Comparative Analysis of the Cytology and Transcriptomes of the Cytoplasmic Male Sterility Line H276A and Its Maintainer Line H276B of Cotton (Gossypium barbadense L.)

 ,
,
Abstract
:1. Introduction
2. Results
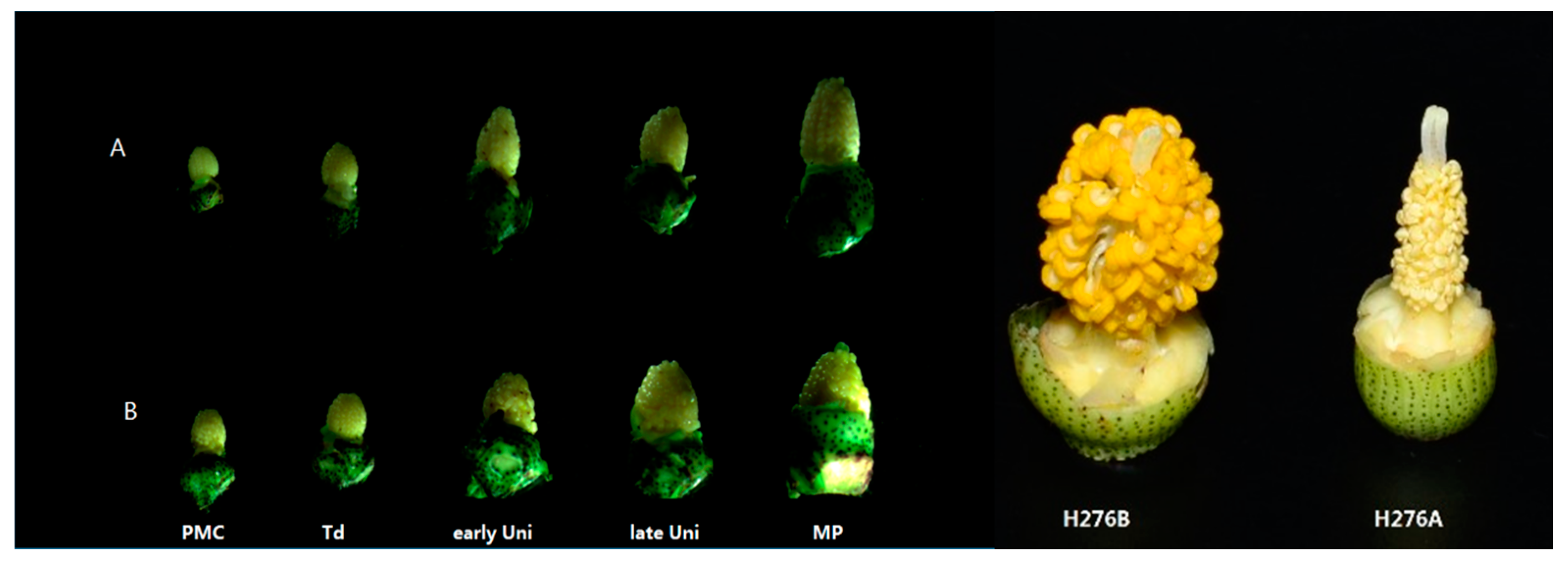
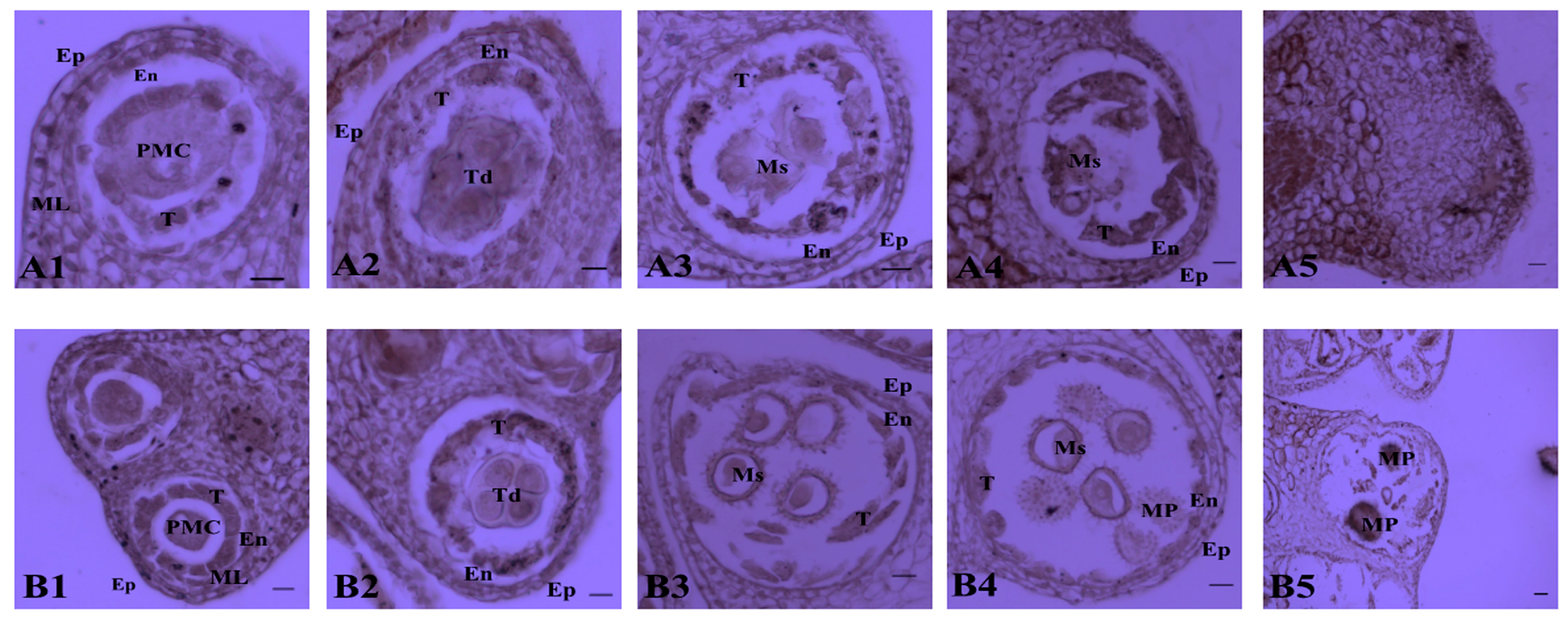
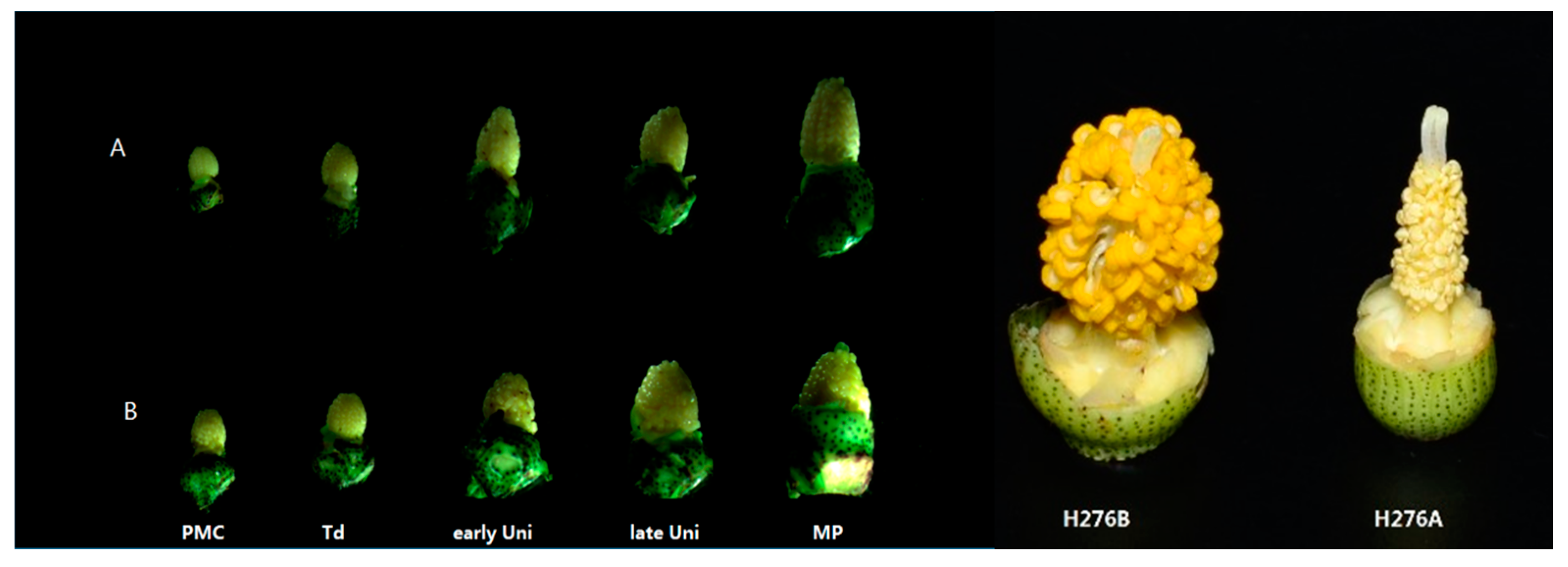
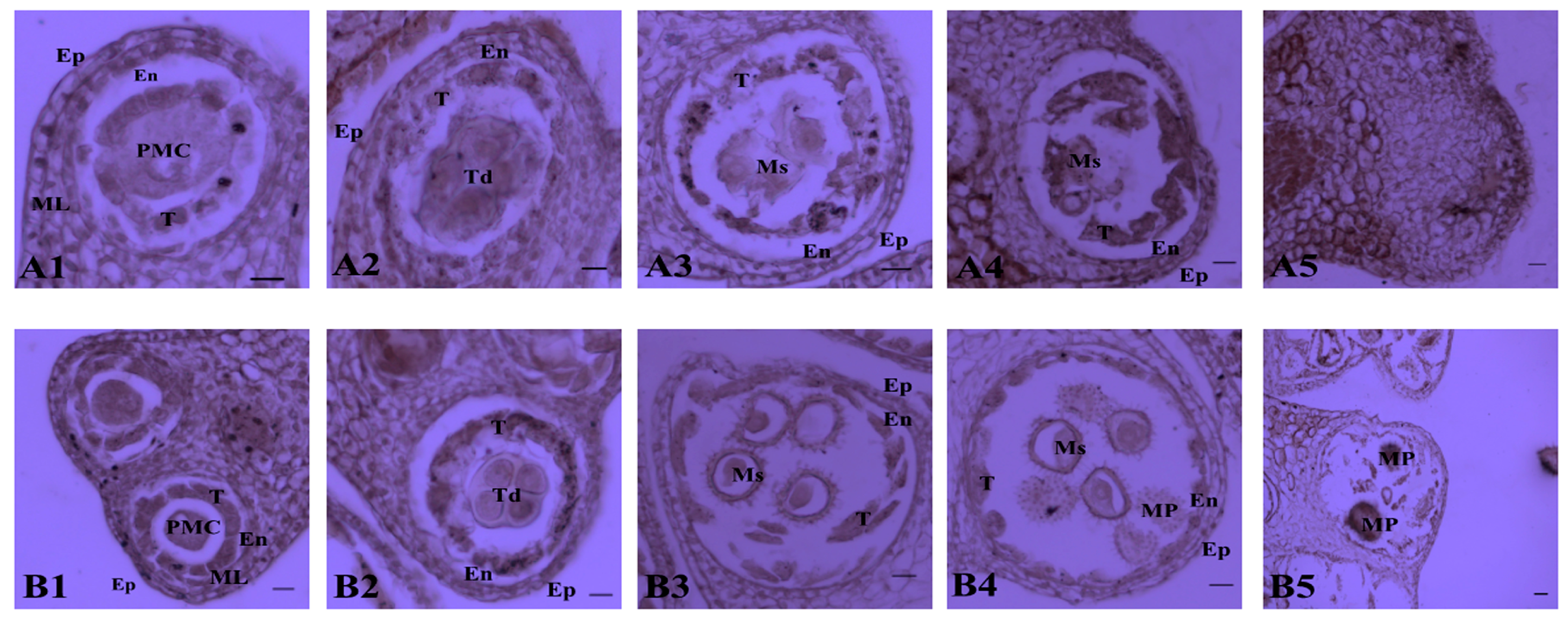
2.1. Determination of the Pollen Abortion Stage in CMS Line H276A
2.2. Transcriptome Sequencing and Genome Mapping
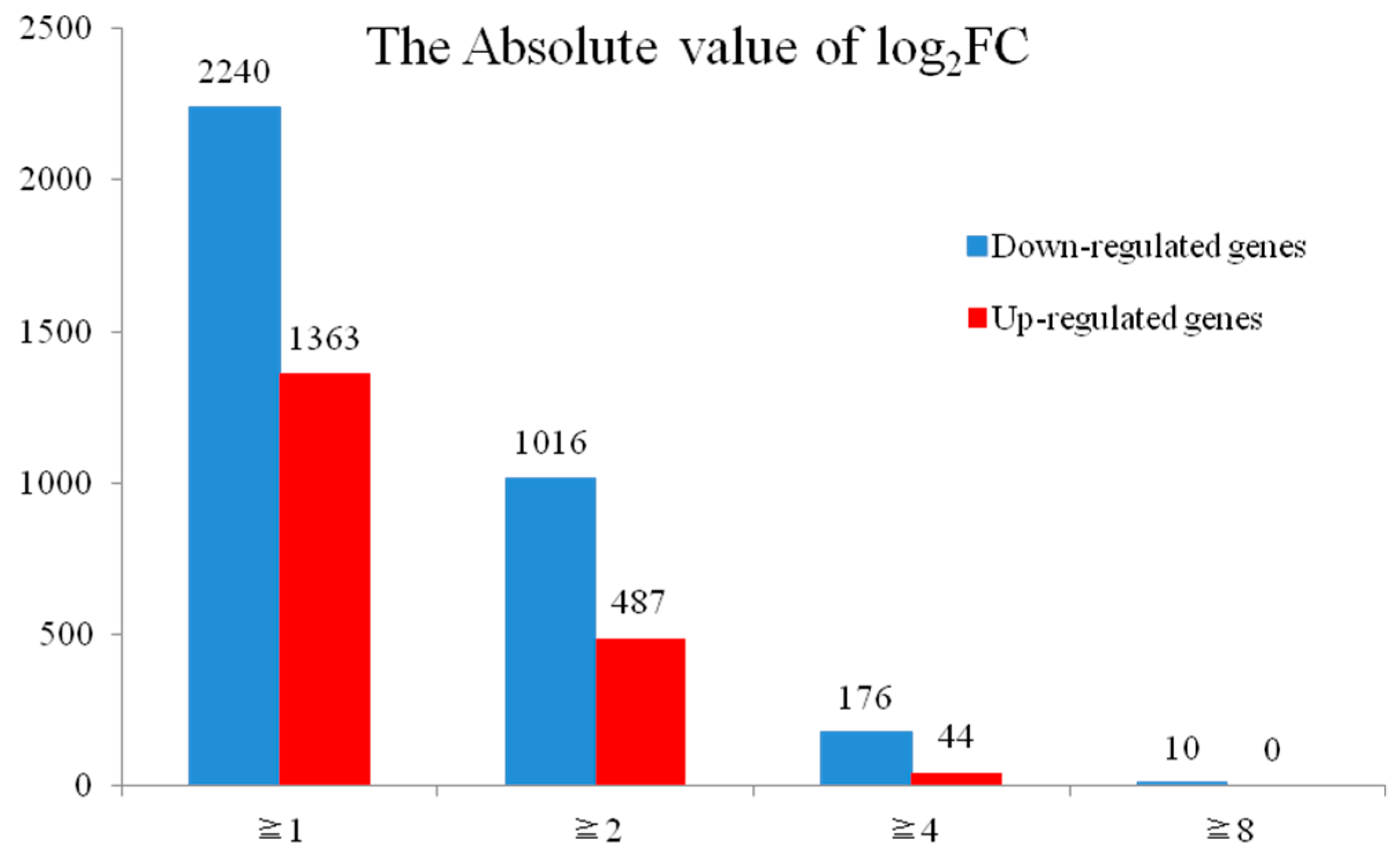
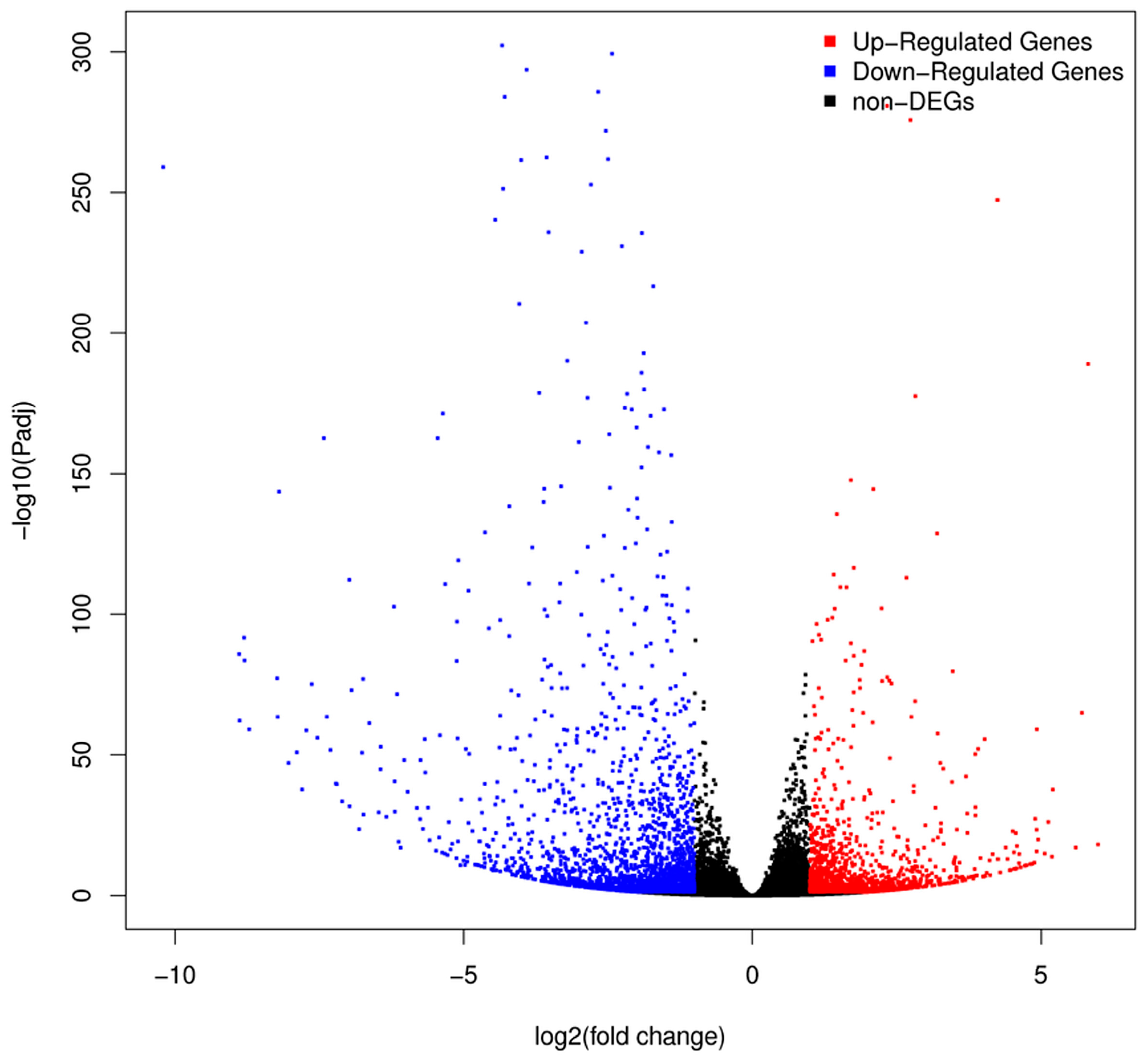
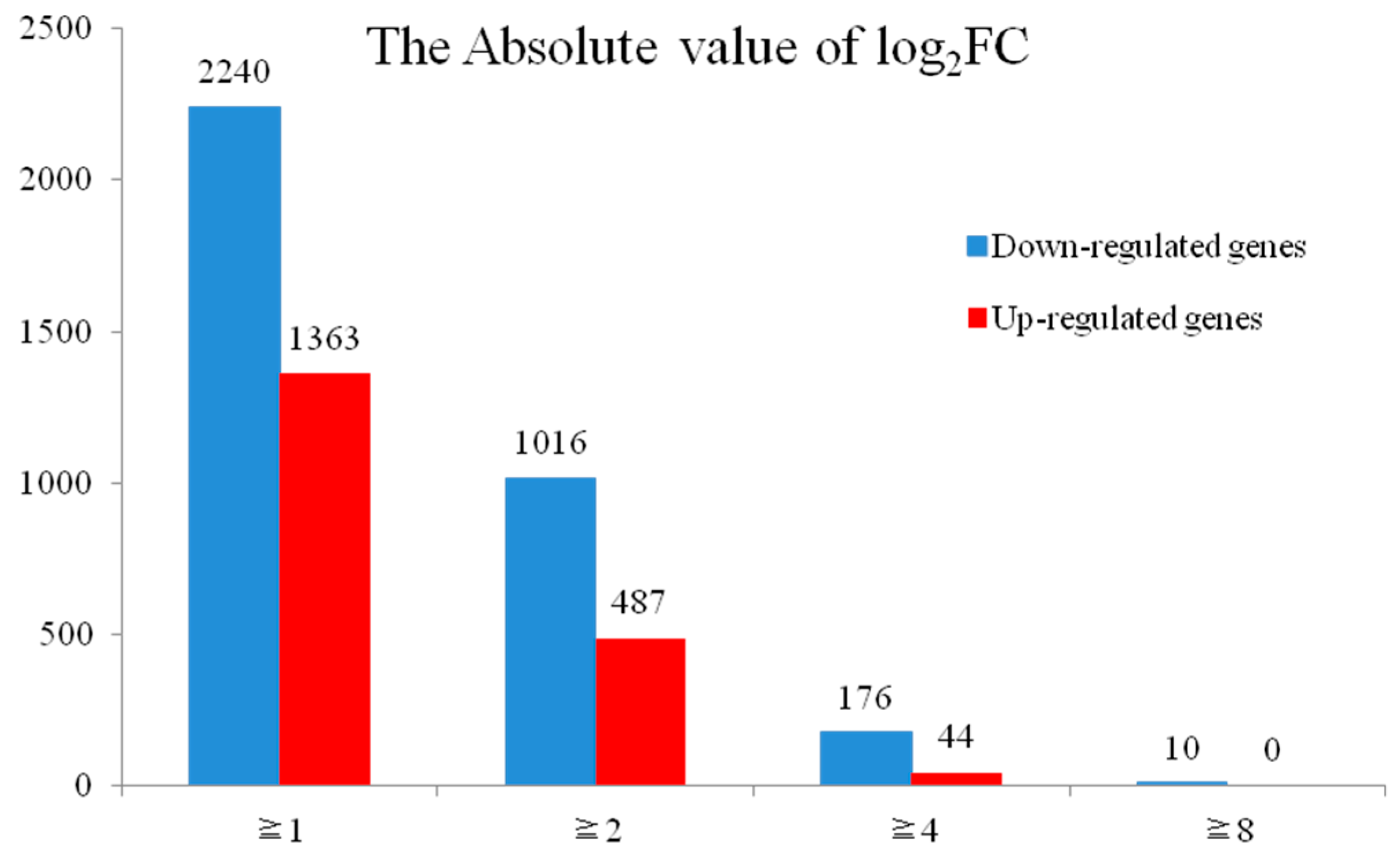
2.3. Analysis of DEGs between H276A and H276B
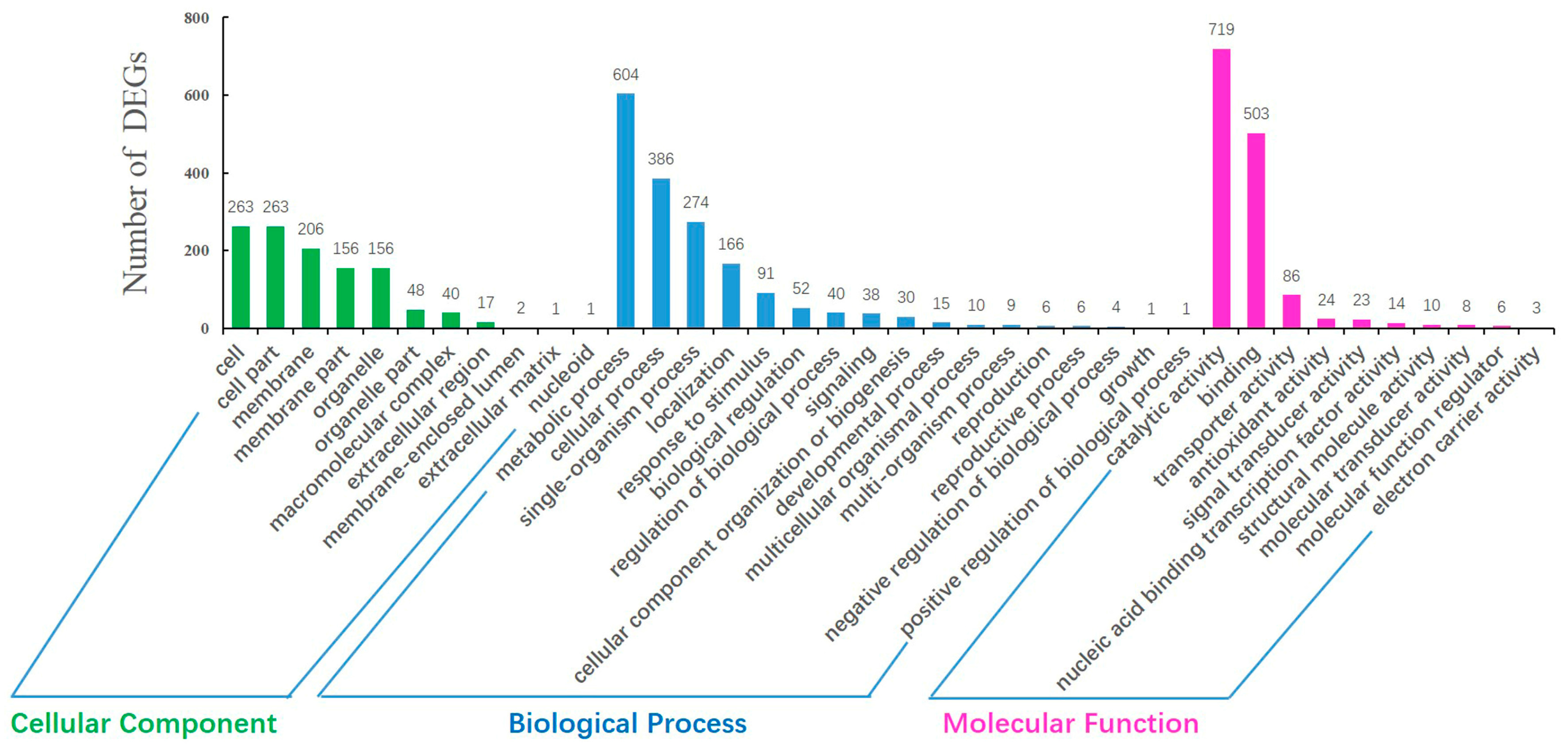
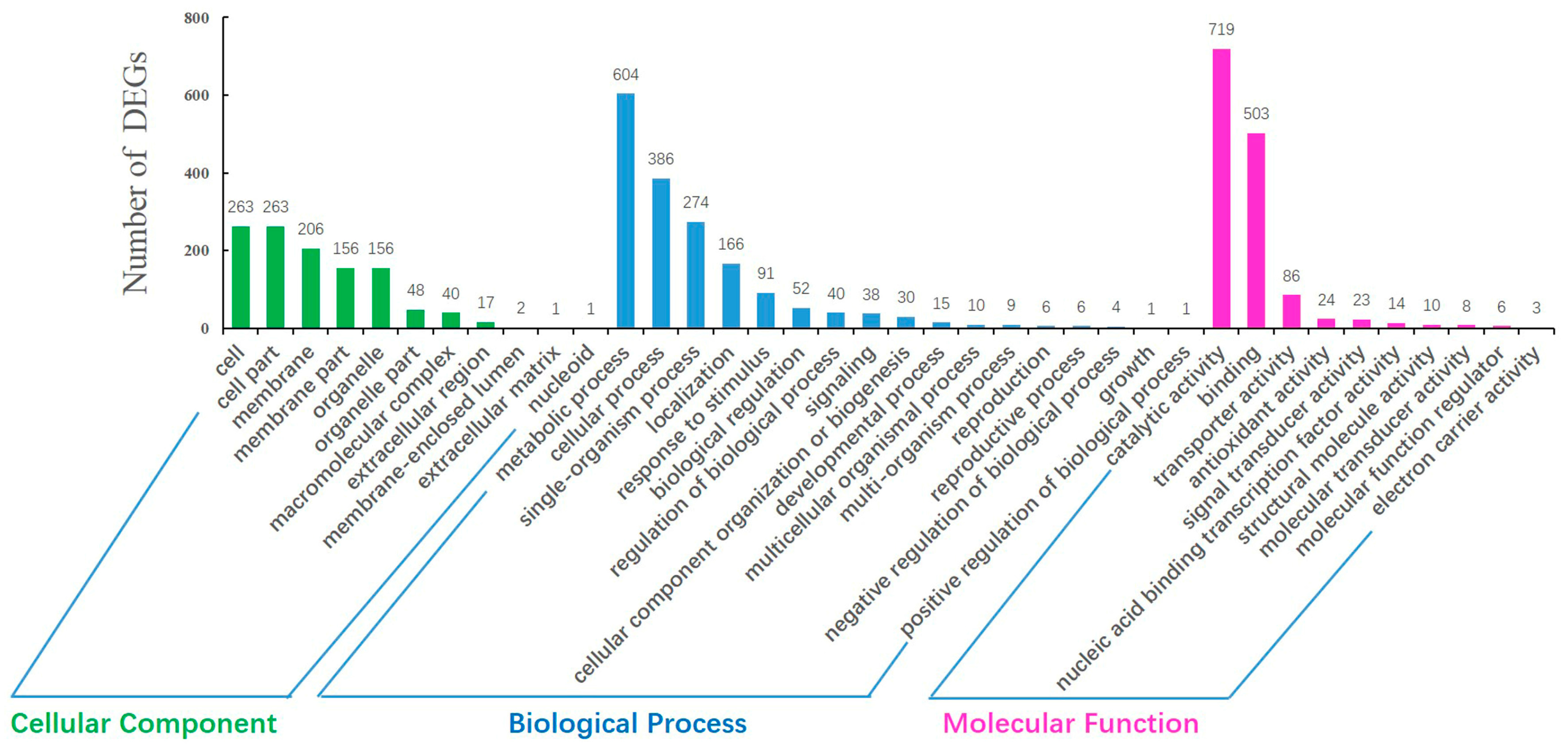
2.4. GO Annotation and Pathway Analysis of DEGs
2.5. Confirmation of DEGs by qRT-PCR
3. Discussion
3.1. Cytological Characteristics of Pollen Abortion
3.2. DEGs Related to the Tricarboxylic Acid (TCA) Cycle and Oxidative Phosphorylation
3.3. DEGs Associated with Pentatricopeptide Repeat (PPR) Proteins
3.4. DEGs Involved in MYB Transcription Factors
3.5. Other DEGs
4. Materials and Methods
4.1. Plant Materials
4.2. Paraffin Sections and Microspore Development Observations
4.3. RNA Extraction, cDNA Library Construction and Illumina Deep Sequencing
4.4. Genome Mapping and Novel Transcript Prediction
4.5. Differential Expression Analysis
4.6. Functional Enrichment Analysis and Transcription Factor Prediction of DEGs
4.7. Reverse-Transcription PCR (RT-PCR) and Quantitative Reverse-Transcription PCR (qRT-PCR)
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CMS | Cytoplasmic male sterility |
| DEGs | Differentially expressed genes |
| qRT-PCR | Quantitative reverse-transcribed PCR |
| PMC | Pollen mother cell |
| RNA-Seq | Transcriptome sequencing |
| TCA | Tricarboxylic acid cycle |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GO | Gene ontology |
References
- Budar, F.; Pelletier, G. Male sterility in plants: Occurrence, determinism, significance and use. C. R. Acad. Sci. Ser. III 2001, 324, 543–550. [Google Scholar] [CrossRef]
