, I.; Blake, A.; Ohayon, J.; Cherup, J.; Maric, D.; Kosa, P.; Wu, T.; Bielekova, B. Insufficient disease inhibition by intrathecal rituximab in progressive multiple sclerosis. Ann. Clin. Transl. Neurol. 2016, 3, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Flach, A.C.; Litke, T.; Strauss, J.; Haberl, M.; Gomez, C.C.; Reindl, M.; Saiz, A.; Fehling, H.J.; Wienands, J.; Odoardi, F.; et al. Autoantibody-boosted T-cell reactivation in the target organ triggers manifestation of autoimmune CNS disease. Proc. Natl. Acad. Sci. USA 2016, 113, 3323–3328. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, L.; Franciotta, D.; Vigo, T.; Grandis, M.; Fiorina, E.; Ghiglione, E.; Roccatagliata, L.; Mancardi, G.L.; Uccelli, A.; Schenone, A. Relapses after treatment with rituximab in a patient with multiple sclerosis and anti myelin-associated glycoprotein polyneuropathy. Arch. Neurol. 2007, 64, 1531–1533. [Google Scholar] [CrossRef] [PubMed]
- Gong, Q.; Ou, Q.; Ye, S.; Lee, W.P.; Cornelius, J.; Diehl, L.; Lin, W.Y.; Hu, Z.; Lu, Y.; Chen, Y.; et al. Importance of cellular microenvironment and circulatory dynamics in B cell immunotherapy. J. Immunol. 2005, 174, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Thaunat, O.; Patey, N.; Gautreau, C.; Lechaton, S.; Fremeaux-Bacchi, V.; Dieu-Nosjean, M.C.; Cassuto-Viguier, E.; Legendre, C.; Delahousse, M.; Lang, P.; et al. B cell survival in intragraft tertiary lymphoid organs after rituximab therapy. Transplantation 2008, 85, 1648–1653. [Google Scholar] [CrossRef] [PubMed]
- Hutt-Fletcher, L.M. Epstein-Barr Virus Entry. J. Virol. 2007, 81, 7825–7832. [Google Scholar] [CrossRef] [PubMed]
- Souza, T.A.; Stollar, B.D.; Sullivan, J.L.; Luzuriaga, K.; Thorley-Lawson, D.A. Peripheral B cells latently infected with Epstein–Barr virus display molecular hallmarks of classical antigen-selected memory B cells. Proc. Natl. Acad. Sci. USA 2005, 102, 18093–18098. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.; Klein, E.; Kashuba, E. Interaction of Epstein-Barr virus (EBV) with human B-lymphocytes. Biochem. Biophys. Res. Commun. 2010, 396, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Ramagopalan, S.V.; Dobson, R.; Meier, U.C.; Giovannoni, G. Multiple sclerosis: Risk factors, prodromes, and potential causal pathways. Lancet Neurol. 2010, 9, 727–739. [Google Scholar] [CrossRef]
- Thacker, E.L.; Mirzaei, F.; Ascherio, A. Infectious mononucleosis and risk for multiple sclerosis: A meta-analysis. Ann. Neurol. 2006, 59, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Munger, K.L.; Lennette, E.T.; Spiegelman, D.; Hernán, M.A.; Olek, M.J.; Hankinson, S.E.; Hunter, D.J. Epstein-barr virus antibodies and risk of multiple sclerosis: A prospective study. JAMA 2001, 286, 3083–3088. [Google Scholar] [CrossRef] [PubMed]
- Serafini, B.; Rosicarelli, B.; Franciotta, D.; Magliozzi, R.; Reynolds, R.; Cinque, P.; Andreoni, L.; Trivedi, P.; Salvetti, M.; Faggioni, A.; et al. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J. Exp. Med. 2007, 204, 2899–2912. [Google Scholar] [CrossRef] [PubMed]
- Willis, S.N.; Stadelmann, C.; Rodig, S.J.; Caron, T.; Gattenloehner, S.; Mallozzi, S.S.; Roughan, J.E.; Almendinger, S.E.; Blewett, M.M.; Brück, W.; et al. Epstein–Barr virus infection is not a characteristic feature of multiple sclerosis brain. Brain 2009, 132, 3318–3328. [Google Scholar] [CrossRef] [PubMed]
- Sargsyan, S.A.; Shearer, A.J.; Ritchie, A.M.; Burgoon, M.P.; Anderson, S.; Hemmer, B.; Stadelmann, C.; Gattenlöhner, S.; Owens, G.P.; Gilden, D.; et al. Absence of Epstein-Barr virus in the brain and CSF of patients with multiple sclerosis. Neurology 2010, 74, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Palanichamy, A.; Jahn, S.; Nickles, D.; Derstine, M.; Abounasr, A.; Hauser, S.L.; Baranzini, S.E.; Leppert, D.; von Budingen, H.C. Rituximab efficiently depletes increased CD20-expressing T cells in multiple sclerosis patients. J. Immunol. 