1. Introduction
By finely harmonizing the processes of differentiation and self-renewal, proliferation, and quiescence in hematopoietic stem cells (HSC), the bone marrow (BM) microenvironment is able to generate and maintain appropriate numbers of hematopoietic progenitor cells (HPC) and mature blood/immune cells while maintaining the HSC pool. This microenvironment, or “niche” [
1], is assumed to be formed by multiple cell types, with a predominant role played by mesenchymal stem cells (MSC), soluble factors, and extracellular components that could even structure a tailored niche for all stem, progenitor, and immune cells [
2,
3,
4].
Early experiments have also suggested an important role for the microenvironment in the pathogenesis of some human diseases [
5], but only recently has it been proven that the microenvironment plays a pivotal role in cancer initiation and progression [
6,
7]. In fact, mouse models have shown that BM microenvironment alterations are sufficient for malignant hematopoietic transformation [
8,
9,
10,
11,
12]. Initial work has also shown that BM survival stimuli are essential for clonal expansion of leukemic progenitors [
13]. More recently, in chronic myeloid leukemia (CML), it has been shown that protein kinase-C-β-dependent signaling in MSC was necessary for leukemia cell survival [
14]. The fact that this signaling is relevant in other hematological malignancies, including acute lymphocytic leukemia (ALL) [
14], suggests that a widespread response for leukemogenesis support can be induced in the BM microenvironment, and that this finding could be generally applied for therapeutic interventions. In addition, it has been demonstrated that niche cells play a protective role in CML and ALL in response to chemotherapeutic agents [
15,
16], implying that by this means survival support and drug resistance could be targeted.
Furthermore, it has recently been shown in transgenic mouse models that leukemic cells alter the niche and normal functioning of stem and progenitor cells (HSPC). Reduced SDF-1 secretion, essential chemokine for HSC homing and retention, and an increase of other chemokines and cytokines by BM stromal cells are responsible for impaired support for HSC and a selective growth advantage for leukemic cells [
17,
18,
19]. Leukemic cells induced an SCF-dependent CD34+ progenitors’ displacement to novel malignant microenvironments [
20]. Also, in ALL, leukemic cell dissemination rebuilds a protective microenvironment to assist in survival during primary chemotherapy [
16]. Intriguingly, in this study it was shown that the niche components are transient and dynamic, suggesting that the in vivo interplay between leukemic cells and the microenvironment could be much more complex than expected.