- Zhang, Z.B.; Zhu, J.; Gao, J.F.; Wang, C.; Li, H.; Li, H.; Zhang, H.Q.; Zhang, S.; Wang, D.M.; Wang, Q.X. Transcription factor AtMYB103 is required for anther development by regulating tapetum development, callose dissolution and exine formation in Arabidopsis. Plant J. 2007, 52, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Du, K.; Liu, Q.; Wu, X.; Jiang, J.; Wu, J.; Fang, Y.; Li, A.; Wang, Y. Morphological Structure and Transcriptome Comparison of the Cytoplasmic Male Sterility Line in Brassica napus (SaNa-1A) Derived from Somatic Hybridization and Its Maintainer Line SaNa-1B. Front. Plant Sci. 2016, 7, 1313. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, C.; Zhang, X.X.; Chen, X.; Liu, J.J.; Jia, X.F.; Jia, S.Q. Transcriptome de novo assembly and analysis of differentially expressed genes related to cytoplasmic male sterility in cabbage. Plant Physiol. Biochem. 2016, 105, 224. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Jo, Y.D.; Kwon, J.K.; Kim, I.T.; Kang, B.C. Characterization and inheritance of a novel thermo-sensitive restoration of cytoplasmic male sterility in Capsicum annuum. Sci. Hortic. 2013, 164, 512–520. [Google Scholar] [CrossRef]
- Wei, W.; Qi, X.; Wang, L.; Zhang, Y.; Wei, H.; Li, D.; Lv, H.; Zhang, X. Characterization of the sesame (Sesamum indicum L.) global transcriptome using Illumina paired-end sequencing and development of EST-SSR markers. BMC Genom. 2011, 12, 451. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, Y.G. Male sterility and fertility restoration in crops. Annu. Rev. Plant Biol. 2014, 65, 579. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Ran, S.; Li, R.; Huang, Z.; Qian, J.; Yu, M.; Zhou, R. Transcriptome de novo assembly and differentially expressed genes related to cytoplasmic male sterility in kenaf (Hibiscus cannabinus L.). Mol. Breed. 2014, 34, 1879–1891. [Google Scholar] [CrossRef]
