Nrf2-Inducers Counteract Neurodegeneration in Frataxin-Silenced Motor Neurons: Disclosing New Therapeutic Targets for Friedreich’s Ataxia

 , and
, and
Abstract
:1. Introduction
2. Results
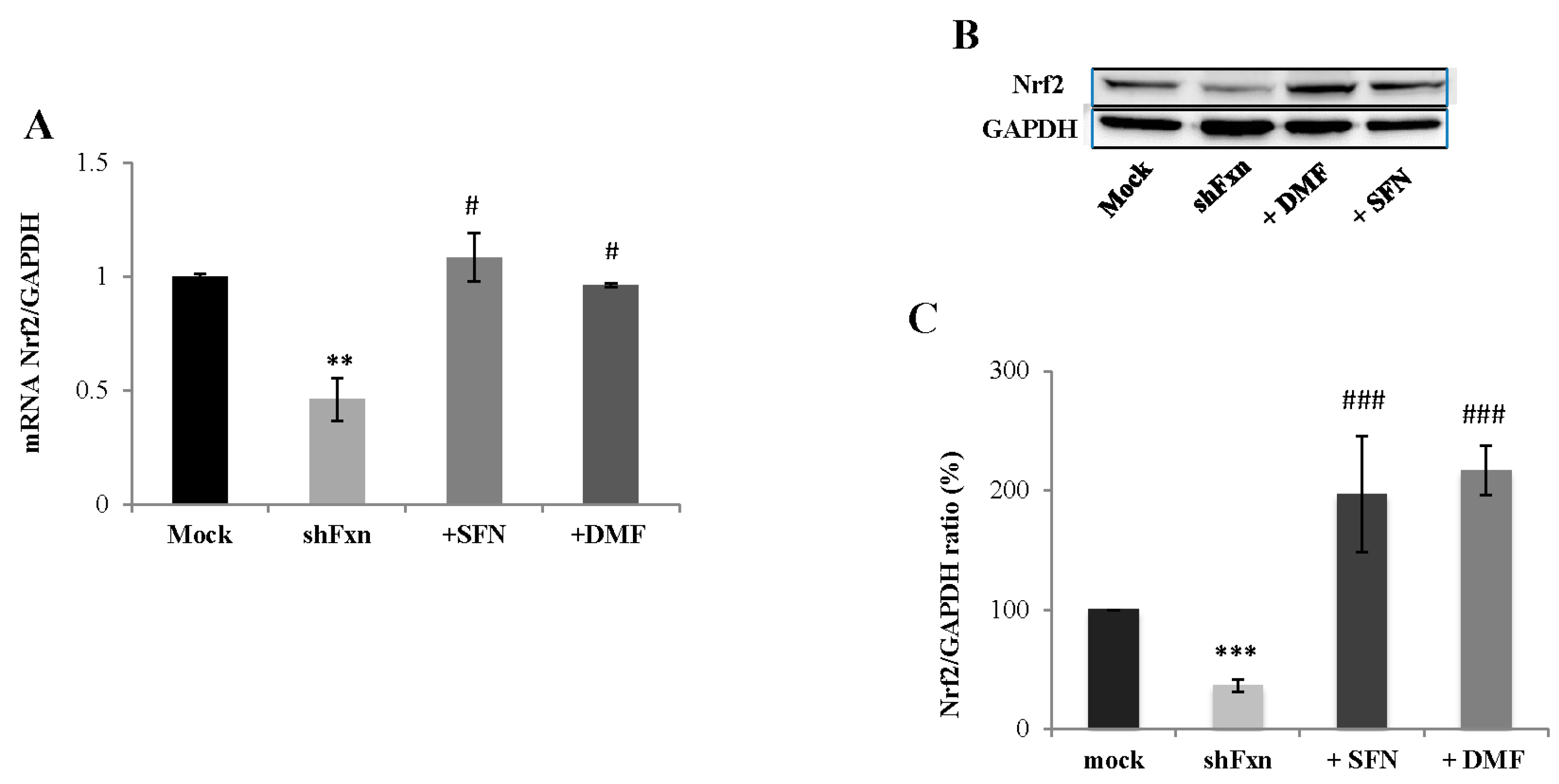
2.1. SFN Upregulates Nrf2 in Frataxin-Silenced Motor Neurons (shFxn)
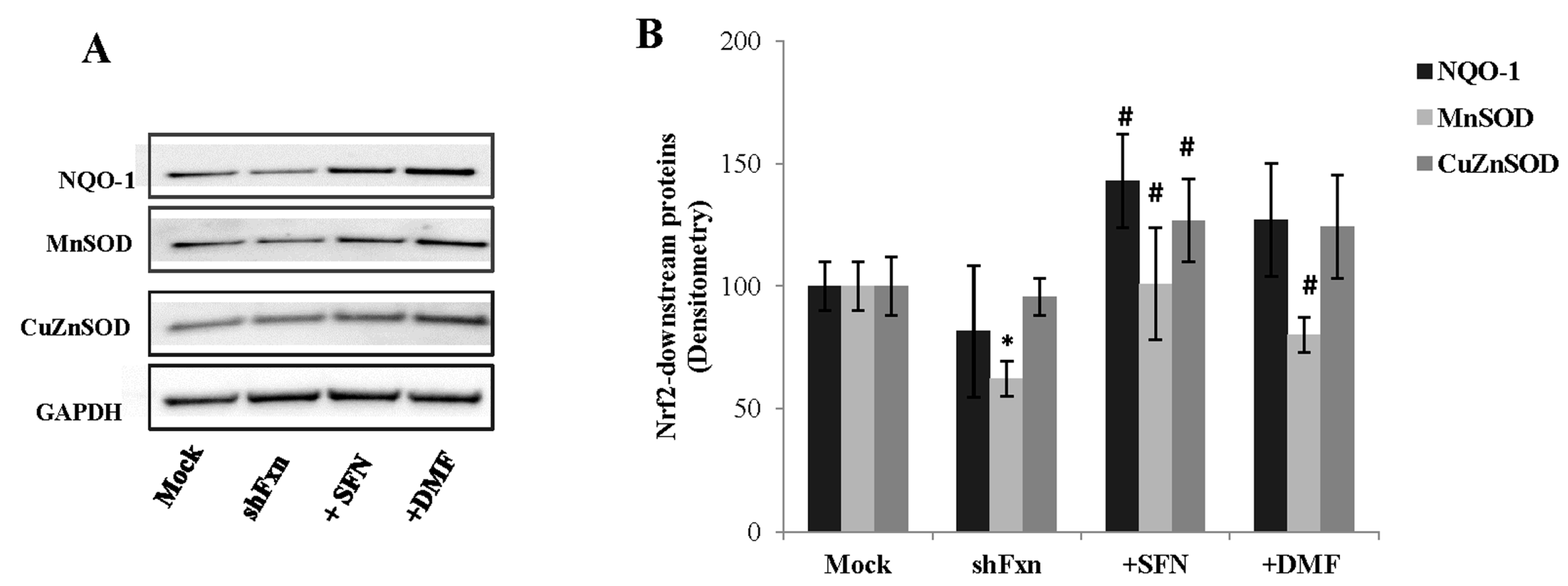
2.2. SFN Activates the Phase II Response in shFxn
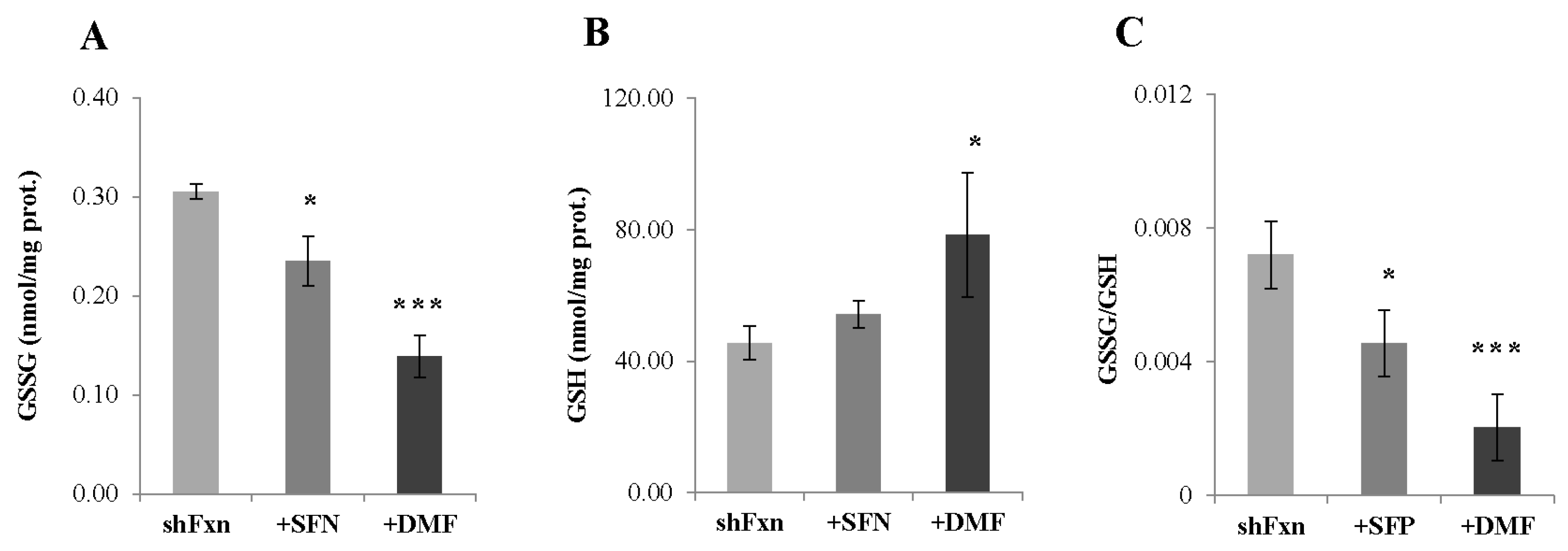
2.3. Nrf2-Inducers Target the Repletion of the Active Form of Glutathione (GSH)
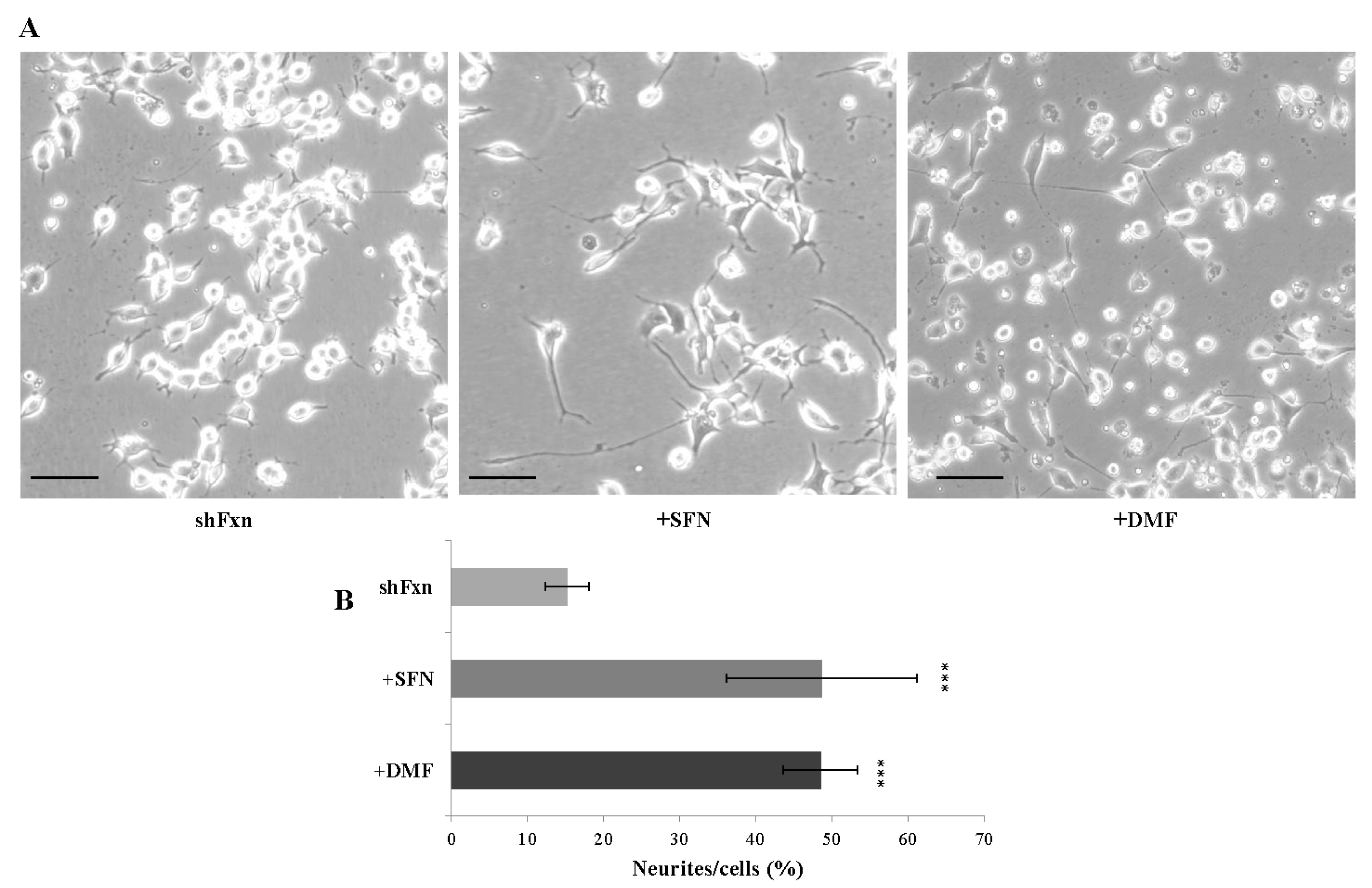
2.4. SFN Triggers Axonal Re-Growth in the shFxn
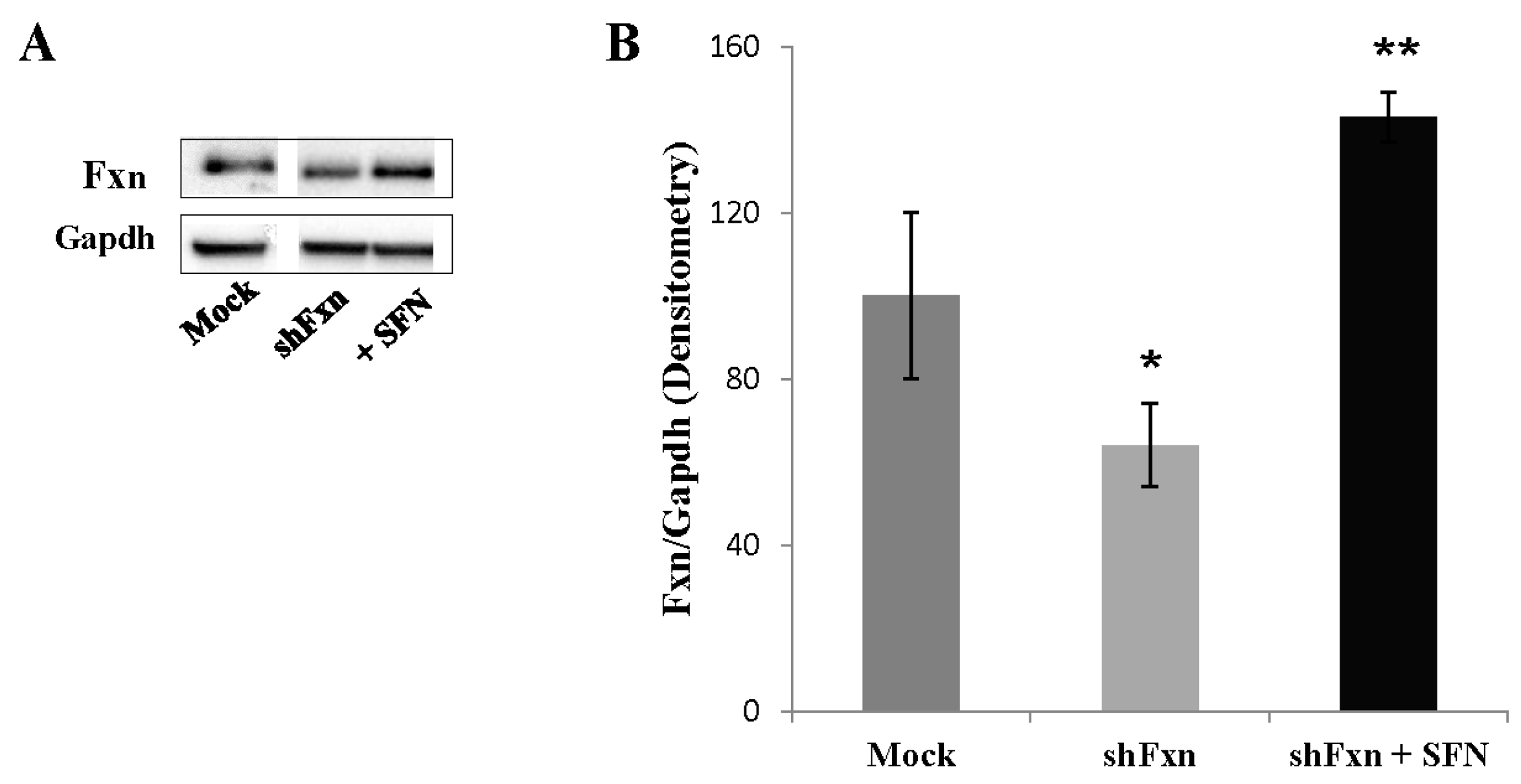
2.5. SFN Increases the Frataxin Expression in shFxn
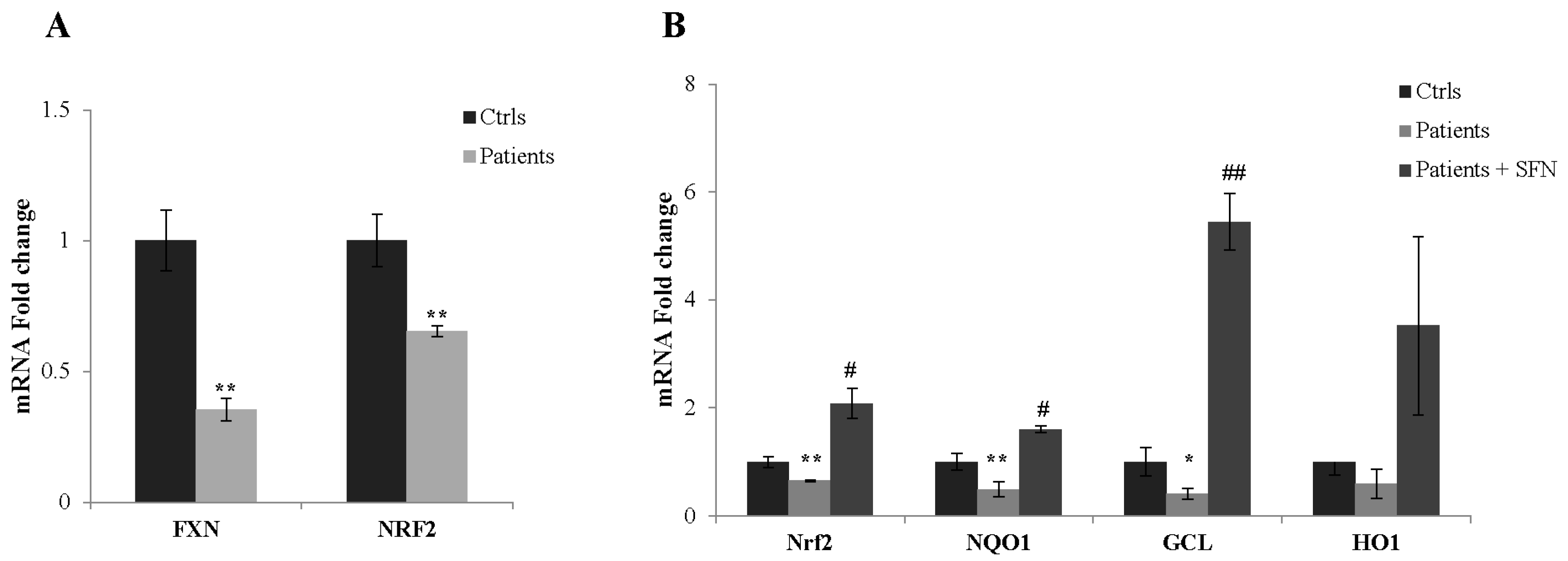
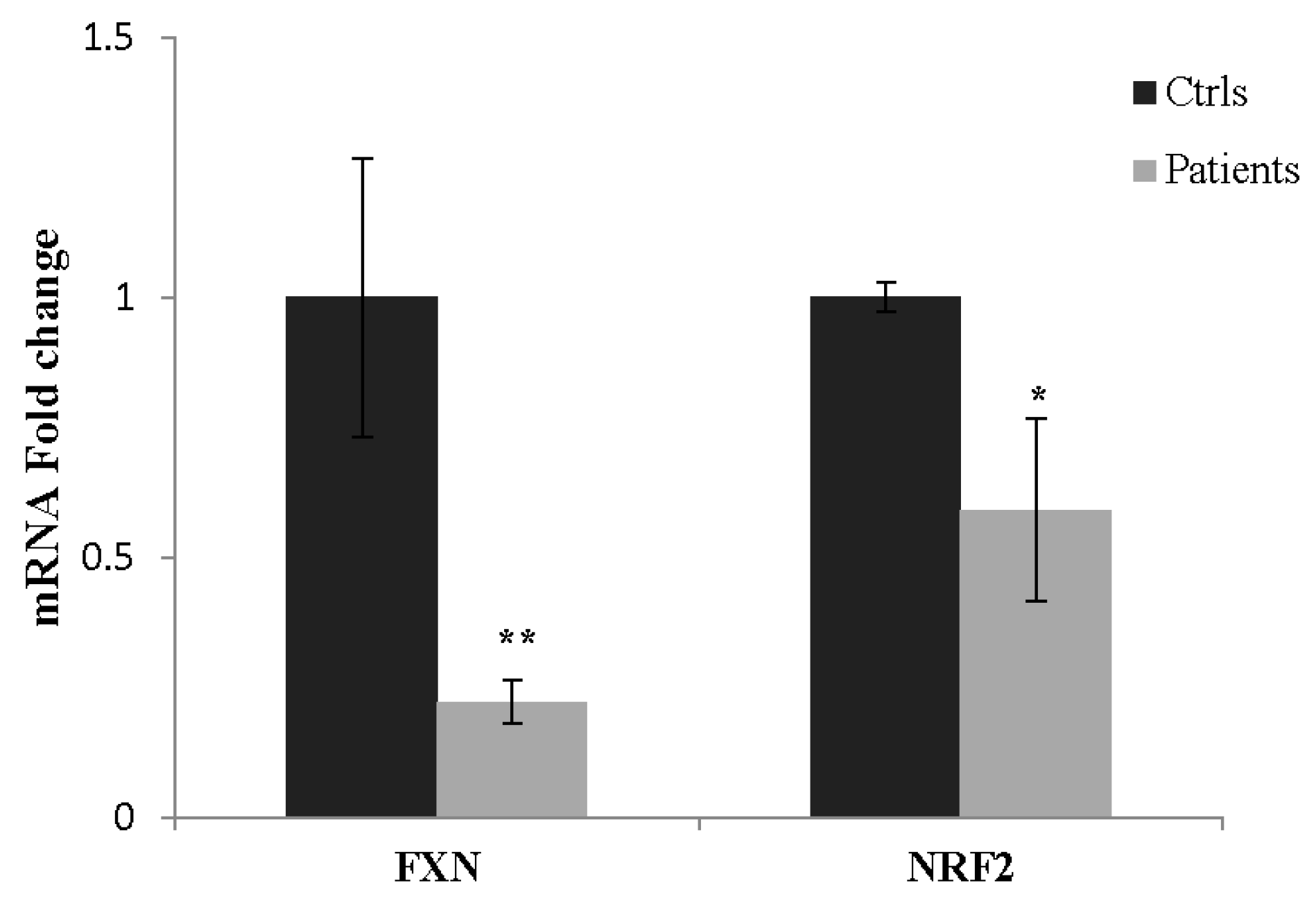
2.6. SFN Restores Nrf2 Transcriptional Activity in Fibroblasts of Patients with FA
2.7. NRF2 as a Potential Biomarker in FA
3. Discussion
4. Materials and Methods
4.1. Stable shFxn Cell Lines Generation and Treatments
4.2. Fibroblasts Cultures
4.3. Blood Sample Collection
4.4. Quantitative Real-Time PCR (qRT-PCR)
4.5. Immunoblot Analysis
4.6. HPLC Analysis of Reduced (GSH) and Oxidized (GSSG) Glutathione Forms
4.7. Neurite Numbers’ and Morphology
4.8. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hayashi, G.; Cortopassi, G. Oxidative stress in inherited mitochondrial diseases. Free Radic. Biol. Med. 2015, 88, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Schoenfeld, R.A.; Hayashi, G.; Napoli, E.; Akiyama, T.; Iodi Carstens, M.; Carstens, E.E.; Pook, M.A.; Cortopassi, G.A. Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich’s ataxia YG8R mouse model. Antioxid. Redox Signal. 