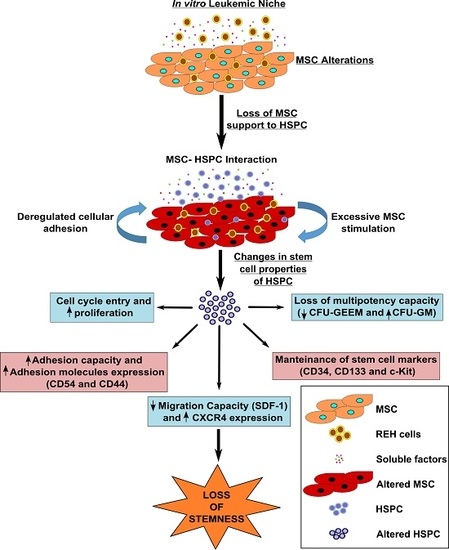
In the context of these new findings using mouse models, an in vitro system where the diverse niche components are better defined would be helpful for elucidating the various mechanisms involved in niche remodeling, HSPC functional failure, and leukemic cell survival and resistance to therapy. This system could be a valuable tool for discovering innovative therapies and for the interrogation of in vivo findings. In the present study, we were interested in finding out if we could recreate in vitro the leukemic microenvironment or niche (LN) in order to study HSPC (CD34+ cells) function. Using a short co-culture period of MSC with an ALL-B cell line or leukemic blasts from an ALL-B patient as models of the LN, we were able to show that HSPC in contact with the LN are affected in different ways, partially resembling what has been reported for HSC in vivo. Our in vitro model could be a valuable system for studying the dynamic and evolving interactions between leukemic cells and niche components.
3. Discussion
Early work has shown that ALL-T cells modify hematopoietic stem cell growth both in vivo and in vitro [
21]. More recently, CML and ALL mouse models have shown that a reduction in SDF-1 levels in BM is responsible for impaired LT-HSC or human CD34+ homing and retention [
17,
18,
19,
20]. This apparently modest change in cytokine production by stromal cells has profound effects on HSC biology. SDF-1 can function as an inhibitor of human LTC-IC cycling [
22], and its reduction in BM contributes to increased cell cycling. Consistent with this, as we have shown here in our in vitro LN model, is that CD34+ cells proliferate more and have increased cell populations in the S/G
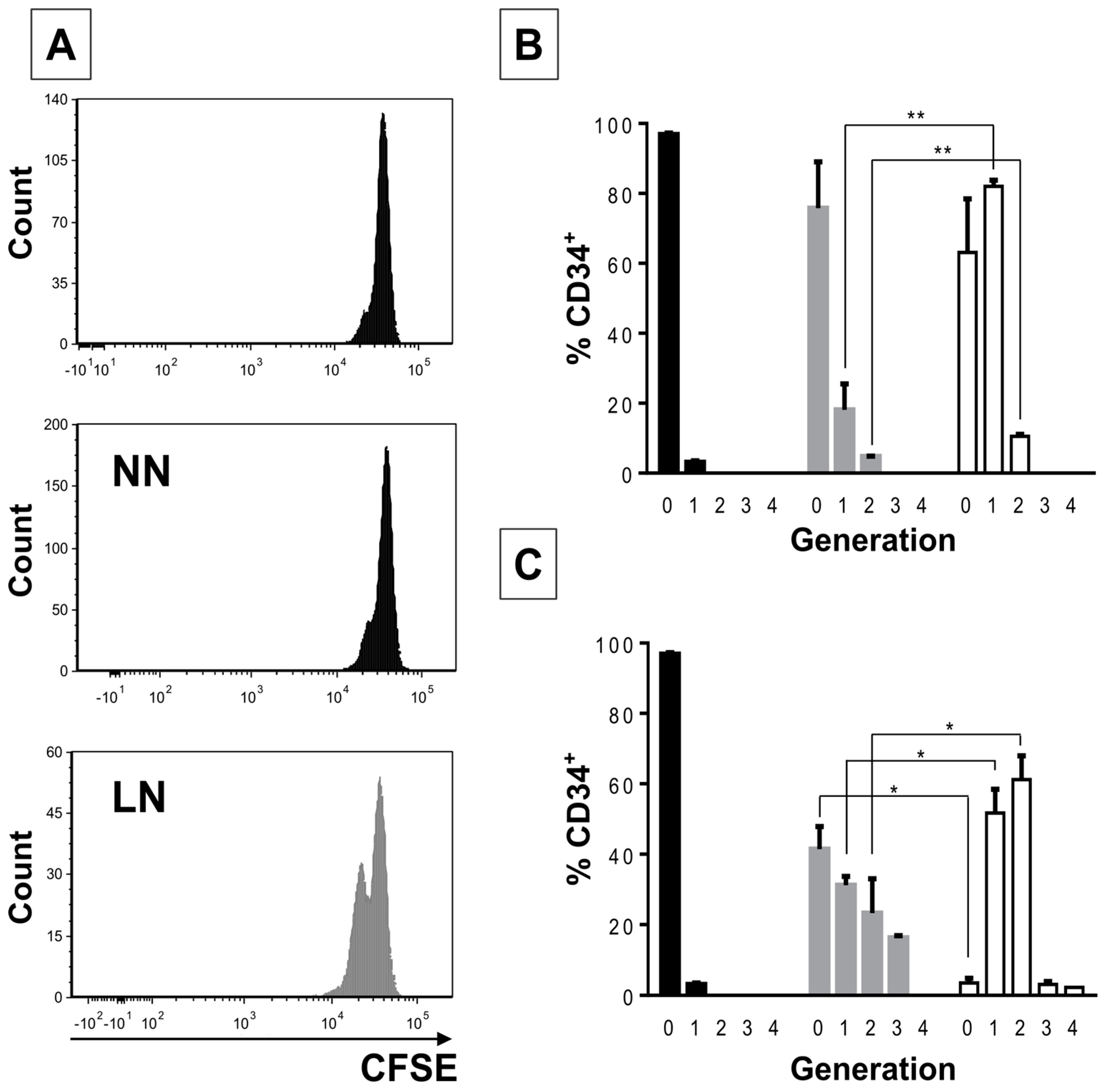
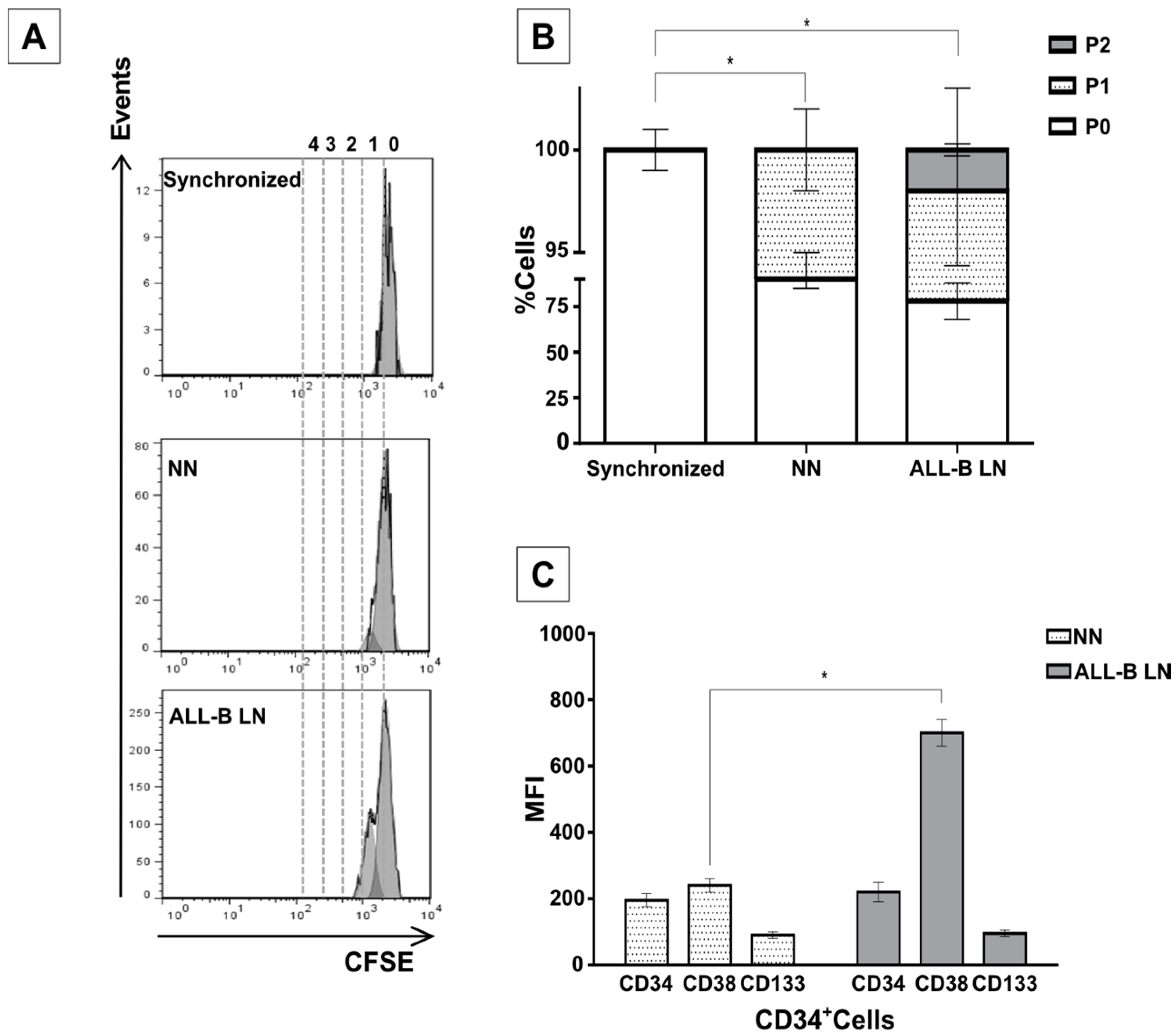
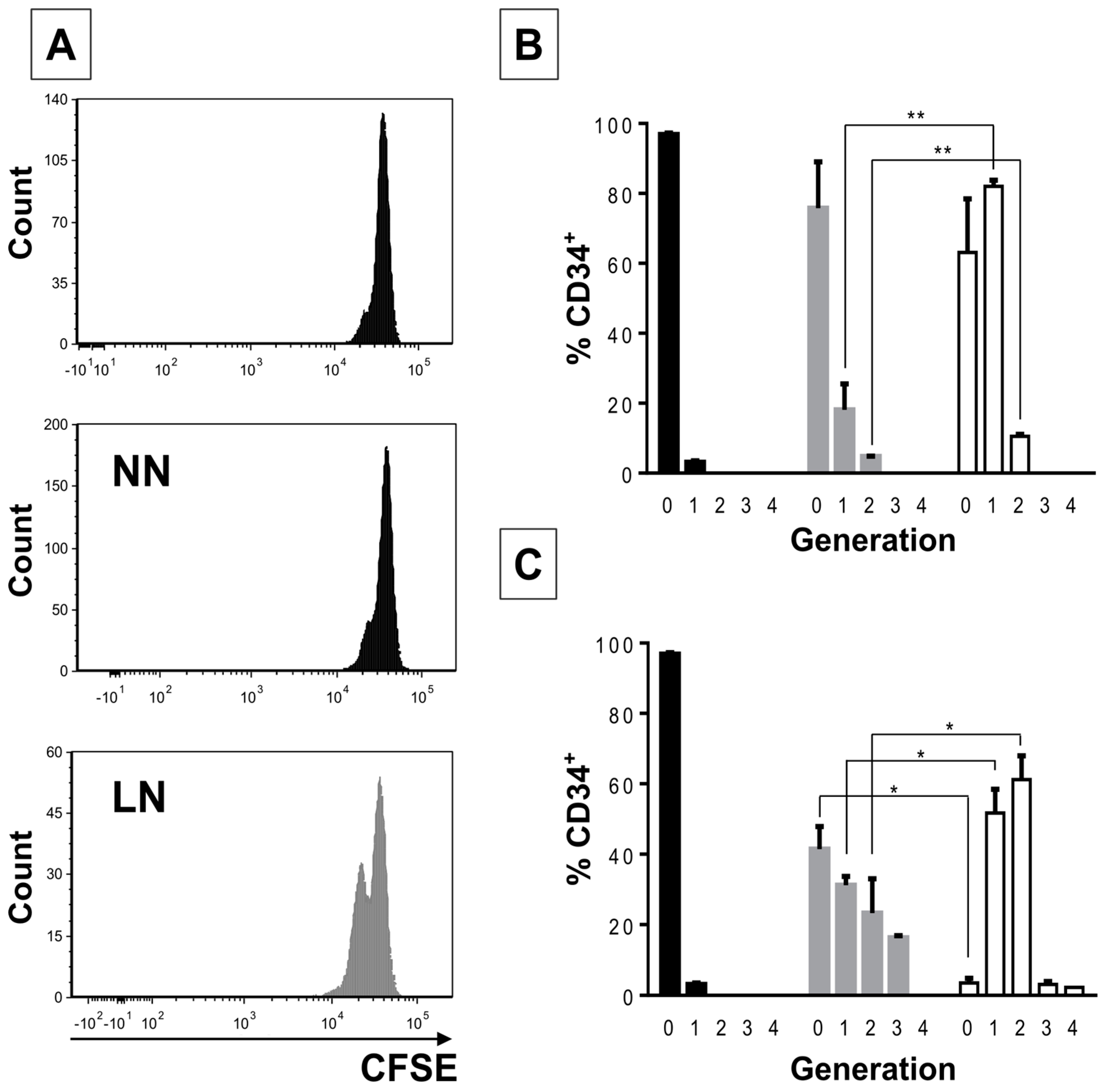
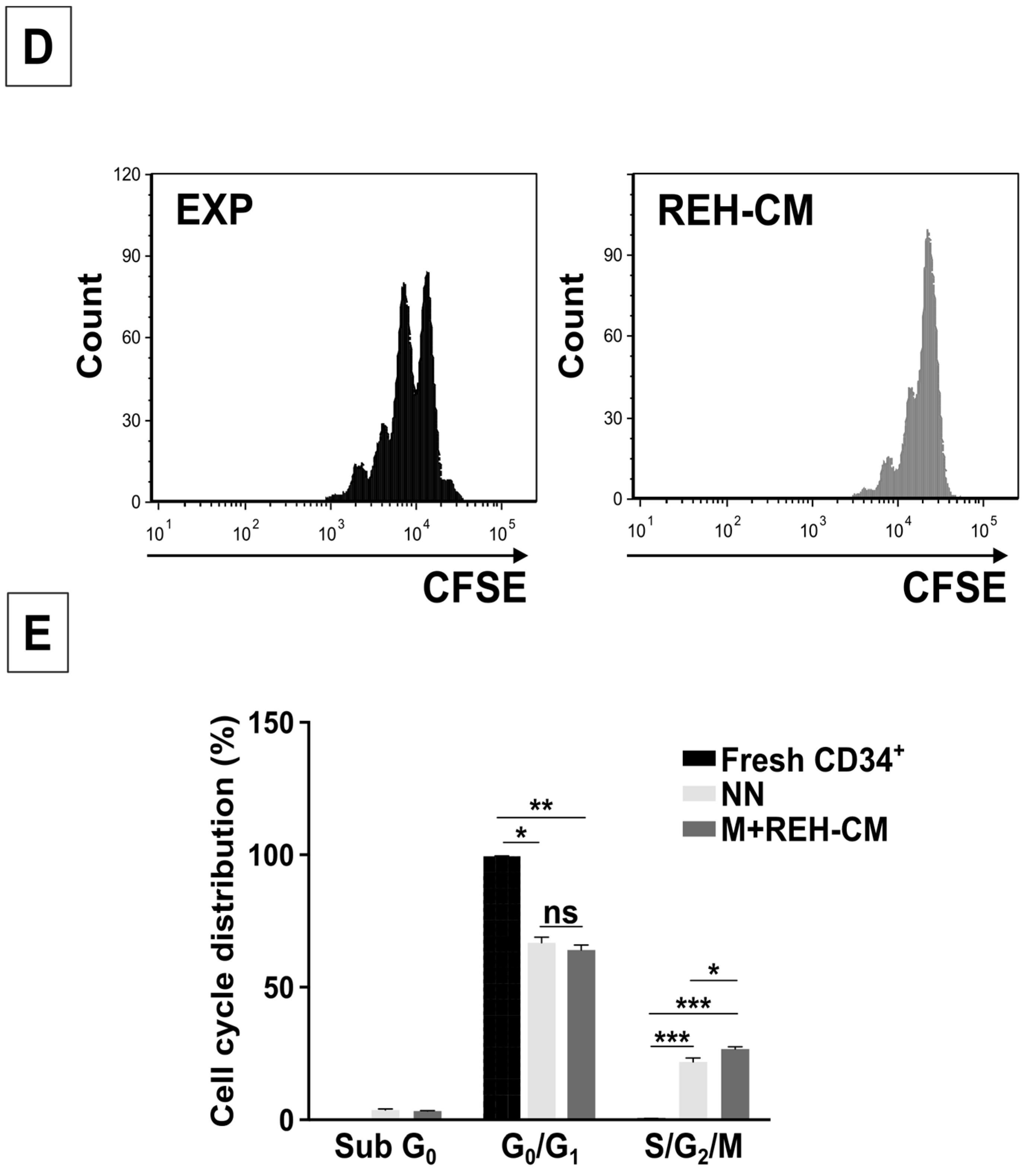
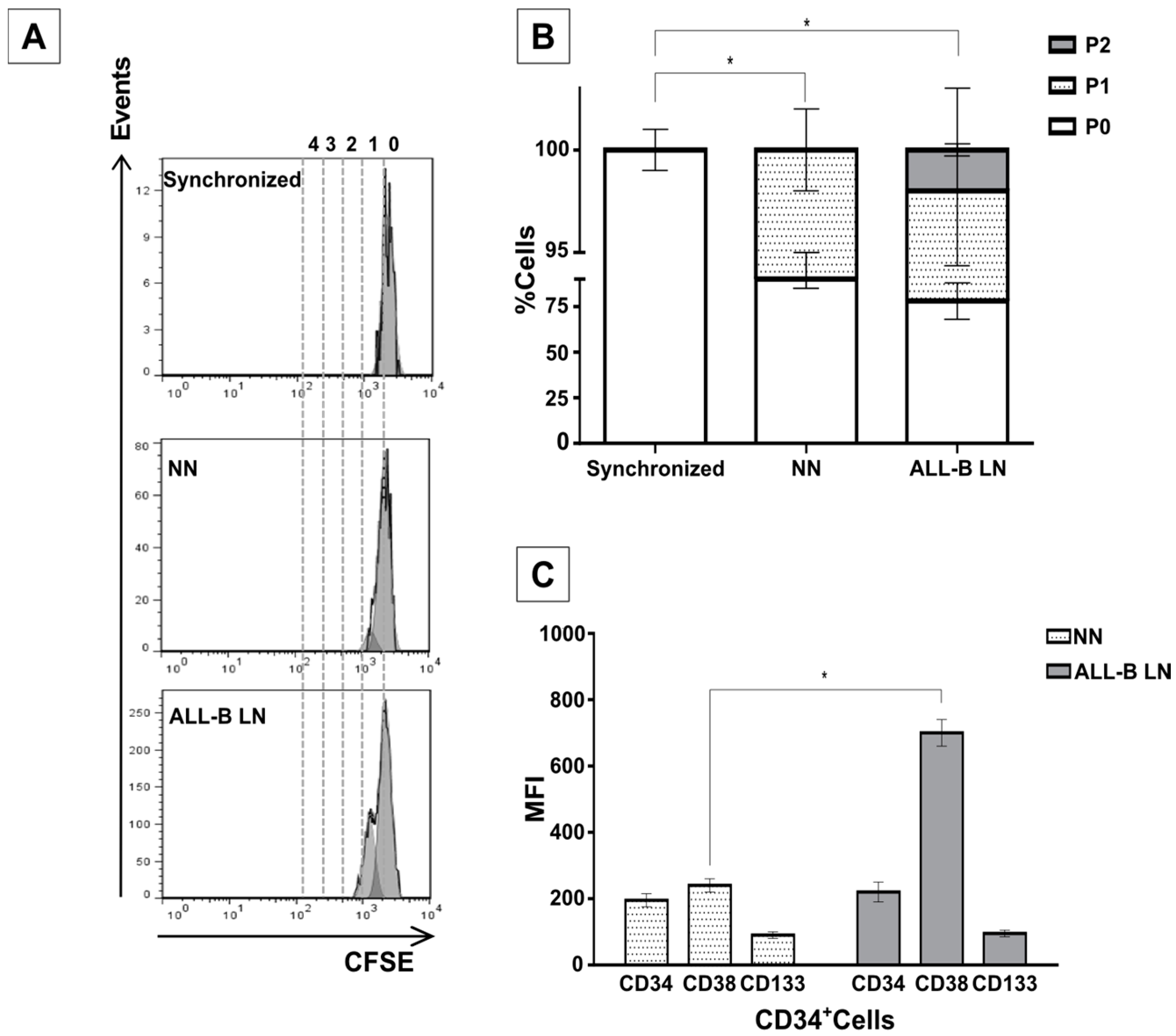
2/M phases of the cell cycle when compared to CD34+ cells in the NN. An even higher cell proliferative response was observed when CD34+ cells were previously incubated with the REH-CM, signifying that soluble factors secreted by the leukemic cells are responsible for this effect. Nevertheless, the CD34+ cell proliferation rate was lower when compared to cells expanded in the presence of early cytokines (SCF, TPO, and Flt3L) and in the absence of MSC. These results showed that MSC previously co-cultured with leukemic cells or with REH-derived CM, partially (LN) or totally (REH-CM) lose their control over the CD34+ cell quiescence.