- Yang, S. Comparative Transcriptome Analysis between Cytoplasmic Male Sterile Line NJCMS1A and Its Maintainer NJCMS1B in Soybean (Glycine max (L.) Merr). PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Liu, Q.; Lan, Y.; Wen, C.; Hong, Z.; Jian, W.; Wang, Y. Transcriptome Sequencing Analyses between the Cytoplasmic Male Sterile Line and Its Maintainer Line in Welsh Onion (Allium fistulosum L.). Int. J. Mol. Sci. 2016, 17, 1058. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Fu, Y.R.; Huang, J.G.; Wu, C.A.; Zheng, C.C. Transcript profiling during the early development of the maize brace root via Solexa sequencing. FEBS J. 2011, 278, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Gong, Y.; Cui, M.; Qi, T.; Guo, L.; Zhang, J.; Xing, C. Molecular characterization of cytoplasmic male sterility conditioned by Gossypium harknessii cytoplasm (CMS-D2) in upland cotton. Euphytica 2011, 181, 17–29. [Google Scholar] [CrossRef]
- Wang, F.; Feng, C.; O’Connell, M.A.; Stewart, J.M.; Zhang, J. RFLP analysis of mitochondrial DNA in two cytoplasmic male sterility systems (CMS-D2 and CMS-D8) of cotton. Euphytica 2010, 172, 93–99. [Google Scholar] [CrossRef]
- Li, Y.Y.; Wang, X.D.; Xu, Y.N. Cytological observat ion of cytoplasmic male-sterile anther of brown cotton. J. Zhejiang Univ. 2002, 28, 11–15. [Google Scholar]
- Yang, J.H.; Huai, Y.; Zhang, M.F. Mitochondrial atpA gene is altered in a new orf220-type cytoplasmic male-sterile line of stem mustard (Brassica juncea). Mol. Biol. Rep. 2009, 36, 273. [Google Scholar] [CrossRef] [PubMed]
- Siedow, J.N.; Umbach, A.L. Plant Mitochondrial Electron Transfer and Molecular Biology. Plant Cell 1995, 7, 821. [Google Scholar] [CrossRef] [PubMed]
- Logan, D.C. The mitochondrial compartment. J. Exp. Bot. 2006, 57, 1225–1243. [Google Scholar] [CrossRef] [PubMed]
- Millar, A.H.; Whelan, J.; Soole, K.L.; Day, D.A. Organization and regulation of mitochondrial respiration in plants. Annu. Rev. Plant Biol. 2011, 62, 79. [Google Scholar] [CrossRef] [PubMed]