2013, 19, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Bürk, K. Friedreich Ataxia: Current status and future prospects. Cerebellum Ataxias 2017, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado, A.; Moreno-Murciano, P.; Pedraza-Chaverri, J. The transcription factor Nrf2 as a new therapeutic target in Parkinson’s disease. Expert Opin. Ther. Targets 2009, 13, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Ishii, T.; Wakabayashi, N.; Yamamoto, M. Regulatory mechanisms of cellular response to oxidative stress. Free Radic. Res. 1999, 31, 319–324. [Google Scholar] [PubMed]
- Johnson, D.A.; Andrews, G.K.; Xu, W.; Johnson, J.A. Activation of the antioxidant response element in primary cortical neuronal cultures derived from transgenic reporter mice. J. Neurochem. 2002, 81, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Calkins, M.J.; Chan, K.; Kan, Y.W.; Johnson, J.A. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J. Biol. Chem. 2003, 278, 12029–12038. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.A.; Johnson, J.A. Nrf2-a therapeutic target for the treatment of neurodegenerative diseases. Free Radic. Biol. Med. 2015, 88, 253–267. [Google Scholar] [CrossRef] [PubMed]
- D’Oria, V.; Petrini, S.; Travaglini, L.; Priori, C.; Piermarini, E.; Petrillo, S.; Carletti, B.; Bertini, E.; Piemonte, F. Frataxin deficiency leads to reduced expression and impaired translocation of NF-E2 Related Factor (Nrf2) in cultured motor neurons. Int. J. Mol. Sci. 2013, 14, 7853–7865. [Google Scholar] [CrossRef] [PubMed]
- Paupe, V.; Dassa, E.P.; Goncalves, S.; Auchère, F.; Lönn, M.; Holmgren, A.; Rustin, P. Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS ONE 2009, 4, 4253–4264. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, G.; Jasoliya, M.; Saccà, F.; Pane, C.; Filla, A.; Marsili, A.; Puorro, G.; Lanzillo, R.; Brescia Morra, V.; Cortopassi, G. Dimethyl Fumarate Mediates Nrf2-dependent Mitochondrial Biogenesis in Mice and Humans. Hum. Mol. Genet. 2017. [Google Scholar] [CrossRef] [PubMed]