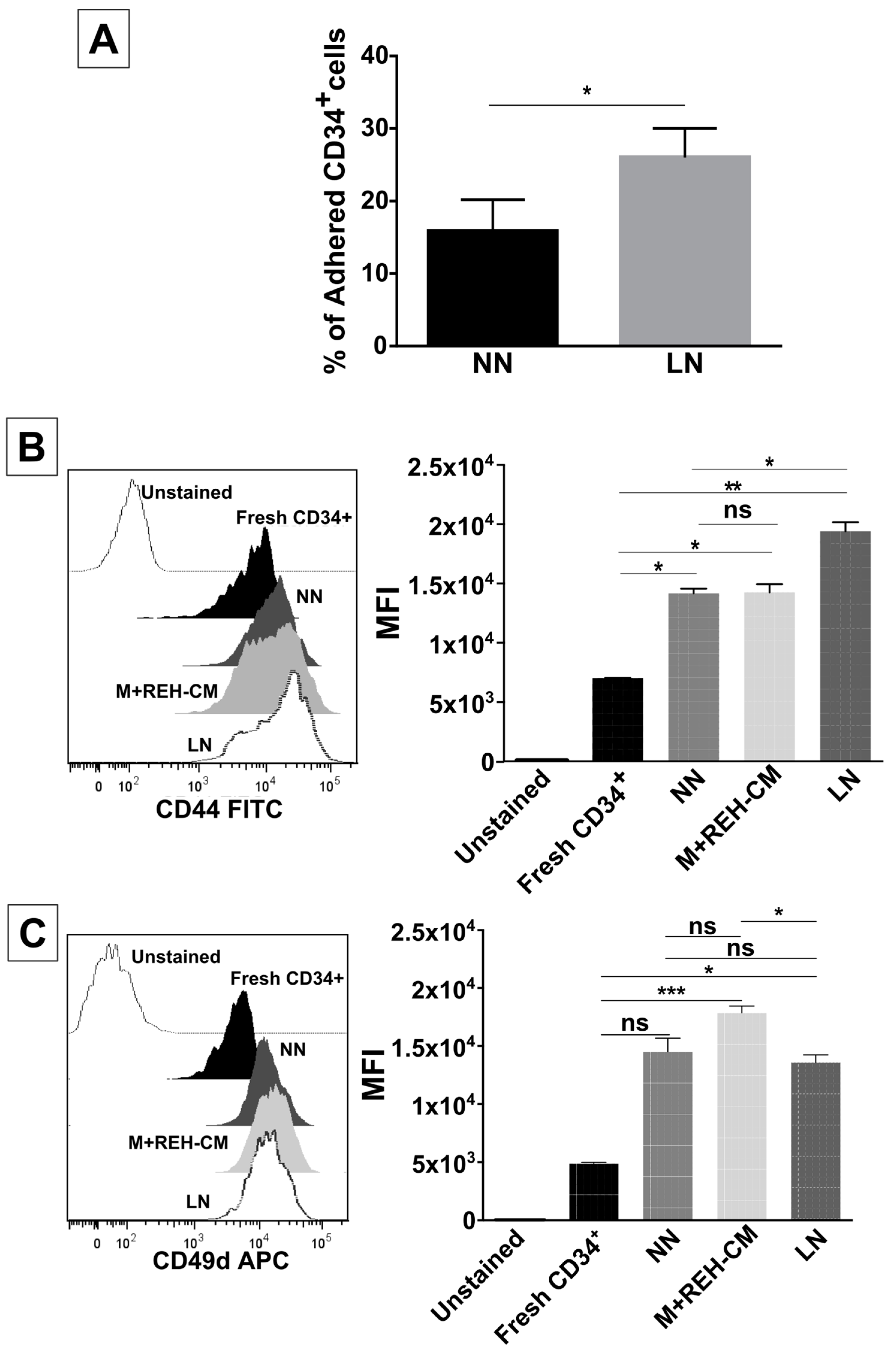
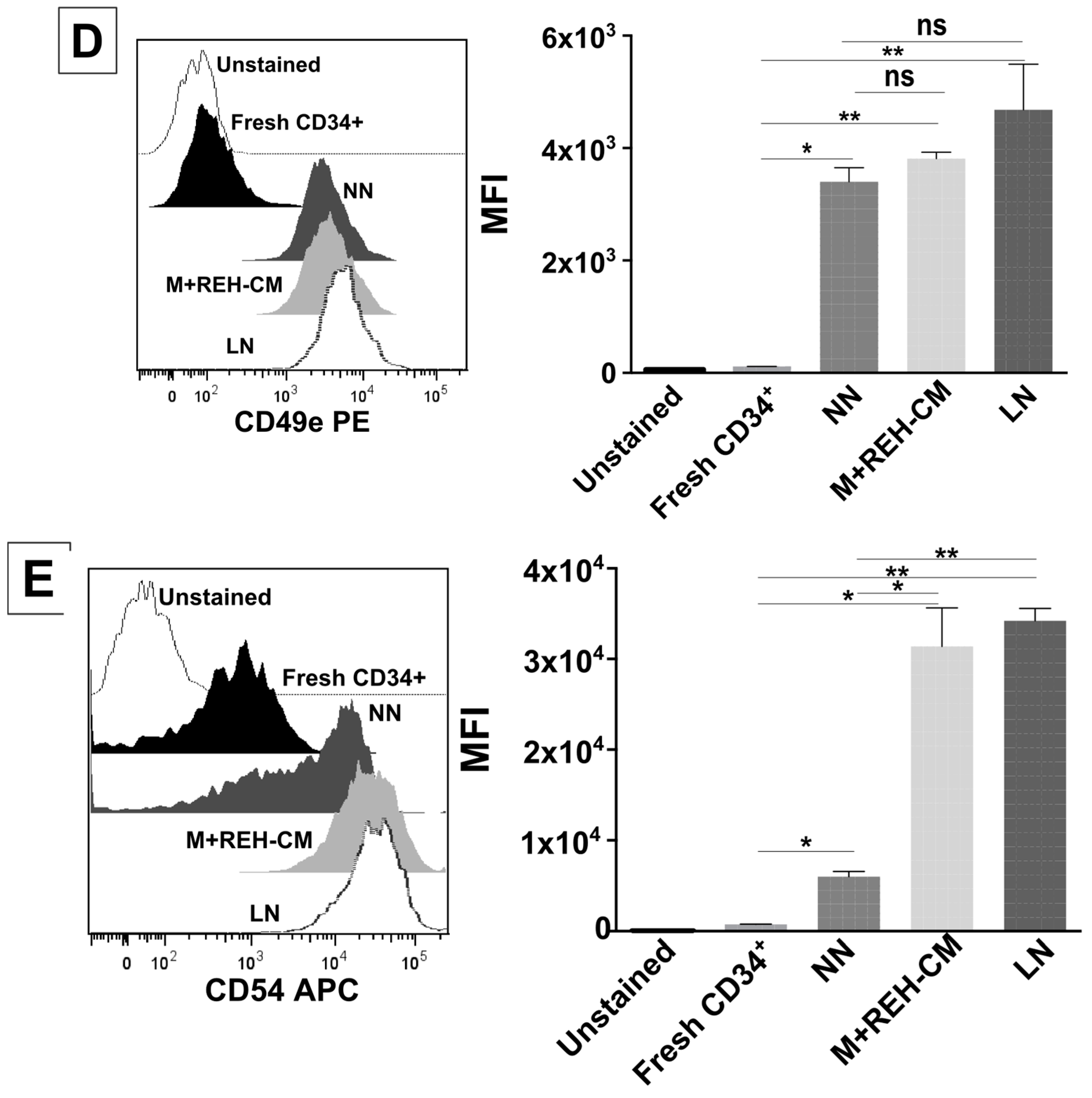
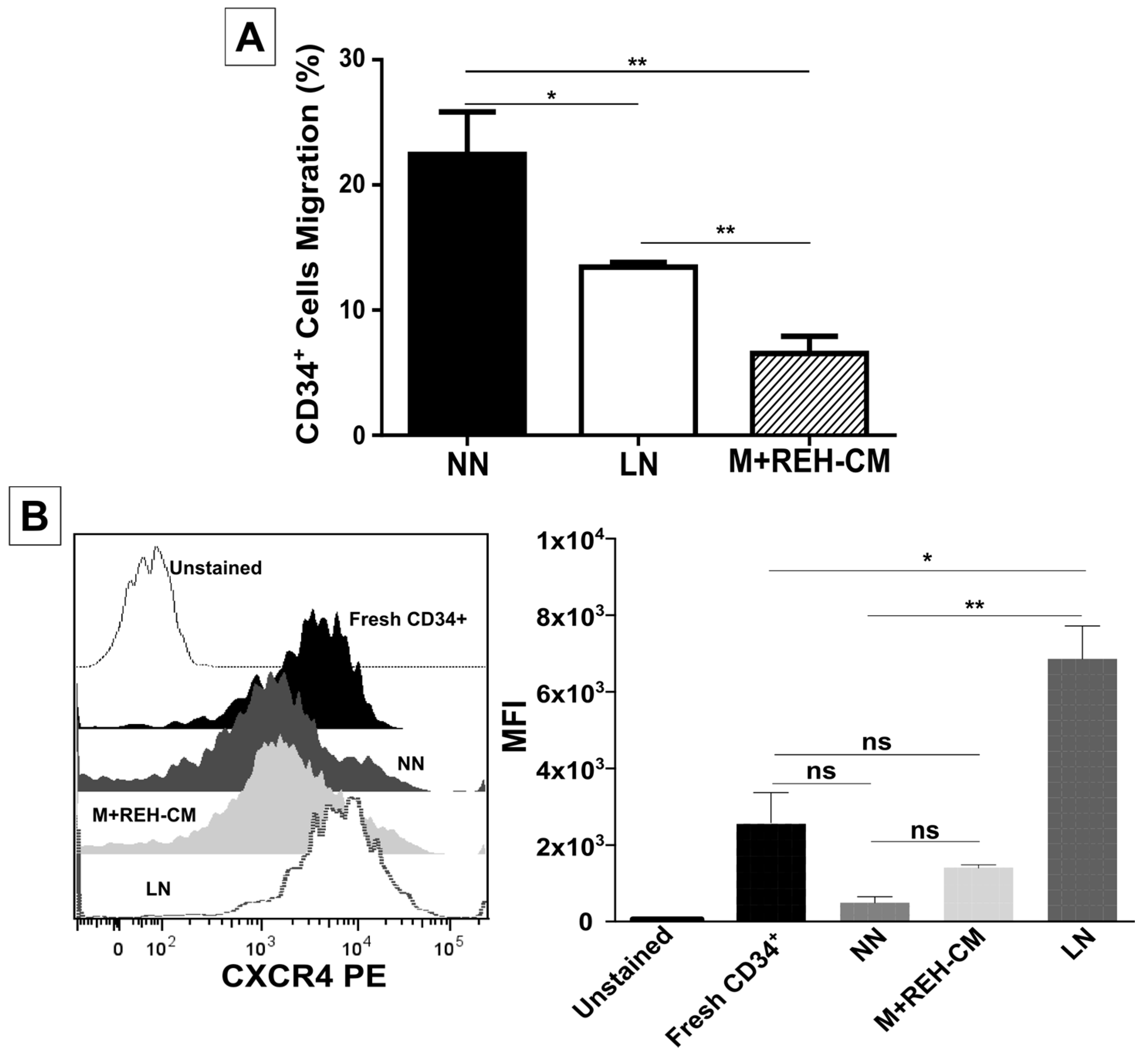
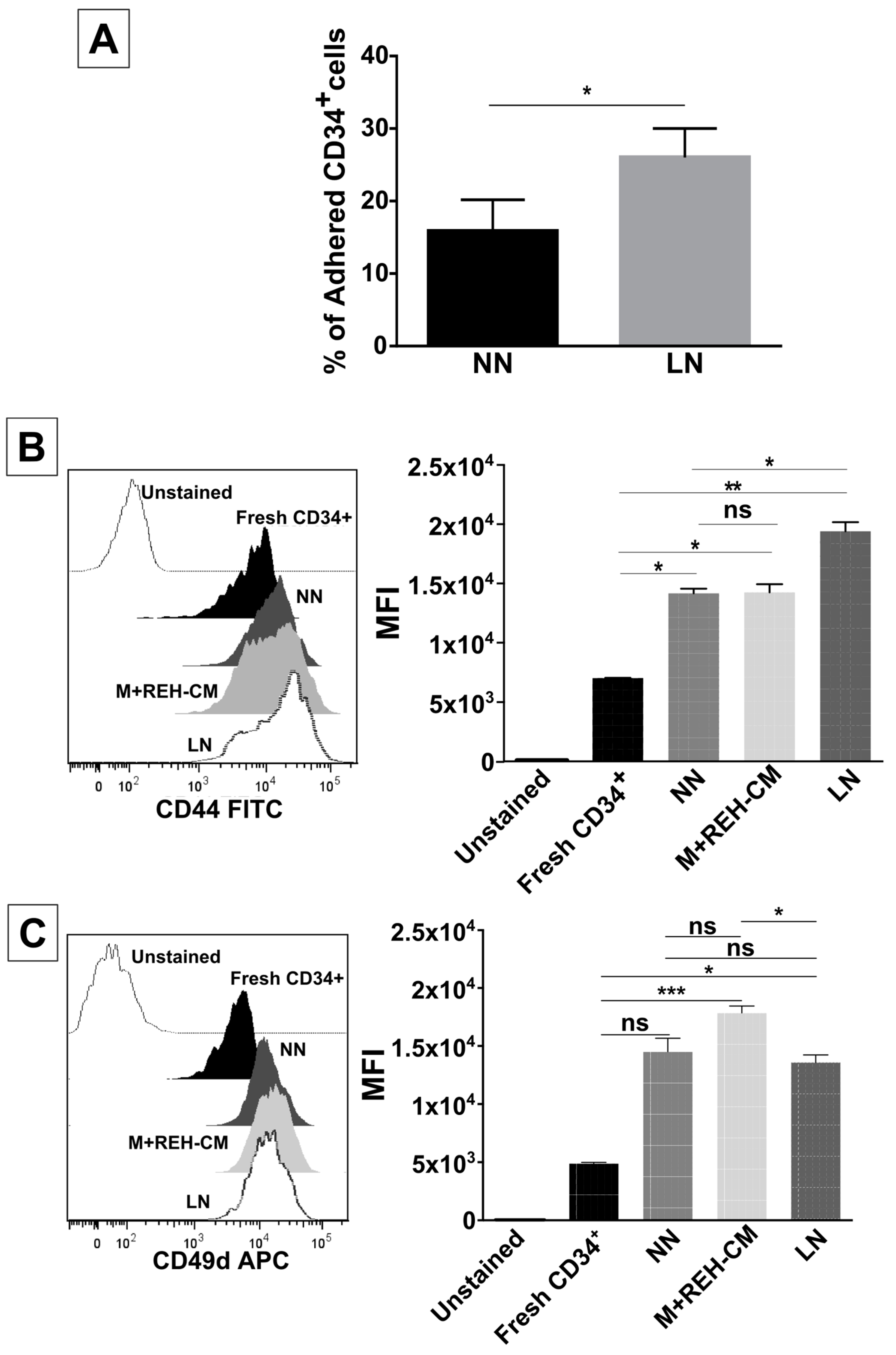
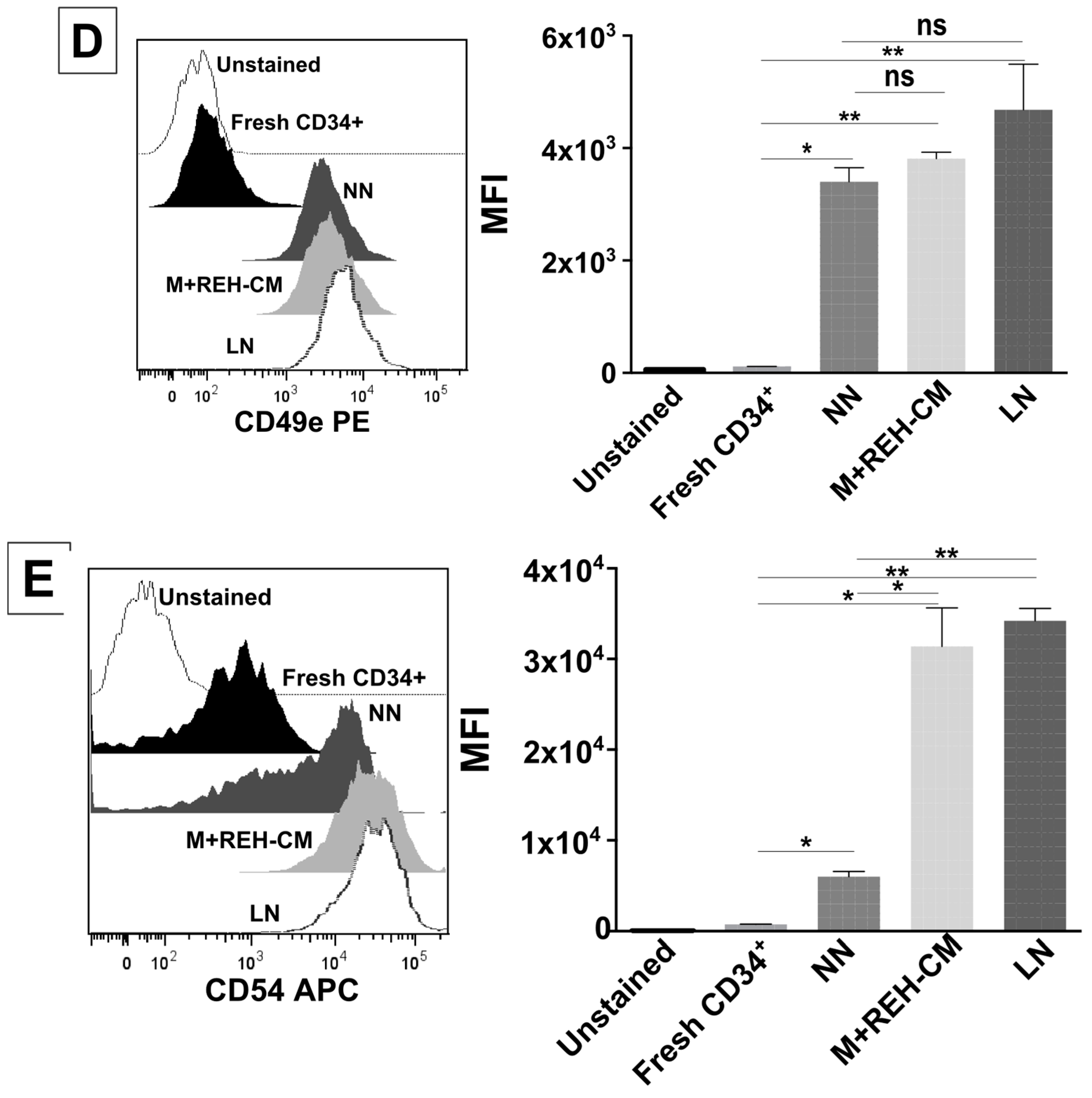
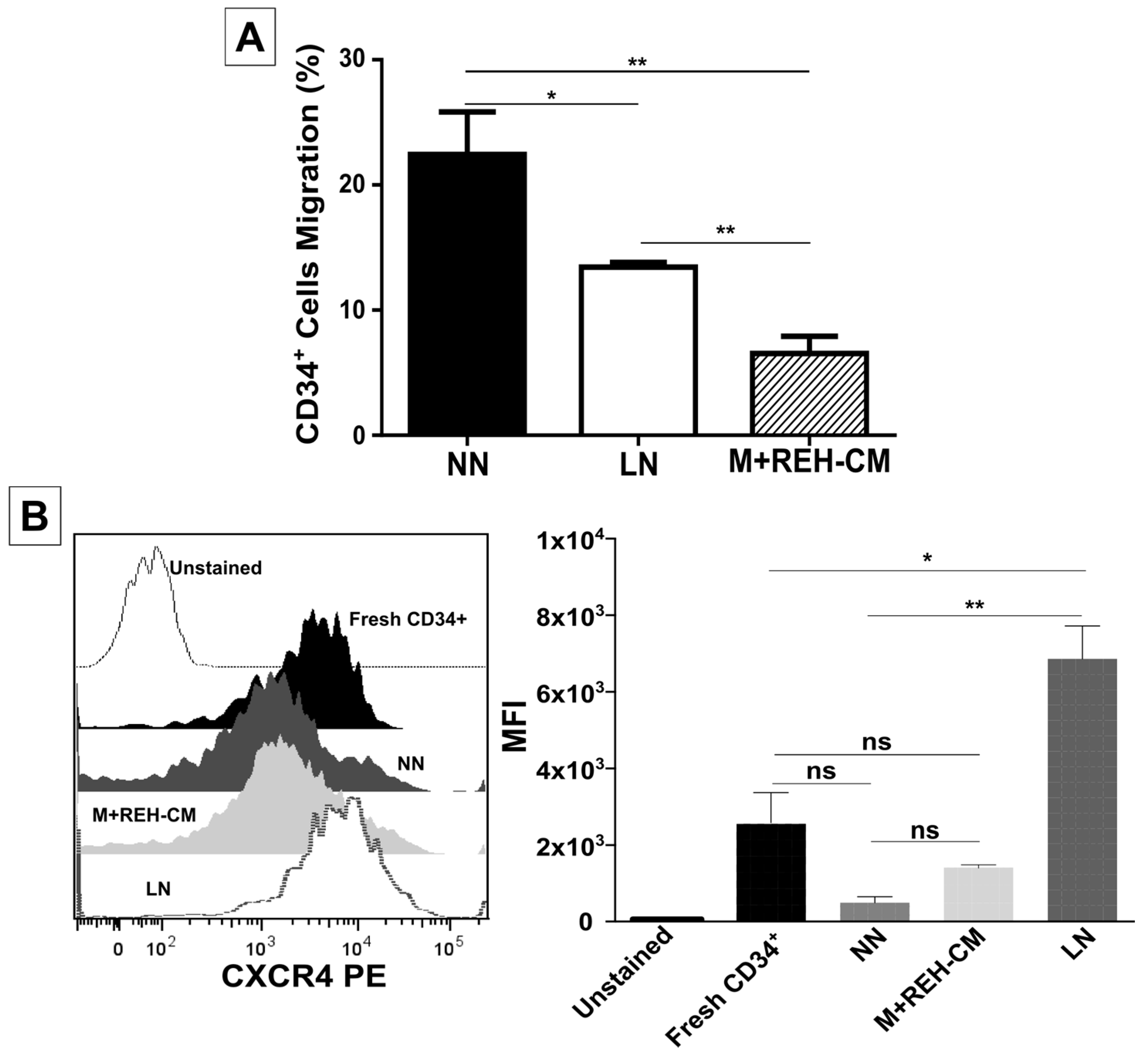
On the other hand, CD34+ cell binding capacity to MSC was enhanced in the LN. This was accompanied by an increased expression of CD44, CD49e, and CD54. Particularly, CD54 (ICAM-1) was highly upregulated in the LN compared to the NN, suggesting its involvement in CD34+ cell adhesion. The M+REH-CM showed the same effect as the LN on this adhesive phenotype, suggesting that soluble factors in the LN are responsible for these alterations. This situation is similar to the phenotype described for MSC from ALL patients having increased adhesion induced by soluble factors [
23]. The specific molecules responsible for the higher cell adhesion to MSC should be further explored. As expected, this higher cell adhesion to MSC in the LN was accompanied by a reduced CD34+ cell migration to SDF-1. Higher CXCR4 expression was observed in CD34+ cell from the LN, suggestive of an inadequate endocytosis