- Ducos, E.; Touzet, P.; Boutry, M. The male sterile G cytoplasm of wild beet displays modified mitochondrial respiratory complexes. Plant J. 2001, 26, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kang, J.G.; Kim, B.D. Isolation and characterization of the cytoplasmic male sterility-associated orf456 gene of chili pepper (Capsicum annuum L.). Plant Mol. Biol. 2007, 63, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.R.; Bentolila, S. Interactions of mitochondrial and nuclear genes that affect male gametophyte development. Plant Cell 2004, 16, S154–S169. [Google Scholar] [CrossRef] [PubMed]
- Lurin, C.; Andrés, C.; Aubourg, S.; Bellaoui, M.; Bitton, F.; Bruyère, C.; Caboche, M.; Debast, C.; Gualberto, J.; Hoffmann, B. Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 2004, 16, 2089. [Google Scholar] [CrossRef] [PubMed]
- Delannoy, E.; Stanley, W.A.; Bond, C.S.; Small, I.D. Pentatricopeptide repeat (PPR) proteins as sequence-specificity factors in post-transcriptional processes in organelles. Biochem. Soc. Trans. 2007, 35, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Koizuka, N.; Imai, R.; Fujimoto, H.; Hayakawa, T.; Kimura, Y.; Kohnomurase, J.; Sakai, T.; Kawasaki, S.; Imamura, J. Genetic characterization of a pentatricopeptide repeat protein gene, orf687, that restores fertility in the cytoplasmic male-sterile Kosena radish. Plant J. 2003, 34, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Akagi, H.; Nakamura, A.; Yokozekimisono, Y.; Inagaki, A.; Takahashi, H.; Mori, K.; Fujimura, T. Positional cloning of the rice Rf-1 gene, a restorer of BT-type cytoplasmic male sterility that encodes a mitochondria-targeting PPR protein. Theor. Appl. Genet. 2004, 108, 1449–1457. [Google Scholar] [CrossRef] [PubMed]
- Uyttewaal, M.; Arnal, N.; Quadrado, M.; Martincanadell, A.; Vrielynck, N.; Hiard, S.; Gherbi, H.; Bendahmane, A.; Budar, F.; Mireau, H. Characterization of Raphanus sativus Pentatricopeptide Repeat Proteins Encoded by the Fertility Restorer Locus for Ogura Cytoplasmic Male Sterility. Plant Cell 2008, 20, 3331. [Google Scholar] [CrossRef] [PubMed]