- Carletti, B.; Piermarini, E.; Tozzi, G.; Travaglini, L.; Torraco, A.; Pastore, A.; Sparaco, M.; Petrillo, S.; Carrozzo, R.; Bertini, E.; et al. Frataxin silencing inactivates mitochondrial Complex I in NSC34 motoneuronal cells and alters glutathione homeostasis. Int. J. Mol. Sci. 2014, 15, 5789–5806. [Google Scholar] [CrossRef] [PubMed]
- Piermarini, E.; Cartelli, D.; Pastore, A.; Tozzi, G.; Compagnucci, C.; Giorda, E.; D’Amico, J.; Petrini, S.; Bertini, E.; Cappelletti, G.; et al. Frataxin silencing alters microtubule stability in motor neurons: Implications for Friedreich’s ataxia. Hum. Mol. Genet. 2016, 25, 4288–4301. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Talalay, P.; Cho, C.G.; Posner, G.H. A major inducer of anticarcinogenic protective enzymes from broccoli: Isolation and elucidation of structure. Proc. Natl. Acad. Sci. USA 1992, 89, 2399–2403. [Google Scholar] [CrossRef] [PubMed]
- Innamorato, N.G.; Rojo, A.I.; Garcia-Yague, A.J.; Yamamoto, M.; de Ceballos, M.L.; Cuadrado, A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J. Immunol. 2008, 181, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Houghton, C.A.; Fassett, R.G.; Coombes, J.S. Sulforaphane: Translational research from laboratory bench to clinic. Nutr. Rev. 2013, 71, 709–726. [Google Scholar] [CrossRef] [PubMed]
- Jazwa, A.; Rojo, A.I.; Innamorato, N.G.; Hesse, M.; Fernandez Ruiz, J.; Cuadrado, A. Pharmacological targeting of the transcription factor Nrf2 at the basal ganglia provides disease modifying therapy for experimental parkinsonism. Antioxid. Redox Signal. 2011, 14, 2347–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Chen, B.; Wang, X.; Wu, L.; Yang, Y.; Cheng, X.; Hu, Z.; Cai, X.; Yang, J.; Sun, X.; et al. Sulforaphane protects against rotenone-induced neurotoxicity in vivo: Involvement of the mTOR, Nrf2, and autophagy pathways. Sci. Rep. 2016, 24, 32206. [Google Scholar] [CrossRef] [PubMed]
