
Splicing and Polyadenylation of Human Papillomavirus Type 16 mRNAs


Abstract
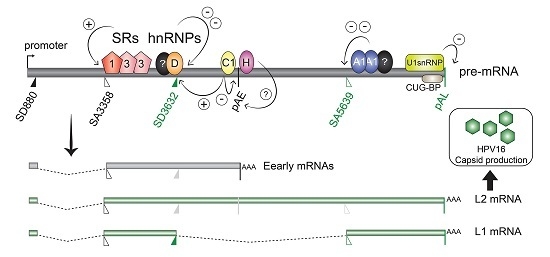
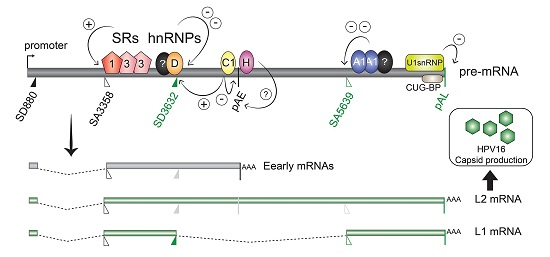
:
1. Introduction
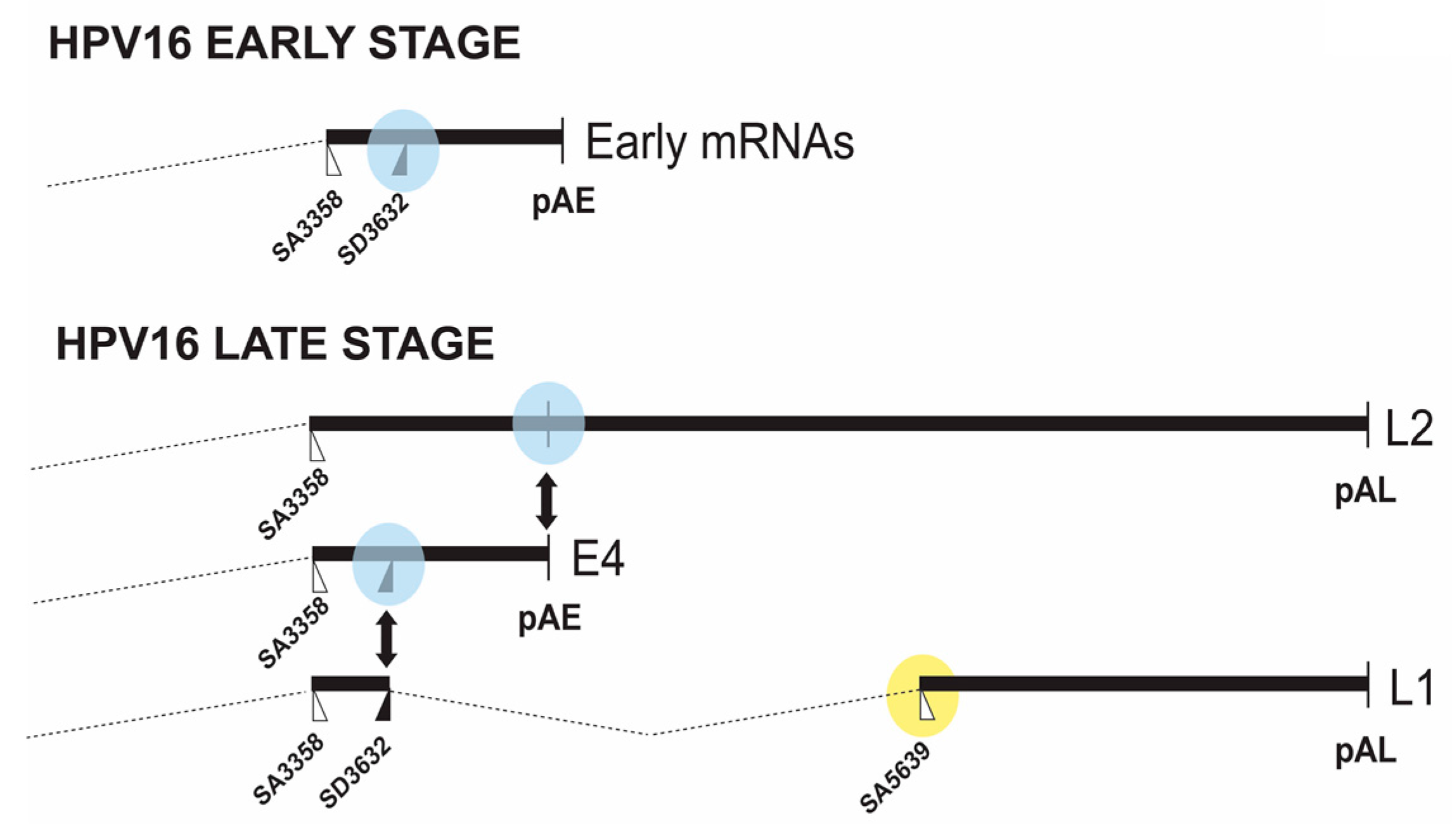
2. HPV16 Gene Regulation at the Levels of RNA Splicing and Polyadenylation
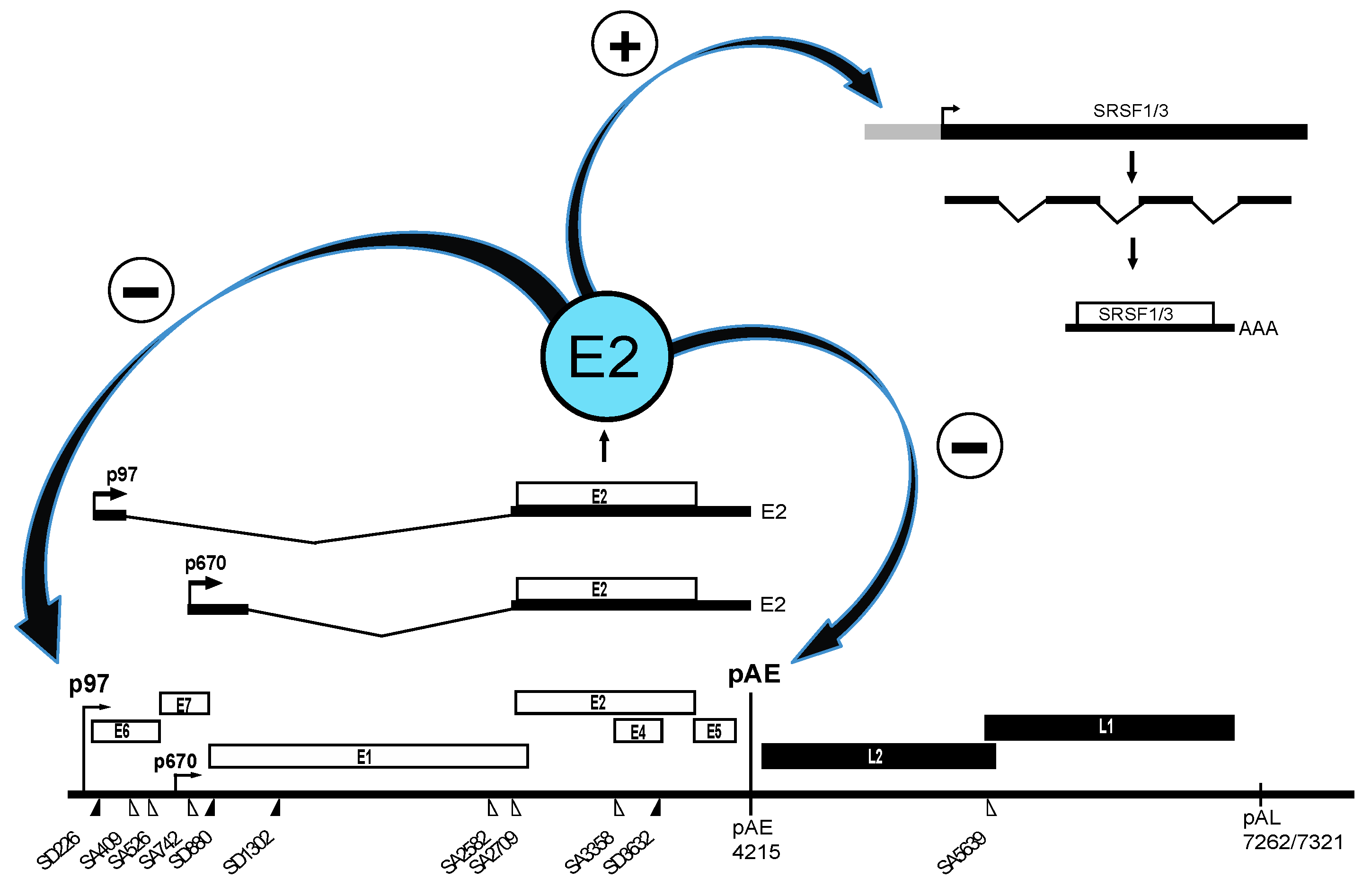
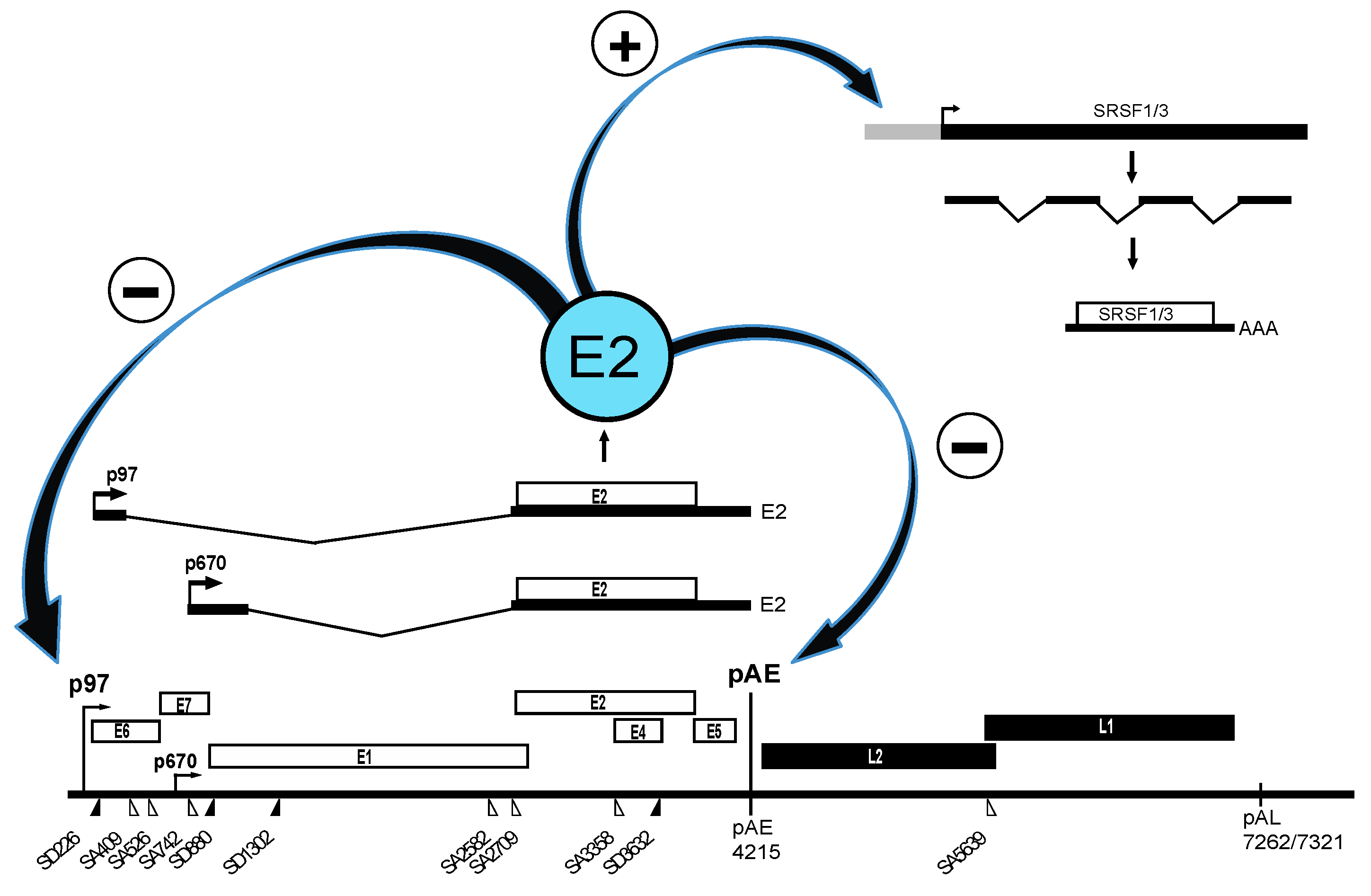
2.1. Upstream Region in HPV16
2.2. Middle Region in HPV16
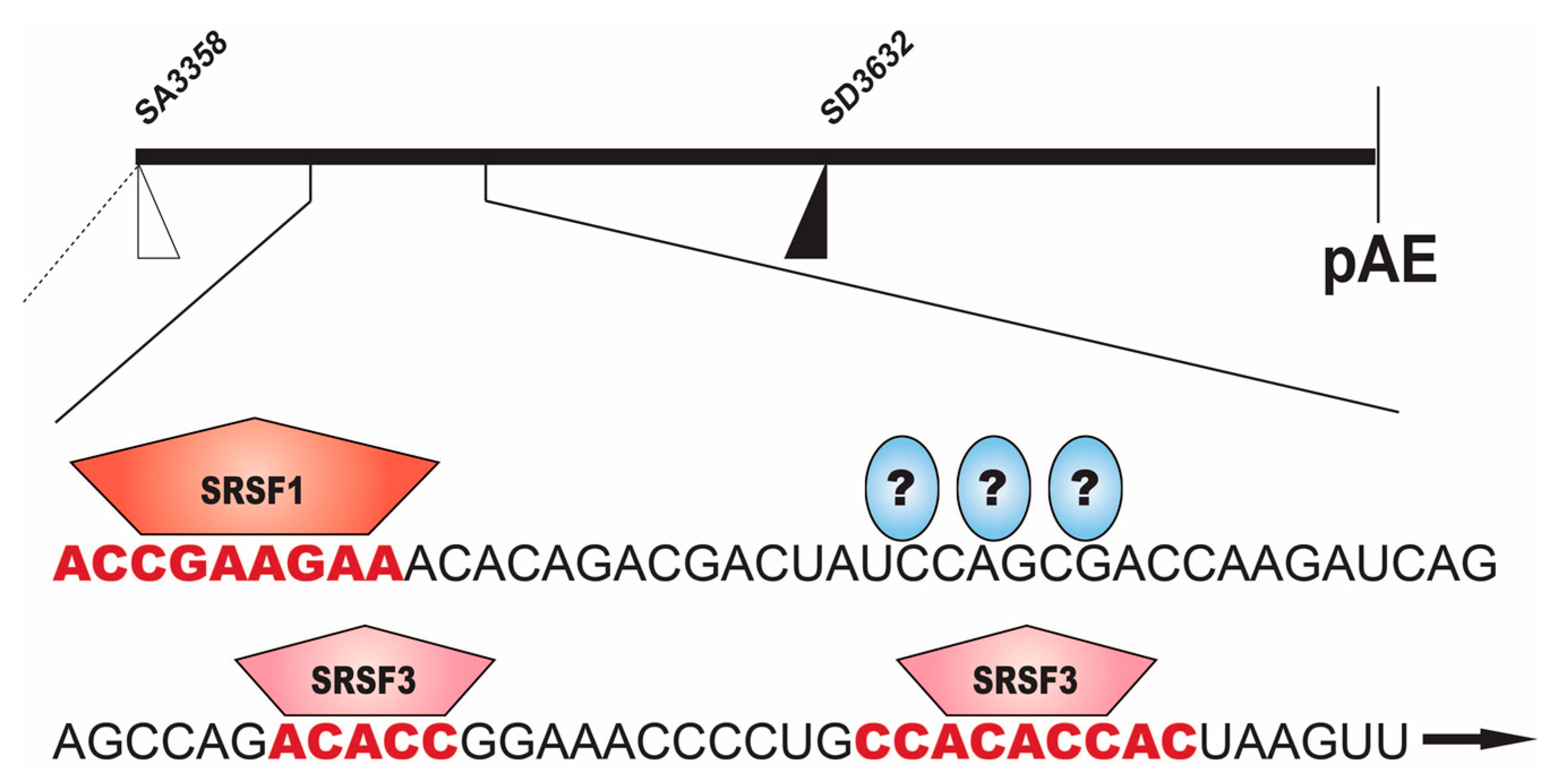
2.2.1. Regulation of HPV16 SA3358
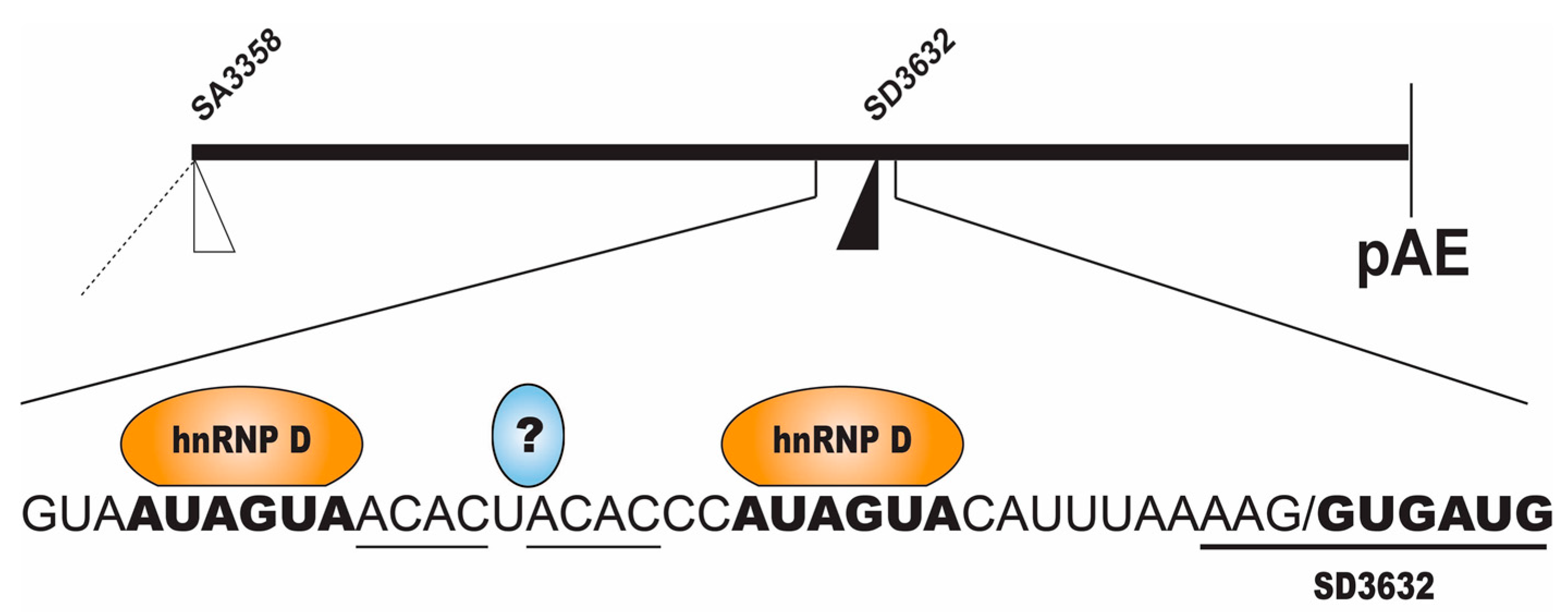
2.2.2. Regulation of HPV16 SD3632
2.2.3. Regulation of HPV16 Early PolyA Signal pAE
2.3. Downstream Region in HPV16
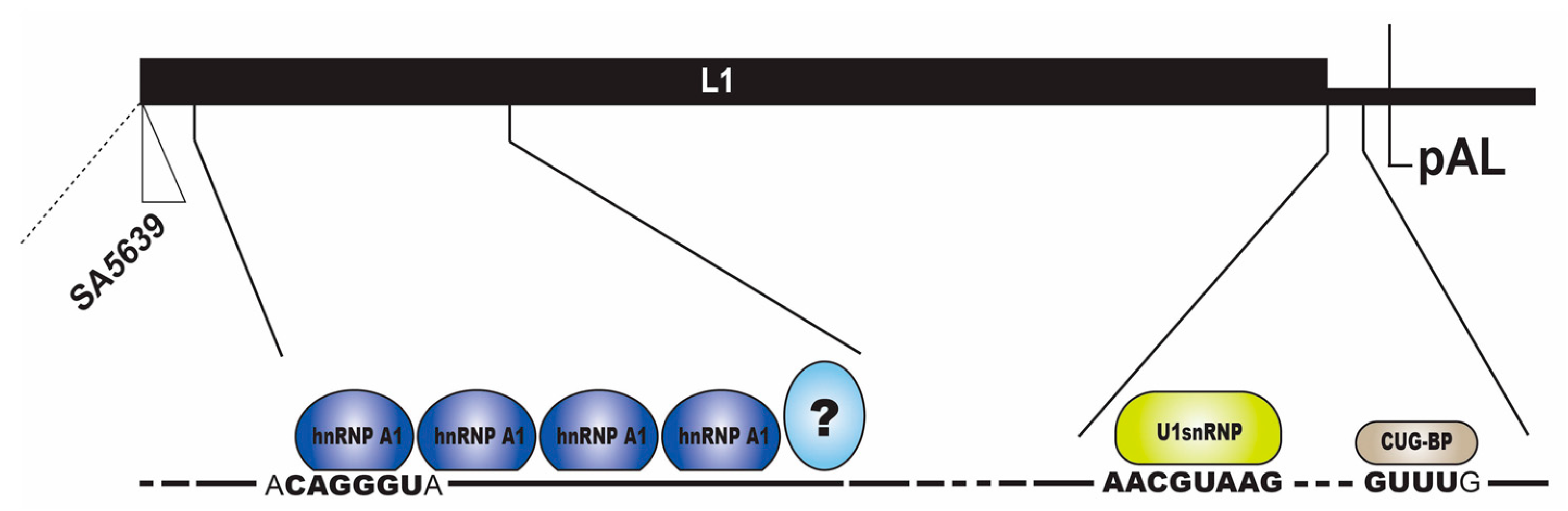
2.3.1. Regulation of HPV16 SA5639
2.3.2. Negative Regulatory Elements in the HPV16 Late Untranslated Region
3. Future Perspectives—Control of HPV16 RNA Processing through Transcription Coupled Transcription and Epigenetics
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Van Doorslaer, K. Evolution of the papillomaviridae. Virology 2013, 445, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.T.; Broker, T.R.; Steinberg, B.M. The natural history of human papillomavirus infections of the mucosal epithelia. APMIS 2010, 118, 422–449. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K.; Tan, Q.; Xirasagar, S.; Bandaru, S.; Gopalan, V.; Mohamoud, Y.; Huyen, Y.; McBride, A.A. The papillomavirus episteme: A central resource for papillomavirus sequence data and analysis. Nucleic Acids Res. 2013, 41, D571–D578. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Bosch, F.X.; Lorincz, A.; Munoz, N.; Meijer, C.J. The causal relation between human papillomavirus and cervical cancer. J. Clin. Pathol. 2002, 55, 244–265. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Schwartz, S. Regulation of human papillomavirus gene expression by splicing and polyadenylation. Nat. Rev. Microbiol. 2013, 11, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Zheng, Z.M. Regulation of bovine papillomavirus type 1 gene expression by RNA processing. Front. Biosci. 2009, 14, 1270–1282. [Google Scholar] [CrossRef]