- Jordan, D.R.; Mace, E.S.; Henzell, R.G.; Klein, P.E.; Klein, R.R. Molecular mapping and candidate gene identification of the Rf2 gene for pollen fertility restoration in sorghum [Sorghum bicolor (L.) Moench]. Theor. Appl. Genet. 2010, 120, 1279. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zhu, Y. The Rice Pentatricopeptide Repeat Protein RF5 Restores Fertility in Hong-Lian Cytoplasmic Male-Sterile Lines via a Complex with the Glycine-Rich Protein GRP162. Plant Cell 2012, 24, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Zhao, M.; Leavitt, J.M.; Lloyd, A.M. Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J. 2008, 53, 814. [Google Scholar] [CrossRef] [PubMed]
- Stracke, R.; Ishihara, H.; Huep, G.; Barsch, A.; Mehrtens, F.; Niehaus, K.; Weisshaar, B. Differential regulation of closely related R2R3-MYB transcription factors controls flavonol accumulation in different parts of the Arabidopsis thaliana seedling. Plant J. 2007, 50, 660–677. [Google Scholar] [CrossRef] [PubMed]
- Higginson, T.; Li, S.F.; Parish, R.W. AtMYB103 regulates tapetum and trichome development in Arabidopsis thaliana. Plant J. 2003, 35, 177. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Qi, T.; Huang, H.; Ren, Q.; Wu, D.; Chang, C.; Peng, W.; Liu, Y.; Peng, J.; Xie, D. The Jasmonate-ZIM Domain Proteins Interact with the R2R3-MYB Transcription Factors MYB21 and MYB24 to Affect Jasmonate-Regulated Stamen Development in Arabidopsis. Plant Cell 2011, 23, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, J.; Du, M.L.; Li, L.; Wang, X.L.; Li, X.B. A Cotton Gene Encoding MYB-Like Transcription Factor is Specifically Expressed in Pollen and is Involved in Regulation of Late Anther/Pollen Development. Plant Cell Physiol. 2013, 54, 893. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, J.P. The Male Gametophyte of Flowering Plants. Plant Cell 1989, 1, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Rato, C.; Monteiro, D.; Hepler, P.K.; Rui, M. Calmodulin activity and cAMP signalling modulate growth and apical secretion in pollen tubes. Plant J. 2004, 38, 887. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Audic, S.; Claverie, J.M. The Significance of Digital Gene Expression Profiles. Genome Res. 1997, 7, 986. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. TIG 2000, 16, 276. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | H276A-1 | H276A-2 | H276A-3 | H276B-1 | H276B-2 | H276B-3 |