- Lastres-Becker, I.; García-Yagüe, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kügler, S.; Rábano, A.; Cuadrado, A. Repurposing the NRF2 Activator Dimethyl Fumarate as Therapy Against Synucleinopathy in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Carletti, B.; Passarelli, C.; Sparaco, M.; Tozzi, G.; Pastore, A.; Bertini, E.; Piemonte, F. Effect of protein glutathionylation on neuronal cytoskeleton: A potential link to neurodegeneration. Neuroscience 2011, 192, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Canning, P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch. Biochem. Biophys. 2017, 617, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Holland, R.; Hawkins, A.E.; Eggler, A.L.; Mesecar, A.D.; Fabris, D.; Fishbein, J.C. Prospective type 1 and type 2 disulfides of Keap1 protein. Chem. Res. Toxicol. 2008, 21, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Cebula, M.; Schmidt, E.E.; Arnér, E.S. TrxR1 as a potent regulator of the Nrf2-Keap1 response system. Antioxid. Redox Signal. 2015, 23, 823–853. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: Role of antioxidant response element-like sequences in the nrf2 promoter. Mol. Cell. Biol. 2002, 22, 2883–2892. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, W.; Su, Z.Y.; Kong, A.N. The complexity of the Nrf2 pathway: Beyond the antioxidant response. J. Nutr. Biochem. 2015, 26, 1401–1413. [Google Scholar] [CrossRef] [PubMed]
- Burchell, V.S.; Gandhi, S.; Deas, E.; Wood, N.W.; Abramov, A.Y.; Plun-Favreau, H. Targeting mitochondrial dysfunction in neurodegenerative disease: Part II. Expert Opin. Ther. Targets 2010, 14, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Flohé, L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal. 2011, 15, 2335–2381. [Google Scholar] [CrossRef] [PubMed]