- Kajitani, N.; Satsuka, A.; Kawate, A.; Sakai, H. Productive lifecycle of human papillomaviruses that depends upon squamous epithelial differentiation. Front. Microbiol. 2012, 3, 152. [Google Scholar] [CrossRef] [PubMed]
- Kajitani, N.; Schwartz, S. RNA binding proteins that control human papillomavirus gene expression. Biomolecules 2015, 5, 758–774. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S. Papillomavirus transcripts and posttranscriptional regulation. Virology 2013, 445, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Howley, P.M.; Schiller, J.C.; Lowy, D.R. Papillomaviruses. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott/The Williams & Wilkins: Philadelphia, PA, USA, 2006; Volume 2, pp. 2299–2354. [Google Scholar]
- Hong, S.; Laimins, L.A. Regulation of the life cycle of HPVs by differentiation and the DNA damage response. Future Microbiol. 2013, 8, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30, F55–F70. [Google Scholar] [CrossRef] [PubMed]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [PubMed]
- Vande Pol, S.B.; Klingelhutz, A.J. Papillomavirus E6 oncoproteins. Virology 2013, 445, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Kadaja, M.; Silla, T.; Ustav, E.; Ustav, M. Papillomavirus DNA replication—From initiation to genomic instability. Virology 2009, 384, 360–368. [Google Scholar] [CrossRef] [PubMed]
- McBride, A.A. The papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Bergvall, M.; Melendy, T.; Archambault, J. The E1 proteins. Virology 2013, 445, 35–56. [Google Scholar] [CrossRef] [PubMed]