|---|---|---|---|---|---|---|
| Total Raw Reads (Mb) | 50.42 | 53.42 | 51.82 | 51.81 | 51.82 | 51.82 |
| Total Clean Reads (Mb) | 40.38 | 44.48 | 44.73 | 44.58 | 44.25 | 44.37 |
| Clean Reads Q20 (%) | 98.06 | 98.01 | 98.39 | 98.38 | 98.39 | 98.34 |
| Total Mapping Ratio (%) | 73.34 | 74.20 | 75.20 | 76.04 | 75.60 | 76.18 |
| Total Gene Number | 57,228 | 57,777 | 57,428 | 56,938 | 56,929 | 56,629 |
| Total Transcript Number | 86,162 | 87,603 | 85,063 | 85,039 | 84,961 | 84,434 |
| Novel Transcript Number | 20,514 | 20,952 | 20,288 | 20,580 | 20,554 | 20,474 |
| Pathway | Pathway ID | Genes with Pathway Annotation | p-Value | Q-Value | |
|---|---|---|---|---|---|
| DEGs (2683) | All Genes (56,363) | ||||
| Carbohydrate Metabolism | |||||
| Ascorbate and aldarate metabolism | ko00053 | 87 (3.24%) | 747 (1.33%) | 2.1883346 × 10−14 | 2.772849 × 10−12 |
| Amino sugar and nucleotide sugar metabolism | Ko00520 | 132 (4.92%) | 1789 (3.17%) | 5.144044 × 10−7 | 1.306587 × 10−5 |
| Galactose metabolism | Ko00052 | 93 (3.47%) | 1264 (2.24%) | 2.652517 × 10−5 | 3.142800 × 10−4 |
| Pentose and glucuronate interconversions | Ko00040 | 119 (4.44%) | 1711 (3.04%) | 2.72211 × 10−5 | 3.142800 × 10−4 |
| Fructose and mannose metabolism | Ko00051 | 48 (1.79%) | 623 (1.11%) | 0.0008306261 | 6.203606 × 10−3 |
| Pentose phosphate pathway | Ko00030 | 44 (1.64%) | 562 (1%) | 0.0009706133 | 6.487784 × 10−3 |
| Glycolysis/gluconeogenesis | Ko00010 | 62 (2.31%) | 894 (1.59%) | 0.00222321 | 1.344513 × 10−2 |
| Global and Overview Map | |||||
| Metabolic pathways | ko01100 | 822 (30.64%) | 14,040 (24.91%) | 3.498244 × 10−12 | 2.221385 × 10−10 |
| Biosynthesis of secondary metabolites | Ko01110 | 464 (17.29%) | 8032 (14.25) | 3.713275 × 10−6 | 5.239844 × 10−5 |
| Biosynthesis of Other Secondary Metabolites | |||||
| Flavonoid biosynthesis | Ko00941 | 35 (1.3%) | 266 (0.47%) | 6.13195 × 10−8 | 2.595859 × 10−6 |
| Stilbenoid, diarylheptanoid and gingerol biosynthesis | Ko00945 | 31 (1.16%) | 240 (0.43%) | 5.103317 × 10−7 | 1.306587 × 10−5 |