- Esteras, N.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 activation in the treatment of neurodegenerative diseases: A focus on its role in mitochondrial bioenergetics and function. Biol. Chem. 2016, 397, 383–400. [Google Scholar] [CrossRef] [PubMed]
- Burton, N.C.; Kensler, T.W.; Guilarte, T.R. In vivo modulation of the Parkinsonian phenotype by Nrf2. Neurotoxicology 2006, 27, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar] [CrossRef] [PubMed]
- Deacon, R.M.J.; Hurley, M.J.; Rebolledo, C.M.; Snape, M.; Altimiras, F.J.; Farías, L.; Pino, M.; Biekofsky, R.; Glass, L.; Cogram, P. Nrf2: A novel therapeutic target in fragile X syndrome is modulated by NNZ2566. Genes Brain Behav. 2017. [Google Scholar] [CrossRef] [PubMed]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Kawalec, P.; Mikrut, A.; Wiśniewska, N.; Pilc, A. The effectiveness of dimethyl fumarate monotherapy in the treatment of relapsing-remitting multiple sclerosis: A systematic review and meta-analysis. Curr. Neuropharmacol. 2014, 12, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jiang, S.; Yan, J.; Li, Y.; Xin, Z.; Lin, Y.; Qu, Y. An overview of the molecular mechanisms and novel roles of Nrf2 in neurodegenerative disorders. Cytokine Growth Factor Rev. 2015, 26, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Nizzardo, M.; Nardini, M.; Ronchi, D.; Salani, S.; Donadoni, C.; Fortunato, F.; Colciago, G.; Falcone, M.; Simone, C.; Riboldi, G.; et al. Beta-lactam antibiotic offers neuroprotection in a spinal muscular atrophy model by multiple mechanisms. Exp. Neurol. 2011, 229, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Kaufman-Szymczyk, A.; Majewski, G.; Lubecka-Pietruszewska, K.; Fabianowska-Majewska, K. The Role of Sulforaphane in Epigenetic Mechanisms, Including Interdependence between Histone Modification and DNA Methylation. Int. J. Mol. Sci. 2015, 16, 29732–29743. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Gustafson, D.L.; Dehn, D.L.; Han, J.Y.; Boonchoong, P.; Berliner, L.J.; Ross, D. NAD(P)H:quinone oxidoreductase 1: Role as a superoxide scavenger. Mol. Pharmacol. 2004, 65, 1238–1247. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.K. Regulation of genes encoding NAD(P)H:quinone oxidoreductases. Free Radic. Biol. Med. 2000, 29, 254–262. [Google Scholar] [CrossRef]
- Morroni, F.; Tarozzi, A.; Sita, G.; Bolondi, C.; Zolezzi Moraga, J.M.; Cantelli-Forti, G.; Hrelia, P. Neuroprotective effect of sulforaphane in 6-hydroxydopamine-lesioned mouse model of Parkinson’s disease. Neurotoxicology 2013, 36, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Y. Chemical and mechanistic approaches to the study of protein tyrosine phosphatases. Acc. Chem. Res. 2003, 36, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Li, L.; Iwamoto, N.; Nakajima-Takagi, Y.; Kaneko, H.; Nakayama, Y.; Eguchi, M.; Wada, Y.; Kumagai, Y.; Yamamoto, M. The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol. Cell. Biol. 2009, 29, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Motohashi, H.; Yamamoto, M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol. Sci. 2013, 34, 340–346. [Google Scholar] [CrossRef] [PubMed]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell. Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Thakor, N.; Xu, E.Y.; Huang, Y.; Chen, C.; Yu, R.; Holcik, M.; Kong, A.N. An internal ribosomal entry site mediates redox-sensitive translation of Nrf2. Nucleic Acids Res. 2010, 38, 778–788. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Su, Z.Y.; Khor, T.O.; Shu, L.; Kong, A.N. Sulforaphane enhances Nrf2 expression in prostate cancer TRAMP C1 cells through epigenetic regulation. Biochem. Pharmacol. 2013, 85, 1398–1404. [Google Scholar] [CrossRef] [PubMed]