and altered CXCR4 signaling. On the contrary, a reduction in CXCR4 expression was evident in the NN, showing an appropriate SDF-1 signaling. The situation with the M+REH-CM was similar, with reduced cell migration and higher CXCR4 expression compared to NN. To our knowledge, the evaluation of CXCR4 expression in HSC in patients with leukemia has not been done. Nevertheless, it has been shown, in an experimentally-induced leukemia model, that HSC increased CXCR4 expression [
24], showing the similarity to our in vitro model. Additionally, it has been shown that BM levels of SDF-1 in pediatric precursors of ALL-B were almost three times lower compared to non-leukemic controls [
25]. As expected, we did not find SDF-1 secretion in the LN. Interestingly, it has been shown in leukemic mouse models that HSC respond better to SCF [
20], and it was suggested that less retention in response to SDF-1 would facilitate displacement in response to this factor or others present in this in vivo setting.
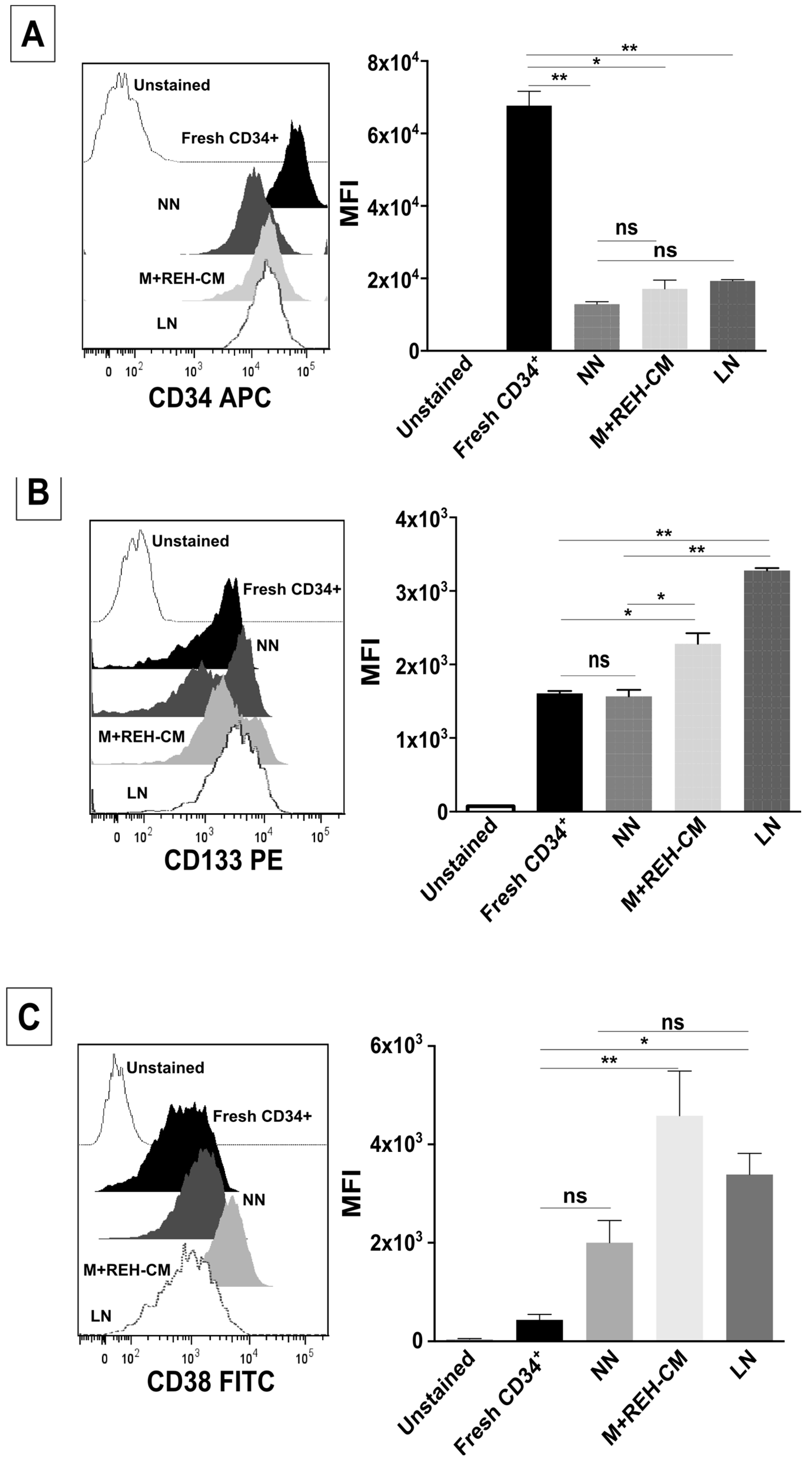
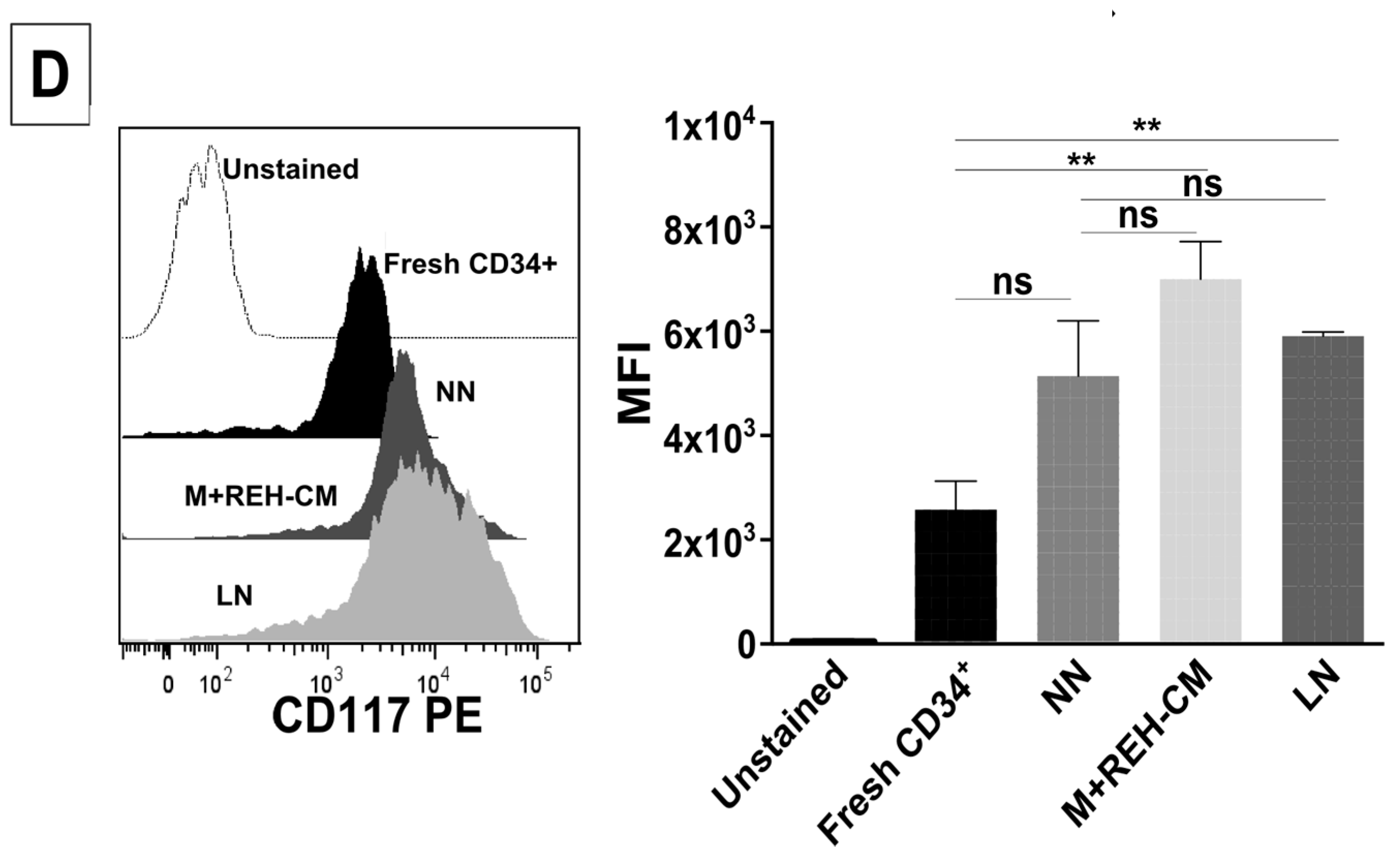
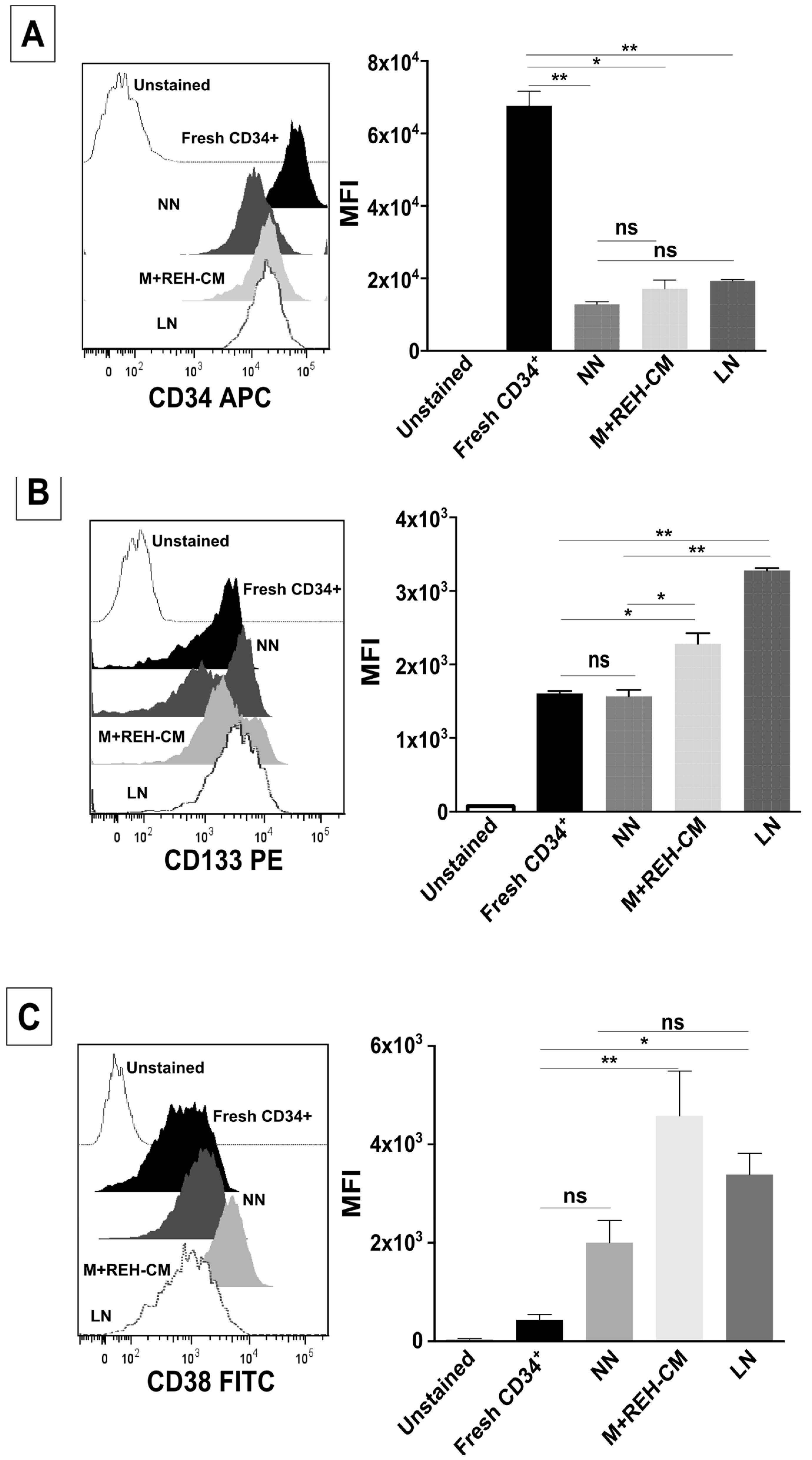