- Thierry, F. Transcriptional regulation of the papillomavirus oncogenes by cellular and viral transcription factors in cervical carcinoma. Virology 2009, 384, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Bernard, H.U. Regulatory elements in the viral genome. Virology 2013, 445, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Cripe, T.P.; Haugen, T.H.; Turk, J.P.; Tabatabai, F.; Schmid, P.G., 3rd; Durst, M.; Gissmann, L.; Roman, A.; Turek, L.P. Transcriptional regulation of the human papillomavirus-16 E6-E7 promoter by a keratinocyte-dependent enhancer, and by viral E2 trans-activator and repressor gene products: Implications for cervical carcinogenesis. EMBO J. 1987, 6, 3745–3753. [Google Scholar] [PubMed]
- Johansson, C.; Somberg, M.; Li, X.; Backström Winquist, E.; Fay, J.; Ryan, F.; Pim, D.; Banks, L.; Schwartz, S. HPV-16 E2 contributes to induction of HPV-16 late gene expression by inhibiting early polyadenylation. EMBO J. 2012, 13, 3212–3227. [Google Scholar] [CrossRef] [PubMed]
- Klymenko, T.; Hernandez-Lopez, H.; MacDonald, A.I.; Bodily, J.M.; Graham, S.V. Human papillomavirus E2 regulates SRSF3 (SRp20) to promote capsid protein expression in infected differentiated keratinocytes. J. Virol. 2016, 90, 5047–5058. [Google Scholar] [CrossRef] [PubMed]
- Mole, S.; Milligan, S.G.; Graham, S.V. Human papillomavirus type 16 E2 protein transcriptionally activates the promoter of a key cellular splicing factor, SF2/ASF. J. Virol. 2009, 83, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Ajiro, M.; Jia, R.; Zhang, L.; Liu, X.; Zheng, Z.M. Intron definition and a branch site adenosine at nt 385 control RNA splicing of HPV16 E6*I and E7 expression. PLoS ONE 2012, 7, e46412. [Google Scholar] [CrossRef] [PubMed]
- Rosenberger, S.; De-Castro Arce, J.; Langbein, L.; Steenbergen, R.D.; Rosl, F. Alternative splicing of human papillomavirus type-16 E6/E6* early mRNA is coupled to EGF signaling via Erk1/2 activation. Proc. Natl. Acad. Sci. USA 2010, 107, 7006–7011. [Google Scholar] [CrossRef] [PubMed]
- Geuens, T.; Bouhy, D.; Timmerman, V. The hnRNP family: Insights into their role in health and disease. Hum. Genet. 2016, 135, 851–867. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Jimenez, F.; Sanchez-Margalet, V. Role of Sam68 in post-transcriptional gene regulation. Int. J. Mol. Sci. 2013, 14, 23402–23419. [Google Scholar] [CrossRef] [PubMed]