| Flavone and flavonol biosynthesis | Ko00944 | 12 (0.45%) | 105 (0.19%) | 0.004277302 | 2.263406 × 10−2 |
| Phenylpropanoid biosynthesis | Ko00940 | 91(3.39%) | 1497 (2.66%) | 0.01086005 | 4.755953 × 10−2 |
| Metabolism of Terpenoids and Polyketides | |||||
| Zeatin biosynthesis | Ko00908 | 24 (0.89%) | 162 (0.29%) | 8.181436 × 10−7 | 1.731737 × 10−5 |
| Limonene and pinene degradation | Ko00903 | 24 (0.89%) | 210 (0.37%) | 7.083635 × 10−5 | 7.496847 × 10−4 |
| Carotenoid biosynthesis | Ko00906 | 21 (0.78%) | 244 (0.43%) | 0.006774023 | 3.308850 × 10−2 |
| Transcription | |||||
| RNA polymerase | Ko03020 | 79 (2.94%) | 962 (1.71%) | 2.302535 × 10−6 | 3.878907 × 10−5 |
| Energy Metabolism | |||||
| Nitrogen metabolism | Ko00910 | 22 (0.82%) | 149 (0.26%) | 2.443406 × 10−6 | 3.878907 × 10−5 |
| Photosynthesis | Ko00195 | 22 (0.82%) | 213 (0.38) | 0.0005760129 | 4.572102 × 10−3 |
| Lipid Metabolism | |||||
| Sphingolipid metabolism | Ko00600 | 33 (1.23%) | 338 (0.6%) | 8.44879 × 10−5 | 8.543074 × 10−4 |
| α-Linolenic acid metabolism | Ko00592 | 27 (1.01%) | 272 (0.48%) | 0.0002803392 | 2.543077 × 10−3 |
| Glycerolipid metabolism | Ko00561 | 65 (2.42%) | 895 (1.59%) | 0.0005644657 | 4.572102 × 10−3 |
| Amino Acid Metabolism | |||||
| Tryptophan metabolism | Ko00380 | 26 (0.97%) | 278 (0.49%) | 0.0008792512 | 6.203606 × 10−3 |
| Metabolism of Other Amino Acids | |||||
| Taurine and hypotaurine metabolism | Ko00430 | 6 (0.22%) | 30 (0.05%) | 0.002561534 | 1.478704 × 10−2 |
| Glycan Biosynthesis and Metabolism | |||||
| Glycosaminoglycan degradation | Ko00531 | 23 (0.86%) | 251 (0.45%) | 0.002192842 | 1.344513 × 10−2 |
| Other glycan degradation | Ko00551 | 42 (1.57%) | 562 (1%) | 0.002931596 | 1.618751 × 10−2 |
| Glycosphingolipid biosynthesis (ganglio series) | Ko00604 | 16 (0.6%) | 164 (0.29%) | 0.005312663 | 2.698833 × 10−2 |
| Glycosphingolipid biosynthesis (globo series) | Ko00603 | 8 (0.3%) | 61 (0.11%) | 0.00811891 | 3.818895 × 10−2 |
| Transport and Catabolism | |||||
| Regulation of autophagy | Ko04140 | 34 (1.27%) | 462 (0.82%) | 0.008579786 | 3.891546 × 10−2 |