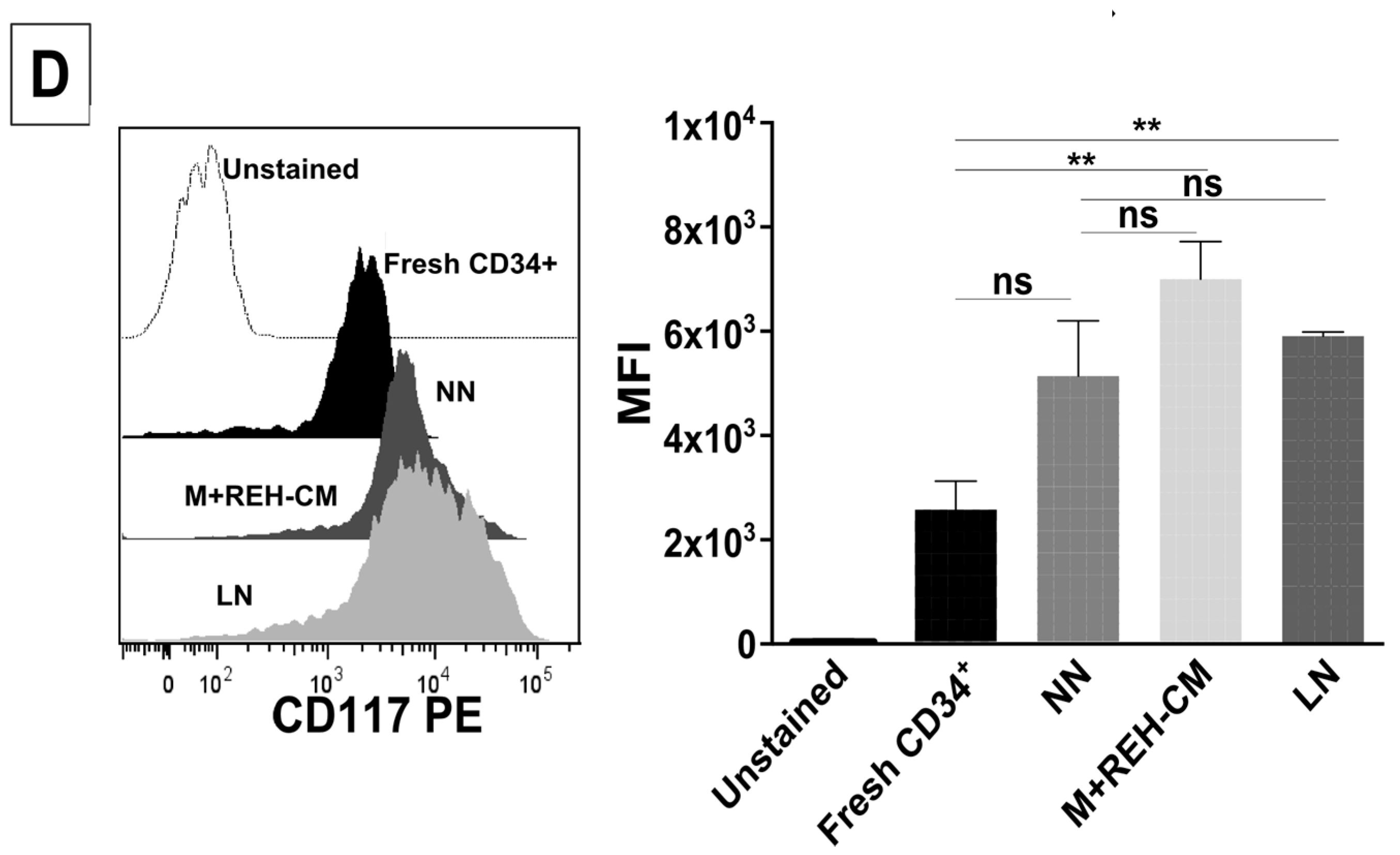
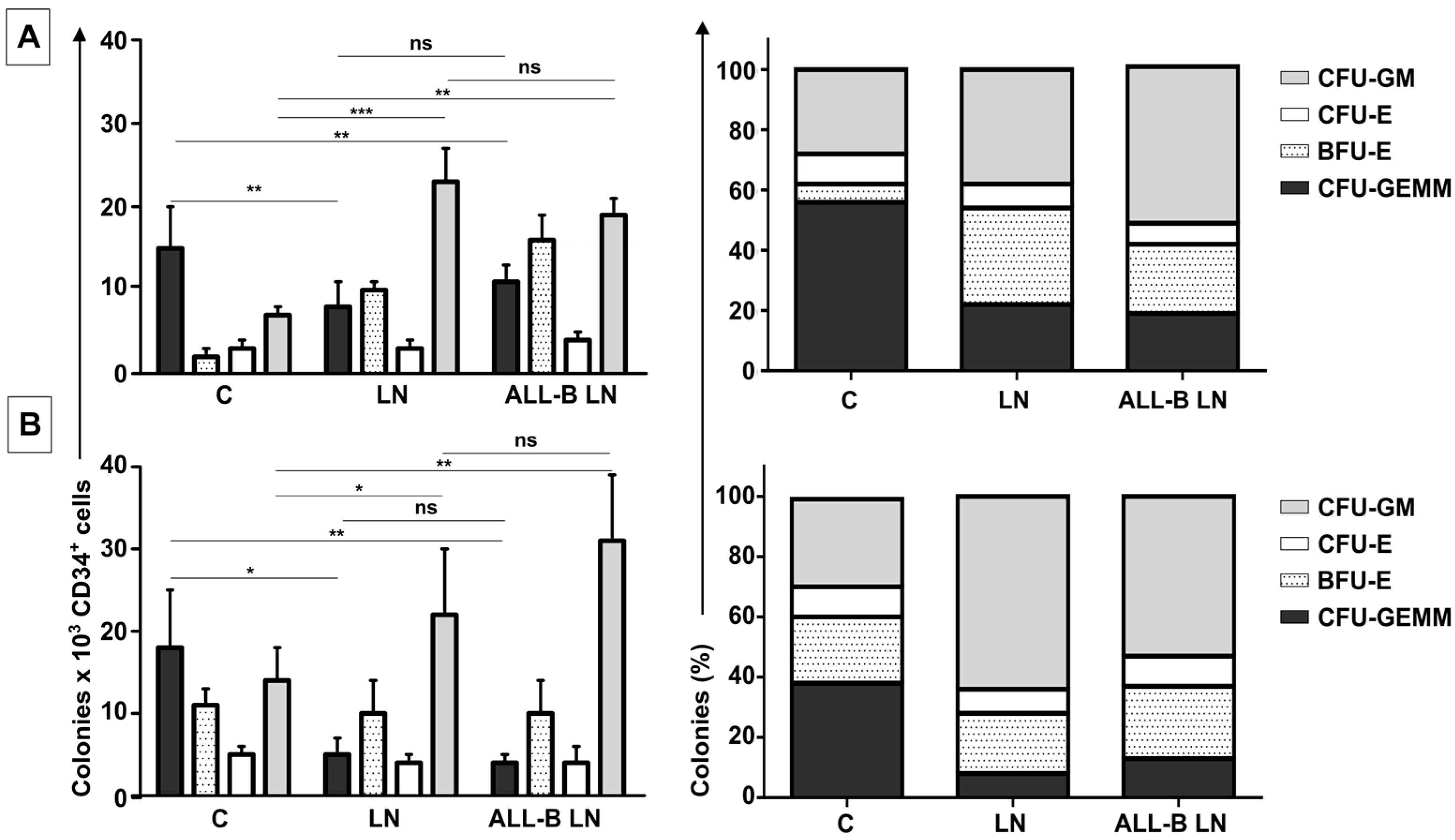
This increased proliferation of CD34+ cells, accompanied by an augmented cell adhesion to MSC and a reduced migration capacity to SDF-1, could also alter the conventional signals arising from HSC-MSC interactions and indirectly affect other HSPC functions. In relation to the expression of primitive markers, it is interesting to point out that the expression of CD34 was similar in the NN and LN (with a slightly increase in the LN), although reduced compared to freshly-isolated cells. Additionally, CD133 was upregulated in the LN and in the M+REH-CM. CD117 (c-kit) expression has been used as an indication of a primitive status and multipotency capabilities [
26]. Compared to freshly isolated cells, CD117 was slightly upregulated with practically no differences between NN, LN, and M+REH-CM. Our in vitro cultures induced a higher expression of the CD38 marker in the different niches compared to freshly isolated cells, but no statistical differences were found between the NN and the LN or M+REH-CM (although a tendency towards a higher expression was noticed in the latter). These results are similar to what has been described for leukemic patients and in a mouse model of AML [
27]. They suggest that signals responsible for a primitive cell state are still present in the LN. They seem to be slightly incomplete for HSPC, but they are possibly suitable for the leukemic cells. To further explore this issue, the differentiation potential into hematopoietic lineages was also evaluated in cells obtained from both the NN and the LN. Here again, an important reduction in the absolute count and the percentage of primitive progenitor colonies (CFU-GEMM) was detected in cells obtained from the LN, accompanied by an increase of CFU-GM. These changes were also similar to cytokine-expanded CD34+ cells when compared to a similar setting of the NN [
26]. This is also consistent with a study in a leukemia mouse model that showed a loss of HSC self-renewal capacity and differentiation [
28]. No important differences were observed in erythroid precursors (BFU-E or CFU-E), a situation that has been described in CLL patients in spite of the occurrence of the disease-related anemia [
29].