- Belaguli, N.S.; Pater, M.M.; Pater, A. Splice sites of human papillomavirus type 16 E6 gene or heterologous gene required for transformation by E7 and accumulation of E7 RNA. J. Med. Virol. 1995, 47, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Urrutia, E.; Valdes, J.; Bonilla-Moreno, R.; Martinez-Salazar, M.; Martinez-Garcia, M.; Berumen, J.; Villegas-Sepulveda, N. A few nucleotide polymorphisms are sufficient to recruit nuclear factors differentially to the intron 1 of HPV-16 intratypic variants. Virus Res. 2012, 166, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Ordonez, R.M.; Espinosa, A.M.; Sanchez-Gonzalez, D.J.; Armendariz-Borunda, J.; Berumen, J. Enhanced oncogenicity of Asian-American human papillomavirus 16 is associated with impaired E2 repression of E6/E7 oncogene transcription. J. Gen. Virol. 2004, 85, 1433–1444. [Google Scholar] [CrossRef] [PubMed]
- Fay, J.; Kelehan, P.; Lambkin, H.; Schwartz, S. Increased expression of cellular RNA-binding proteins in HPV-induced neoplasia and cervical cancer. J. Med. Virol. 2009, 81, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Dalstein, V.; Waterboer, T.; Clavel, C.; Gissman, L.; Pawlita, M. Diagnosing cervical cancer and high-grade precursors by HPV-16 transcription patterns. Cancer Res. 2010, 70, 249–256. [Google Scholar] [CrossRef]
- Somberg, M.; Schwartz, S. Multiple ASF/SF2 sites in the HPV-16 E4-coding region promote splicing to the most commonly used 3′-splice site on the HPV-16 genome. J. Virol. 2010, 84, 8219–8230. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Johansson, C.; Cardoso-Palacios, C.; Mossberg, A.; Dhanjal, S.; Bergvall, M.; Schwartz, S. Eight nucleotide substitutions inhibit splicing to HPV-16 3′-splice site SA3358 and reduce the efficiency by which HPV-16 increases the life span of primary human keratinocytes. PLoS ONE 2013, 8, e72776. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Liu, X.; Tao, M.; Kruhlak, M.; Guo, M.; Meyers, C.; Baker, C.C.; Zheng, Z.M. Control of the papillomavirus early-to-late switch by differentially expressed SRp20. J. Virol. 2009, 83, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Rio, D.C. Mechanisms and regulation of alternative pre-mRNA splicing. Annu. Rev. Biochem. 2015, 84, 291–323. [Google Scholar] [CrossRef] [PubMed]
- Somberg, M.; Li, X.; Johansson, C.; Orru, B.; Chang, R.; Rush, M.; Fay, J.; Ryan, F.; Schwartz, S. SRp30c activates human papillomavirus type 16 L1 mRNA expression via a bimodal mechanism. J. Gen. Virol. 2011, 92, 2411–2421. [Google Scholar] [CrossRef] [PubMed]
- Ajiro, M.; Tang, S.; Doorbar, J.; Zheng, Z.M. Serine/arginine-rich splicing factor 3 and heterogeneous nuclear ribonucleoprotein A1 regulate alternative RNA splicing and gene expression of human papillomavirus 18 through two functionally distinguishable cis elements. J. Virol. 2016, 90, 9138–9152. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Johansson, C.; Glahder, J.; Mossberg, A.K.; Schwartz, S. Suppression of HPV-16 late L1 5′-splice site SD3632 by binding of hnRNP D proteins and hnRNP A2/B1 to upstream AUAGUA RNA motifs. Nucleic Acids Res. 2013, 22, 10488–10508. [Google Scholar] [CrossRef] [PubMed]
- Rush, M.; Zhao, X.; Schwartz, S. A splicing enhancer in the E4 coding region of human papillomavirus type 16 is required for early mRNA splicing and polyadenylation as well as inhibition of premature late gene expression. J. Virol. 2005, 79, 12002–12015. [Google Scholar] [CrossRef] [PubMed]
- Andrew, E.M.; DiMaio, D. Hierarchy of polyadenylation site usage by bovine papillomavirus in transformed cells. J. Virol. 1993, 67, 7705–7710. [Google Scholar]
- Zhao, X.; Öberg, D.; Rush, M.; Fay, J.; Lambkin, H.; Schwartz, S. A 57 nucleotide upstream early polyadenylation element in human papillomavirus type 16 interacts with hFip1, CstF-64, hnRNP C1/C2 and PTB. J. Virol. 2005, 79, 4270–4288. [Google Scholar] [CrossRef] [PubMed]