| Gene ID | Protein Identity | Fold Change | |
|---|---|---|---|
| RNA-Seq | qRT-PCR | ||
| LOC107957220 | Glycine-rich cell wall structural protein-like | 4.92 | 5.02 |
| LOC107903993 | PPR protein At2g01860-like | 2.81 | 0.70 |
| LOC107893809 | MADS-box CMB1 | 2.81 | 1.65 |
| LOC107928908 | Hyoscyamine 6-dioxygenase-like | 2.73 | 2.71 |
| LOC107940910 | Ethylene responsive element binding protein 3 | 2.59 | 2.63 |
| LOC107916224 | Cell wall/vacuolar inhibitor of fructosidase 1-like | 1.96 | 2.11 |
| LOC107919764 | Transcription factor MYB86 | −3.21 | −3.60 |
| LOC107908692 | Pentatricopeptide repeat-containing protein At3g11460 | −3.38 | −4.38 |
| LOC107940452 | Flavonol synthase/flavanone 3-hydroxylase-like | −3.86 | −1.19 |
| LOC107941000 | Tetraketide α-pyrone reductase 1 | −3.94 | −3.50 |
| LOC107961143 | Anther-specific protein LAT52 | −4.03 | −2.74 |
| LOC107945055 | Chalcone synthase | −5.15 | −5.72 |
| LOC107900663 | Bifunctional pinoresinol-lariciresinol reductase 2 | −5.67 | −3.43 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kong, X.; Liu, D.; Liao, X.; Zheng, J.; Diao, Y.; Liu, Y.; Zhou, R. Comparative Analysis of the Cytology and Transcriptomes of the Cytoplasmic Male Sterility Line H276A and Its Maintainer Line H276B of Cotton (Gossypium barbadense L.). Int. J. Mol. Sci. 2017, 18, 2240. https://doi.org/10.3390/ijms18112240
Kong X, Liu D, Liao X, Zheng J, Diao Y, Liu Y, Zhou R. Comparative Analysis of the Cytology and Transcriptomes of the Cytoplasmic Male Sterility Line H276A and Its Maintainer Line H276B of Cotton (Gossypium barbadense L.). International Journal of Molecular Sciences. 2017; 18(11):2240. https://doi.org/10.3390/ijms18112240
Chicago/Turabian StyleKong, Xiangjun, Dongmei Liu, Xiaofang Liao, Jie Zheng, Yong Diao, Yiding Liu, and Ruiyang Zhou. 2017. "Comparative Analysis of the Cytology and Transcriptomes of the Cytoplasmic Male Sterility Line H276A and Its Maintainer Line H276B of Cotton (Gossypium barbadense L.)" International Journal of Molecular Sciences 18, no. 11: 2240. https://doi.org/10.3390/ijms18112240