Remarkably, HSPC alterations were also observed when the leukemic microenvironment was established by incubation of MSC with primary leukemic cells isolated from an ALL-B patient. Increased proliferation, reduced clonogenic capacity, significant upregulation of CD38, and primitive marker (CD34 and CD133) expression maintenance were also confirmed in this system. This is a valuable finding, since further insight into the mechanisms involved in these interactions could be perfectly performed with the REH cell line. This would certainly facilitate further HSPC evaluation in this in vitro leukemic microenvironment.
Altogether, these results support the concept that the control imposed on stem cells and progenitors cells by normal MSC is lost or severely reduced in the LN. Paradoxically, in spite of a higher adherence to MSC, HSPC from the LN proliferate extensively, differentiate abnormally, and lose their clonogenic capacity. This is very similar to the findings described by Welner et al. [
30], who demonstrated that CML caused normal mouse HSPC to divide more readily, altered their differentiation, and reduced their reconstitution and self-renewal potential.
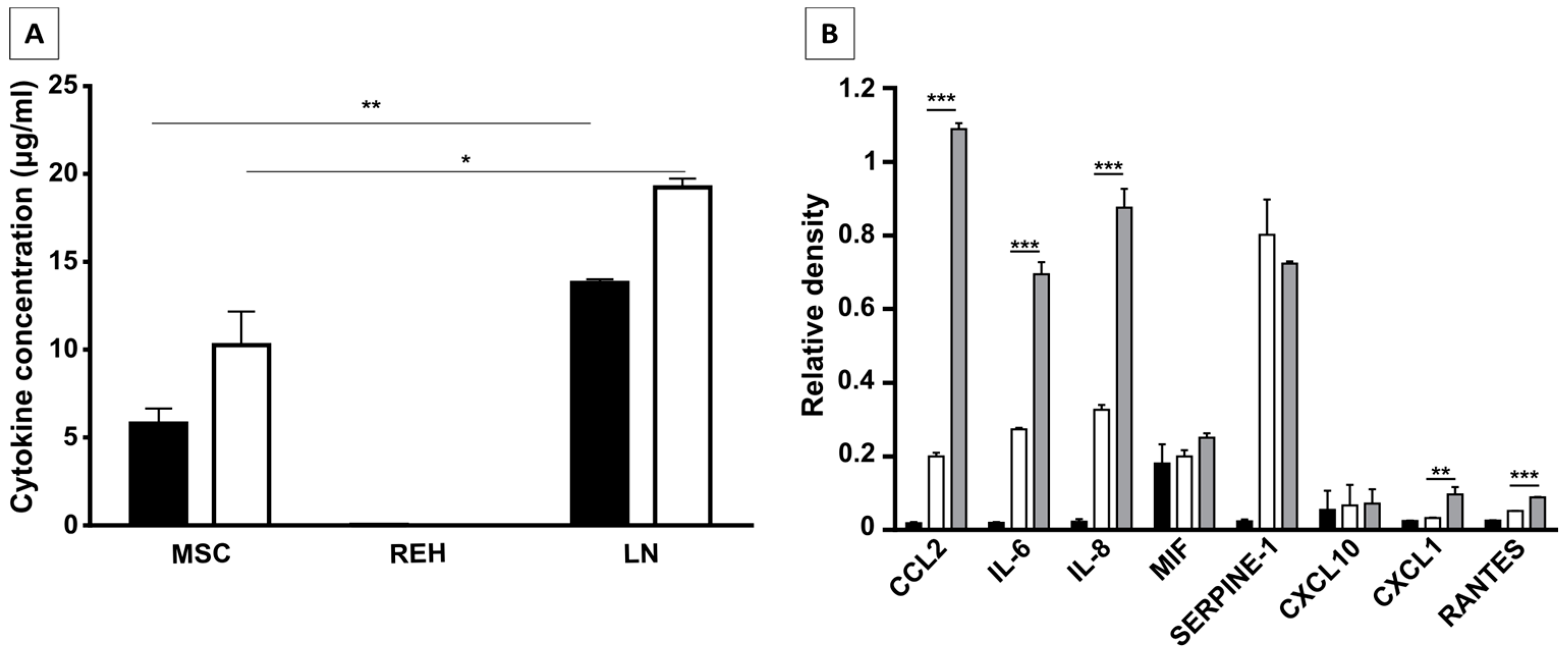
Knowing that in normal circumstances both cell-cell interactions and soluble factors are responsible for HSC homeostasis, in our in vitro model of the LN it seems to be clear that the cytokines secreted in the LN, either by the REH cells, by the MSC co-cultured with REH cells (or both), play a major role in HSPC malfunction. In fact, most of the changes observed in the LN could be replicated by incubation of MSC with the REH-conditioned medium (M+REH-CM), suggesting that paracrine stimulation is responsible for the CD34+ cell alterations. The fact that some soluble factors (CCL2, IL-6, IL8, CXCL1, and RANTES) are synergistically increased in the LN argues in favor that the phenotypic and functional alterations observed in the CD34+ cells is either directly through soluble factors secreted from the REH cell and/or indirectly through REH-CM-induced secretion by MSC. In particular the role of CCL2, IL-6, and IL-8 merits further study; the fact that IL-8 and CCL2 are upregulated in MSC from ALL patients [
23], and that plasma levels of CCL2, IL-8, and IL-6 are increased in children at ALL diagnosis [
23,
31], show their relevance in vivo and validates in part our in vitro LN. The leukemic-induced alterations of the HSPC found here could be equated with the observed reduced hematopoiesis, increased progenitors and differentiated cells, and, in general, bone marrow failure in patients with ALL [
6,
20,
32]. On the other hand, we have shown here that signals responsible for maintaining a primitive phenotype persist in the LN, being certainly used by the leukemic cells for their own benefit. Finally, this in vitro system will allow us to understand the different functional consequences of these multiple interactions (cell–cell and cell-soluble factors) occurring in the LN, also producing valuable information related to the changes occurring in MSC and leukemic cells, with meaningful implications for novel therapeutic approaches.
4. Materials and Methods
4.1. BM Mesenchymal Stem Cell Isolation and Characterization
BM aspirates from the iliac crest of healthy donors aged 5–12 years were obtained after their parents had signed an informed consent and the ethical committees of the participant institutions had given approval. Samples were collected in a sterile tube containing 0.25% EDTA (GIBCO-Invitrogen, Grand Island, NY, USA), and mononuclear cells (MNC) were isolated by Ficoll density gradient centrifugation (Histopaque d = 1.077 g/cm3, Sigma-Aldrich, St. Louis, MO, USA). MNC were plated at a density of 106 cells/cm2 in Iscove’s Modified Dulbecco’s Medium (IMDM) Glutamax-I (GIBCO-Life Technologies, Grand Island, NY, USA) supplemented with 1% sodium pyruvate (GIBCO-Life Technologies, Grand Island, NY, USA), 1% Minimum Essential Medium (MEM) non-essential amino acid solution 100X (GIBCO-Life Technologies, Grand Island, NY, USA), and 10% fetal bovine serum (FBS, GIBCO-Life Technologies, Grand Island, NY, USA). After obtaining 90% cell confluence, adherent cells were detached by treatment with 0.25% Trypsin (Sigma-Aldrich, St. Louis, MO, USA) and 1 mM EDTA. Cells were characterized by means of immunophenotyping and differentiation assays (see below) and were used for the different experiments in passages 3–5.
After the third passage, adherent cells were trypsinized and labeled with the following monoclonal antibodies: Fluorescein isothiocyanate (FITC) mouse anti-human CD73 (clone AD2, BD Pharmingen, San Jose, CA, USA), Allophycocyanin (APC) mouse anti-human CD105 (clone SN6, Invitrogen, Frederick, MD, USA), FITC mouse anti-human CD90 (clone F15-42-1, Abcam, Cambridge, MA, USA), and FITC anti-human CD44 (clone MEM-85, Invitrogen, Frederick, MD, USA). Additionally, the leucocyte-specific antibody PerCP mouse anti-human CD45 (clone 2D1, BD Biosciences, San Jose, CA, USA) and the APC mouse anti-human CD34 (clone 581, BD Pharmingen, San Jose, CA, USA) were used. Data were acquired using a FACSAria II flow cytometer (Becton Dickinson Biosciences, San Jose, CA, USA). FACS Diva software, CellQUEST PRO software, FlowJo, and Paint-A-Gate software (Pro v1.0, Becton Dickinson Biosciences, Sunnyvale, CA, USA) were used for data analysis.
Furthermore, the osteogenic, adipogenic, and chondrogenic differentiation capacities were determined using specific stainings and optical microscopy examination, as previously described [
26]. Third-passage 2 × 10
4 MSC were cultured in a 24-well plate in IMDM until they reached confluence. For adipogenic differentiation, cells were cultured for three days alternately in an induction medium (MEMα supplemented with 10% FBS, 1 mM dexamethasone, 0.5 mM isobutylmethylxanthine, 200 μM indomethacin, and 10 μg/mL insulin; all reagents were from Sigma Aldrich, St. Louis, MO, USA) or in a maintenance medium (MEMα, supplemented with 10% FBS and 10 μg/mL insulin) for two weeks. Osteogenic differentiation was induced by cell incubation in MEMα supplemented with 10% FBS, 100 nM dexamethasone, 0.2 mM ascorbic-2-phosphate, and 10 mM β-glycerophosphate (all reagents were from Sigma Aldrich, St. Louis, MO, USA) for two weeks. For chondrogenic differentiation, cells were plated and cultured in a chondrogenic induction medium (MEMα and 10 ng/mL TGFβ-1, Sigma Aldrich, St. Louis, MO, USA), also for two weeks. Then the cells were washed three times with PBS (1X), followed by fixation with formalin solution (Sigma Aldrich, St. Louis, MO, USA), and were stained with 0.35% Oil Red O solution (Sigma Aldrich, St. Louis, MO, USA) or alkaline phosphatase (using an AP staining kit, EMD Millipore Corporation, Billerica, MA, USA), or with 0.1% Safranin O (Sigma Aldrich, St. Louis, MO, USA). Cells were examined with an inverted microscope (Eclipse Model TS-100, Nikon, Konan, Minato-ku, Tokyo, Japan) and photographed with a Power Shot A460 Zoom Browser EX software (Canon, Melville, NY, USA).