- Terhune, S.S.; Hubert, W.G.; Thomas, J.T.; Laimins, L.A. Early polyadenylation signals of human papillomavirus type 31 negatively regulate capsid gene expression. J. Virol. 2001, 75, 8147–8157. [Google Scholar] [CrossRef] [PubMed]
- Terhune, S.S.; Milcarek, C.; Laimins, L.A. Regulation of human papillomavirus 31 polyadenylation during the differentiation-dependent life cycle. J. Virol. 1999, 73, 7185–7192. [Google Scholar] [PubMed]
- Oberg, D.; Collier, B.; Zhao, X.; Schwartz, S. Mutational inactivation of two distinct negative RNA elements in the human papillomavirus type 16 L2 coding region induces production of high levels of L2 in human cells. J. Virol. 2003, 77, 11674–11684. [Google Scholar] [CrossRef] [PubMed]
- Oberg, D.; Fay, J.; Lambkin, H.; Schwartz, S. A downstream polyadenylation element in human papillomavirus type 16 encodes multiple GGG-motifs and interacts with hnRNP H. J. Virol. 2005, 79, 9254–9269. [Google Scholar] [CrossRef] [PubMed]
- Dhanjal, S.; Kajitani, N.; Glahder, J.; Mossberg, A.K.; Johansson, C.; Schwartz, S. Heterogeneous nuclear ribonucleoprotein C proteins interact with the human papillomavirus type 16 (HPV16) early 3′-untranslated region and alleviate suppression of HPV16 late L1 mRNA splicing. J. Biol. Chem. 2015, 290, 13354–13371. [Google Scholar] [CrossRef] [PubMed]
- Simone, L.E.; Keene, J.D. Mechanisms coordinating ELAV/Hu mRNA regulons. Curr. Opin. Genet. Dev. 2013, 23, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Manley, J.L. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 2017, 18, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Somberg, M.; Zhao, X.; Fröhlich, M.; Evander, M.; Schwartz, S. PTB induces HPV-16 late gene expression by interfering with splicing inhibitory elements at the major late 5′-splice site SD3632. J. Virol. 2008, 82, 3665–3678. [Google Scholar] [CrossRef] [PubMed]