- Johansson, K.; Cebula, M.; Rengby, O.; Dreij, K.; Carlström, K.E.; Sigmundsson, K.; Piehl, F.; Arnér, E.S. Cross Talk in HEK293 Cells between Nrf2, HIF, and NF-κB Activities upon Challenges with Redox Therapeutics Characterized with Single-Cell Resolution. Antioxid. Redox Signal. 2017, 26, 229–246. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.M.; Li, Y.; Catalano, M.J.; Laciak, A.R.; Singh, H.; Seiner, D.R.; Reilly, T.J.; Tanner, J.J.; Gates, K.S. Inactivation of protein tyrosine phosphatases by dietary isothiocyanates. Bioorg. Med. Chem. Lett. 2015, 25, 4549–4552. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. Protein tyrosine phosphatases: From genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Cherubini, F.; Serio, D.; Guccini, I.; Fortuni, S.; Arcuri, G.; Condò, I.; Rufini, A.; Moiz, S.; Camerini, S.; Crescenzi, M.; et al. Src inhibitors modulate frataxin protein levels. Hum. Mol. Genet. 2015, 24, 4296–4305. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Tozzi, G.; Gaeta, L.M.; Bertini, E.; Serafini, V.; Di Cesare, S.; Bonetto, V.; Casoni, F.; Carrozzo, R.; Federici, G.; et al. Actin glutathionylation increases in fibroblasts of patients with Friedreich’s ataxia: A potential role in the pathogenesis of the disease. J. Biol. Chem. 2003, 278, 42588–42595. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Piemonte, F.; Locatelli, M.; Lo Russo, A.; Gaeta, L.M.; Tozzi, G.; Federici, G. Determination of blood total, reduced, and oxidized glutathione in pediatric subjects. Clin. Chem. 2001, 47, 1467–1469. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Age | Sex | Genetics | Onset | Duration | SARA | 8 m | 9HPTdom | PATA | Weight | Height |
|---|---|---|---|---|---|---|---|---|---|---|---|
| FV | 14 | F | 780/1114 | 5 | 9 | 12.5 | 6.9 | 48.0 | -- | 28 | 155 |
| BD | 9 | F | 1116/1116 | 1 | 8 | 10 | 6.8 | 44.0 | 17 | 30 | 130 |
| MA | 15 | M | 999/766 | 8 | 7 | 12 | 9.3 | 47.7 | 24 | 63 | 159 |
| DG | 32 | M | c.410G>T(p.Gly137Val)4/281 | 23 | 9 | 8.5 | 5.3 | 36.3 | 33 | 70 | 174 |