4.2. CD34+ Isolation from Umbilical Cord Blood (UCB)
UCB samples from normal full-term deliveries were collected after obtaining informed consent and in accordance with the guidelines approved by the Ethics Committee of the Faculty of Medicine, Universidad Nacional de Colombia. MNC were isolated as described above, and CD34+ cells (HSPC) were purified through immunomagnetic selection using a MACS CD34+ isolation kit (Miltenyi Biotec, Auburn, CA, USA). Cell purity (>90%) was evaluated by flow cytometry using allophycocyanin (APC)-conjugated mouse anti-human CD34 antibody (Clone AC136, Miltenyi Biotec, Auburn, CA, USA), and cell viability (>95%) by trypan blue dye exclusion.
4.3. Establishment of a Leukemic Niche (LN)
The REH cell line was obtained from the American Type Culture Collection (ATCC; Rockville, MD, USA) and was characterized by flow cytometry for the presence of CD44, CD133, CD38, CD45, and CD19 (not shown). BM-MSC at 80% confluence were co-cultured for three days with the 3–5 × 104 REH cells for the establishment of the leukemic niche (LN). After REH cells addition MSC stop dividing and did not increase cell number. After three days, the majority of REH cells were removed by gently pipetting with cold PBS (1X) and PBS plus 1 mM EDTA (1X). Freshly isolated CD34+ at a density of up to 100,000 cells/mL were added to cultured MSC-REH cells. In order to distinguish between the few REH cells remaining and the CD34+ cells, the latter were labeled in some experiments with 5 μM CFSE, as described below. The distinction between these two cell populations could also be achieved by CD44 or CD34 expression evaluation, since these cell surface markers are absent in the REH cell line. Simultaneously, freshly isolated CD34+ cells from the same UCB were layered over MSC that had reached 80% confluence, for the establishment of the so-called normal niche (NN). CD34+ cell evaluation in the NN or the LN was done after three days in the co-cultures. Some HSPC evaluations were also performed in a LN set by incubation of MSC with a REH-conditioned medium (M+REH-CM) without REH cells; in this case, freshly prepared REH-CM was added twice to cultured MSC for the establishment of the M+REH-CM leukemic microenvironment. For establishing the LN with primary leukemic cells (ALL-B-LN), we obtained BM mononuclear cells from a patient with ALL-B (having an infiltration of blasts in the BM > 96%) after signing the informed consent and we proceeded as described above for the LN.
4.4. REH-Conditioned Medium Preparation
2.5 × 105 REH cells/mL were cultured in RPMI 1640 (Invitrogen Corporation, Carlsbad, CA, USA) supplemented with 1% sodium pyruvate, 1% MEM non-essential amino acid solution 100X and 1% FBS for 24 h at 37 °C and 5% CO2. Next, REH cells were centrifuged at 500× g for 7 min, and the medium was collected and filtered through a 0.22 µm pore membrane filter (Corning Incorporated Pittston, Pittston, PA, USA) and used fresh in all experiments.
4.5. CD34+ Cell Division Determination by Carboxyfluorescein Diacetate Succinimidyl Ester Labeling
CD34+ cells were labeled for 10 min at 37 °C with 5 μM carboxyfluorescein diacetate succinimidyl ester (CFSE; CellTrace™ CFSE Cell Proliferation Kit, Invitrogen, Eugene, OR, USA) in 0.1% bovine serum albumin (BSA, GIBCO-Invitrogen, Grand Island, NY, USA)-supplemented phosphate-buffered saline (PBS 1X). CFSE-labeled cells were suspended in RPMI 1640 (Invitrogen Corporation, Carlsbad, CA, USA) with 10% FBS for 5 min in ice and washed three times with PBS (1X) for 7 min at 400× g and 20 °C, resuspended in RPMI 1640 with 10% FBS, and further incubated 20 min at 37 °C. CFSE labeling was confirmed using fluorescence microscopy (Axiovert C-40 CFL, Carl Zeiss, Thornwood, NY, USA). A fraction of the CD34+ cells were FBS-deprived for 48 h (synchronized cells) to establish non-proliferating cells (the highest CFSE mean fluorescence intensity).
4.6. CD34+ Expansion Cultures
Purified and fresh CD34+ cells were incubated with the early cytokines TPO, SCF, and FLT3L (ProSpec, East Brunswick, NJ, USA) at a concentration of 50 ng/mL. After three days in expansion medium, cells were harvested and evaluated for cell proliferation.
4.7. Cell Cycle Analysis of CD34+
Purified and fresh CD34+ cells and CD34+ obtained from both the NN and the LN after co-culturing with MSC for the days were fixed with 4% formaldehyde and permeabilized with 0.1% Triton X-100 (Sigma Aldrich, St. Louis, MO, USA). Cells were incubated with 2′-(4-ethoxyphenyl)(-5-(4-methyl-1-piperazinyl)-2,5′-bi-1H-benzimidazole trihydrochloride trihydrate (Hoechst 33342, Invitrogen, Eugene, OR, USA) (2n DNA content, G0/G1; >2n DNA content, S-G2-M) for 45 min at 37 °C. Finally, the supernatants were removed and cells were resuspended in PBS (1X) for flow cytometry evaluation (FACSAria™ II, Becton Dickinson Biosciences, San Jose, CA, USA). FCS Express Flow Cytometry Data Analysis Software v5.0 (De Novo Software, Glendale, CA, USA) were used for data analysis.
4.8. CD34+ Cell Adhesion to MSC
For cell adhesion assays to MSC, 6 × 104 MSC/mL were cultured for 24 h at 37 °C and 5% CO2 (80% of confluence). Next, 1 × 105 CD34+ cells, previously stained with CFSE, obtained from both the NN and the LN, were co-cultured with MSC for 6 h. Non-adhered cells were carefully removed by two gentle washes with PBS (1X). Adherent cells were further incubated with warmed culture medium RPMI 1640 supplemented with 10% FBS, 1% sodium pyruvate, and 1% non-essential amino acid solution and counted in a fluorescence microscope.
4.9. Expression of Adhesion Molecules in CD34+ Cells after Co-Culture
CD34+ cells were harvested by repeated pipetting after three days in the niches, washed in PBS (1X), and stained with monoclonal antibodies for FACS analysis: APC-conjugated mouse anti-human CD49d (clone 9F10, BD Pharmingen, San Jose, CA, USA), PE-conjugated mouse anti-human CD49e (clone IIA1, BD Pharmingen, San Jose, CA, USA), FITC-conjugated mouse anti-human CD44 (clone MEM-85, Invitrogen, Frederick, MD, USA), and APC-conjugated mouse anti-human CD54 (clone REA266, Miltenyi Biotec, Auburn, CA, USA). Reliable discrimination between the few MSC present and CD34+ cells was possible using their different forward-scatter and side-scatter signals. Dead cells were excluded during acquisition and analysis (gate: intermediate forward-scatter and low side-scatter). FlowJo (v10.0, FlowJo, LLC, Ashland, OR, USA) and Paint-A-Gate software (Pro v1.0, Becton Dickinson Biosciences, Sunnyvale, CA, USA) were used for data analysis.
4.10. Expression of Primitive Markers in CD34+ after Co-Culture
CD34+ cells from NN or LN were washed in PBS (1X), and stained with monoclonal antibodies for FACS analysis: APC-conjugated mouse anti-human CD34 (clone AC136, Miltenyi Biotec, Auburn, CA, USA), PE-conjugated mouse anti-human CD133 (clone AC133, Miltenyi Biotec, Auburn, CA, USA), FITC-conjugated mouse anti-human CD38 (clone IB6, Miltenyi Biotec, Auburn, CA, USA), and PE-conjugated mouse anti-human CD117 (clone AC126, Miltenyi Biotec, Auburn, CA, USA), and analyzed as described above.
4.11. CD34+ Cell Clonogenic Capacity
Detection and quantification of human hematopoietic progenitors in the NN and the different settings of the LN were performed via colony-forming cell (CFC) assays. Cells (0.5–1 × 10
3/1.1 mL) were cultured for 10–15 days in HSC-CFU, complete with EPO medium (StemMACS HSC-CFU Media, Miltenyi Biotec, Auburn, CA, USA). Colonies of erythroid progenitors (CFU-E and BFU-E), granulocyte-macrophage progenitors (CFU-GM, CFU-G, and CFU-M), and multi-potential granulocyte, erythroid, macrophage, and megakaryocyte progenitors (CFU-GEMM) were morphologically characterized and quantified by inverted microscopy, as described previously [
26].
4.12. CD34+ Migration Assays
UCB-CD34+ cell migration assays were performed in Costar transwell (Corning Costar, Tewksbury, MA, USA), 6.5 mm diameter, and 5 µm pore size. Inserts were incubated for 1 h at 37 °C with the migration buffer (RPMI 1640/2% BSA). CD34+ cells (1 × 105) isolated from the NN or the LN were added to the upper chamber. Lower chambers were filled with 600 µL of the migration buffer. For chemotaxis assays, human recombinant stromal cell-derived factor-1 (hrSDF-1a, Miltenyi Biotec, Auburn, CA, USA) at 100 ng/mL was added to the lower chamber. After 4 h at 37 °C and 5% CO2 incubation, cells that migrated to the lower chamber were collected and counted. Migrated cell percentage was calculated by dividing the number of migrated cells by the number of total input cells ×100. Percentages of migrated cells from at least four different CB samples were used for each paired analysis (FACS acquisition was performed in duplicate in some experiments).
4.13. Identification and Quantification of Cytokines
The concentration of six pro-inflammatory cytokines (IL-1β, TNF-α, IL-12-p70, IL-6, IL-8, and IL-10) present in the cell culture supernatants was determined using a human inflammatory cytokine kit BD™ Cytometric Bead Array (CBA) (BD Biosciences, San Jose, CA, USA) following instructions of the manufacturer. Supernatants were obtained from MSC cultured in supplemented IMDM, REH cells cultured in supplemented RPMI 1640, and co-cultures of MSC with REH cells, after three days of culture. Supernatants were centrifuged at 500× g for 7 min and filtered through a 0.22 µm pore membrane filter. A FACS flow cytometer was used to analyze samples. FlowJo and FCAP Array v3.0 Software (Becton Dickinson Biosciences, San Jose, CA, USA) were used for data analysis. Additionally, the Proteome Profiler Array/Human Cytokine Array (R&D Systems, Minneapolis, MN, USA), detecting more than 36 cytokines and growth factors, was used according to manufacturer’s instructions. Supernatants were obtained as above, except that they were collected after one day of culture.
4.14. Statistical Analysis
Cell proliferation, adhesion, and supernatant characterization data were analyzed using unpaired Student’s t-test. Cell cycle, cell surface, and primitive marker expression, migration, and colony forming unit data were analyzed using the Kruskal-Wallis test (non-parametric one-way ANOVA). The media comparison was made with Dunn’s multiple comparison test. GraphPad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA, USA) was used for mathematical calculations and graphics. Results were considered significant when p < 0.05.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}