2014, 193, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Holley, J.E.; Bremer, E.; Kendall, A.C.; de Bruyn, M.; Helfrich, W.; Tarr, J.M.; Newcombe, J.; Gutowski, N.J.; Eggleton, P. CD20+inflammatory T-cells are present in blood and brain of multiple sclerosis patients and can be selectively targeted for apoptotic elimination. Mult. Scler. Relat. Disord. 2014, 3, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Schuh, E.; Berer, K.; Mulazzani, M.; Feil, K.; Meinl, I.; Lahm, H.; Krane, M.; Lange, R.; Pfannes, K.; Subklewe, M.; et al. Features of Human CD3+CD20+ T Cells. J. Immunol. 2016, 197, 1111–1117. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Gallagher, S.; Monson, N.L.; Herbst, R.; Wang, Y. Inebilizumab, a B Cell-Depleting Anti-CD19 Antibody for the Treatment of Autoimmune Neurological Diseases: Insights from Preclinical Studies. J. Clin. Med. 2016, 5, 107. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Wildemann, B.; Jarius, S.; Orellano, B.; Sasidharan, S.; Weber, M.S.; Stuve, O. Immunopathogenesis of neuromyelitis optica. Adv. Immunol. 2014, 121, 213–242. [Google Scholar] [PubMed]
- Herbst, R.; Wang, Y.; Gallagher, S.; Mittereder, N.; Kuta, E.; Damschroder, M.; Woods, R.; Rowe, D.C.; Cheng, L.; Cook, K.; et al. B-cell depletion in vitro and in vivo with an afucosylated anti-CD19 antibody. J. Pharmacol. Exp. Ther. 2010, 335, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Agius, M.; Klodowska-Duda, G.; Maciejowski, M.; Potemkowski, A.; Sweeny, S.; Li, J.; Yao, W.; Patra, K.; Ratchford, J.N.; Katz, E.; et al. Safety and tolerability of MEDI-551 in patients with relapsing forms of multiple sclerosis: Results from a phase 1 randomised, placebo-controlled, escalating intravenous and subcutaneous dose study. Mult. Scler. J. 2015, 21, 235–236. [Google Scholar]
- Kappos, L.; Hartung, H.P.; Freedman, M.S.; Boyko, A.; Radu, E.W.; Mikol, D.D.; Lamarine, M.; Hyvert, Y.; Freudensprung, U.; Plitz, T.; et al. Atacicept in multiple sclerosis (ATAMS): A randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol. 2014, 13, 353–363. [Google Scholar] [CrossRef]