| Mouse Genes | Sequence (5′→3′) | |
| FXN | Fw-CCTGGCCGAGTTCTTTGAAG | Rv-GCCAGATTTGCTTGTTTGG |
| NRF2 | Fw-TGGAGGCAGCCATGACTGA | Rv-CTGCTTGTTTTCGGTATTAAGACACT |
| TBP | Fw-CTCTGACCACTGCACCGTT | Rv-CTGCAGCAAATCGCTTGGG |
| GAPDH | Fw-CCTCGTCCCGTAGACAAAATG | Rv-TGAAGGGGTCGTTGATGGC |
| Human Genes | Sequence (5′→3′) | |
| FXN | Fw-CCTTGCAGACAAGCCATACA | Rv-CCACTGGATGGAGAAGATAG |
| Nrf2 | Fw-ACACGGTCCACAGCTCATC | Rv-TGTCAATCAAATCCATGTCCTG |
| GCL | Fw-TTGCCTCCTGCTGTGTGATG | Rv-ATCATTGTGAGTCAACAGCTGTATGTC |
| HO-1 | Fw-CTCAACATCCAGCTCTTTGAG | Rv-AATCTTGCACTTTGTTGCTGGC |
| NQO1 | Fw-GACATCACAGGTAAACTGAAGG | Rv-GCAGGGGGAACTGGAATATC |
| TBP | Fw-CCGAAACGCCGAATATAATCC | Rv-AAATCAGTGCCGTGGTTCGT |
| GAPDH | Fw-GATGACATCAAGAAGGTGGTG | Rv-GTCATACCAGGAAATGAGCTTG |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrillo, S.; Piermarini, E.; Pastore, A.; Vasco, G.; Schirinzi, T.; Carrozzo, R.; Bertini, E.; Piemonte, F. Nrf2-Inducers Counteract Neurodegeneration in Frataxin-Silenced Motor Neurons: Disclosing New Therapeutic Targets for Friedreich’s Ataxia. Int. J. Mol. Sci. 2017, 18, 2173. https://doi.org/10.3390/ijms18102173
Petrillo S, Piermarini E, Pastore A, Vasco G, Schirinzi T, Carrozzo R, Bertini E, Piemonte F. Nrf2-Inducers Counteract Neurodegeneration in Frataxin-Silenced Motor Neurons: Disclosing New Therapeutic Targets for Friedreich’s Ataxia. International Journal of Molecular Sciences. 2017; 18(10):2173. https://doi.org/10.3390/ijms18102173
Chicago/Turabian StylePetrillo, Sara, Emanuela Piermarini, Anna Pastore, Gessica Vasco, Tommaso Schirinzi, Rosalba Carrozzo, Enrico Bertini, and Fiorella Piemonte. 2017. "Nrf2-Inducers Counteract Neurodegeneration in Frataxin-Silenced Motor Neurons: Disclosing New Therapeutic Targets for Friedreich’s Ataxia" International Journal of Molecular Sciences 18, no. 10: 2173. https://doi.org/10.3390/ijms18102173