- Weber, M.S.; Menge, T.; Lehmann-Horn, K.; Kronsbein, H.C.; Zettl, U.; Sellner, J.; Hemmer, B.; Stuve, O. Current treatment strategies for multiple sclerosis—Efficacy versus neurological adverse effects. Curr. Pharm. Des. 2012, 18, 209–219. [Google Scholar] [CrossRef] [PubMed]
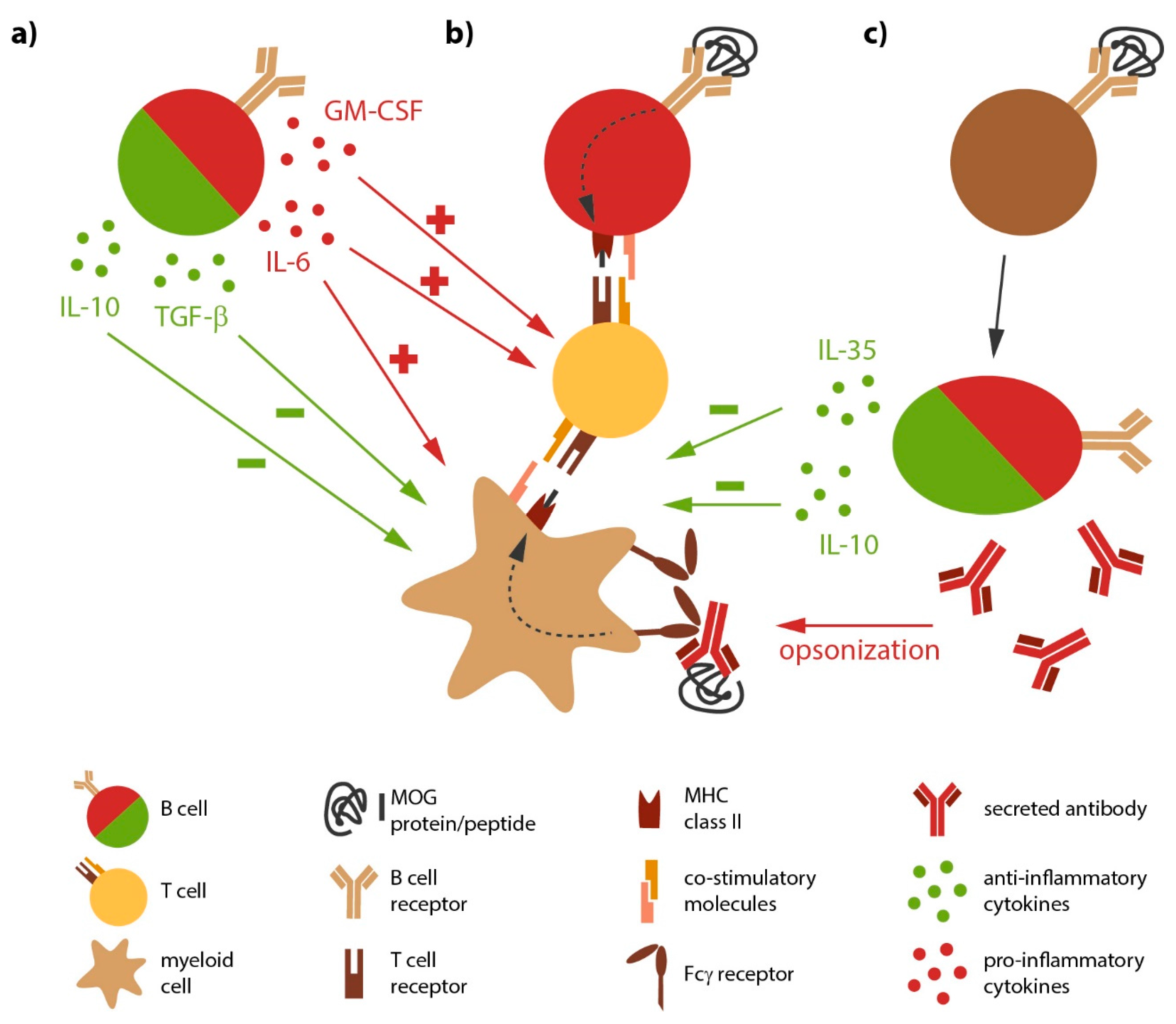

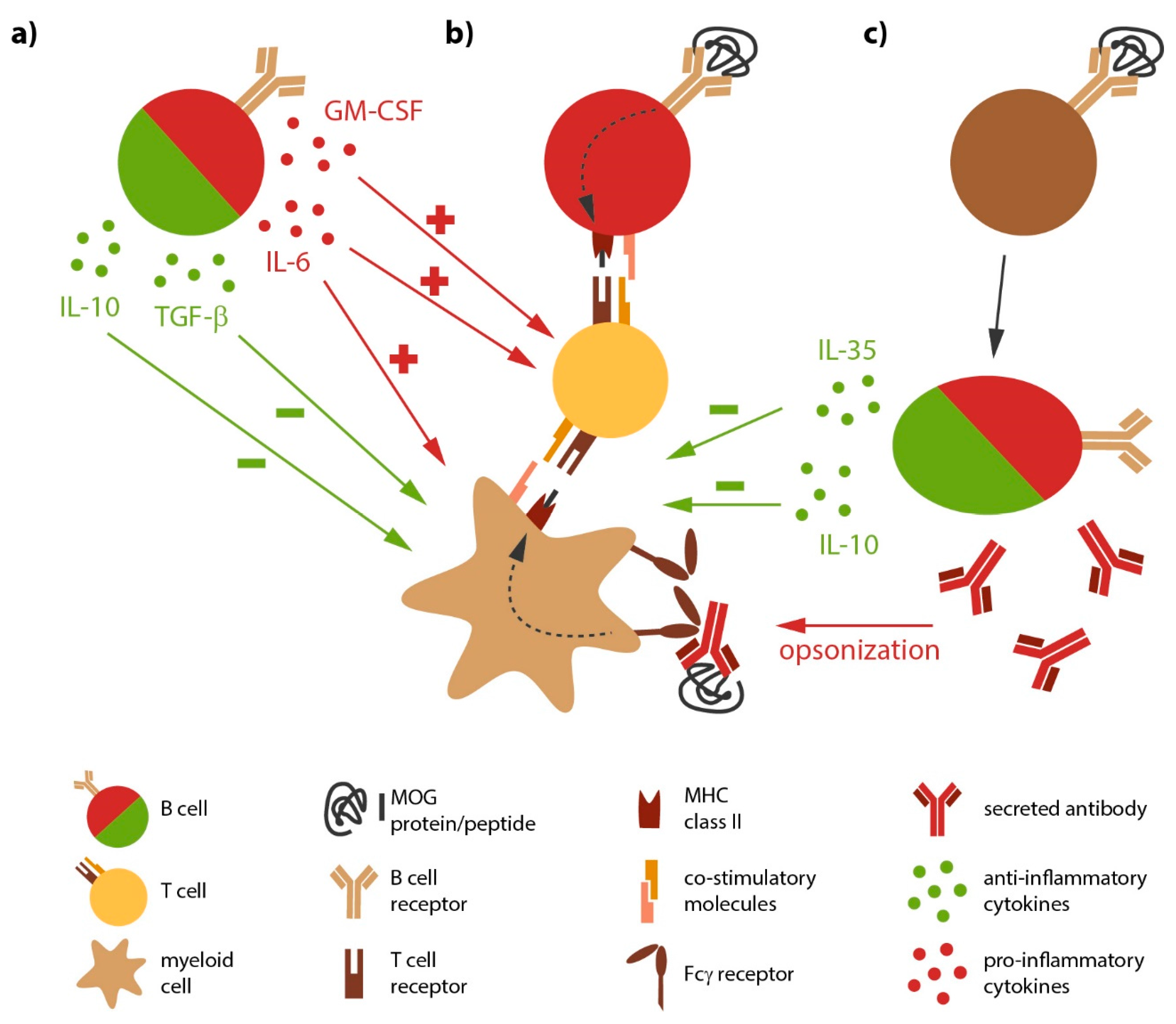{kind=link}
| Rituximab | Ocrelizumab | Ofatumumab | |
|---|---|---|---|
| origin/chimerism | chimeric IgG1 | humanized IgG1 | fully human IgG1 |
| administration | i.v. | i.v. | s.c. or i.v. |
| dosage | variable | induction with 2 × 300 mg, 600 mg every 24 weeks | Variable every 4 weeks |
| mechanism of action | CDC > ADCC | CDC < ADCC | CDC |
| immunogenicity | ++ | + | (+) |
| targeted epitope | CD20 pos. 165–182 | CD20 pos. 165–182 | CD20 pos. 146–160 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lehmann-Horn, K.; Kinzel, S.; Weber, M.S. Deciphering the Role of B Cells in Multiple Sclerosis—Towards Specific Targeting of Pathogenic Function. Int. J. Mol. Sci. 2017, 18, 2048. https://doi.org/10.3390/ijms18102048
Lehmann-Horn K, Kinzel S, Weber MS. Deciphering the Role of B Cells in Multiple Sclerosis—Towards Specific Targeting of Pathogenic Function. International Journal of Molecular Sciences. 2017; 18(10):2048. https://doi.org/10.3390/ijms18102048
Chicago/Turabian StyleLehmann-Horn, Klaus, Silke Kinzel, and Martin S. Weber. 2017. "Deciphering the Role of B Cells in Multiple Sclerosis—Towards Specific Targeting of Pathogenic Function" International Journal of Molecular Sciences 18, no. 10: 2048. https://doi.org/10.3390/ijms18102048
APA StyleLehmann-Horn, K., Kinzel, S., & Weber, M. S. (2017). Deciphering the Role of B Cells in Multiple Sclerosis—Towards Specific Targeting of Pathogenic Function. International Journal of Molecular Sciences, 18(10), 2048. https://doi.org/10.3390/ijms18102048