- Soeda, E.; Ferran, M.C.; Baker, C.C.; McBride, A.A. Repression of HPV16 early region transcription by the E2 protein. Virology 2006, 351, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V. Human papillomavirus E2 protein: Linking replication, transcription, and RNA processing. J. Virol. 2016, 90, 8384–8388. [Google Scholar] [CrossRef] [PubMed]
- Demeret, C.; Desaintes, C.; Yaniv, M.; Thierry, F. Different mechanisms contribute to the E2-mediated transcriptional repression of human papillomavirus type 18 viral oncogenes. J. Virol. 1997, 71, 9343–9349. [Google Scholar] [PubMed]
- Lai, M.C.; Teh, B.H.; Tarn, W.Y. A human papillomavirus E2 transcriptional activator. The interactions with cellular splicing factors and potential function in pre-mRNA processing. J. Biol. Chem. 1999, 274, 11832–11841. [Google Scholar] [CrossRef] [PubMed]
- Bodaghi, S.; Jia, R.; Zheng, Z.M. Human papillomavirus type 16 E2 and E6 are RNA-binding proteins and inhibit in vitro splicing of pre-mRNAs with suboptimal splice sites. Virology 2009, 386, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Gauson, E.J.; Windle, B.; Donaldson, M.M.; Caffarel, M.M.; Dornan, E.S.; Coleman, N.; Herzyk, P.; Henderson, S.C.; Wang, X.; Morgan, I.M. Regulation of human genome expression and RNA splicing by human papillomavirus 16 E2 protein. Virology 2014, 468, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Fay, J.; Lambkin, H.; Schwartz, S. Identification of a 17-nucleotide splicing enhancer in HPV-16 L1 that counteracts the effect of multiple hnRNP A1-binding splicing silencers. Virology 2007, 369, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Rush, M.; Schwartz, S. Identification of an hnRNP A1 dependent splicing silencer in the HPV-16 L1 coding region that prevents premature expression of the late L1 gene. J. Virol. 2004, 78, 10888–10905. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Schwartz, S. Inhibition of HPV-16 L1 expression from L1 cDNAs correlates with the presence of hnRNP A1 binding sites in the L1 coding region. Virus Genes 2008, 36, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Collier, B.; Öberg, D.; Zhao, X.; Schwartz, S. Specific inactivation of inhibitory sequences in the 5′ end of the human papillomavirus type 16 L1 open reading frame results in production of high levels of L1 protein in human epithelial cells. J. Virol. 2002, 76, 2739–2752. [Google Scholar] [CrossRef] [PubMed]
- Graham, S.V. Papillomavirus 3′ UTR regulatory elements. Front. Biosci. 2008, 13, 5646–5663. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Rush, M.; Carlsson, A.; Schwartz, S. The presence of inhibitory RNA elements in the late 3′-untranslated region is a conserved property of human papillomaviruses. Virus Res. 2007, 125, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Cumming, S.A.; McPhillips, M.G.; Veerapraditsin, T.; Milligan, S.G.; Graham, S.V. Activity of the human papillomavirus type 16 late negative regulatory element is partly due to four weak consensus 5′ splice sites that bind a U1 snRNP-like complex. J. Virol. 2003, 77, 5167–5177. [Google Scholar] [CrossRef] [PubMed]
- Furth, P.A.; Choe, W.T.; Rex, J.H.; Byrne, J.C.; Baker, C.C. Sequences homologous to 5′ splice sites are required for the inhibitory activity of papillomavirus late 3′ untranslated regions. Mol. Cell. Biol. 1994, 14, 5278–5289. [Google Scholar] [CrossRef] [PubMed]
- Goraczniak, R.; Gunderson, S.I. The regulatory element in the 3′ untranslated region of human papillomavirus 16 inhibits expression by binding CUG binding protein 1. J. Biol. Chem. 2008, 283, 2286–2296. [Google Scholar] [CrossRef] [PubMed]
- Furth, P.A.; Baker, C.C. An element in the bovine papillomavirus late 3′ untranslated region reduces polyadenylated cytoplasmic RNA levels. J. Virol. 1991, 65, 5806–5812. [Google Scholar] [PubMed]
- Barksdale, S.K.; Baker, C.C. The human immunodeficiency virus type 1 Rev protein and the Rev-responsive element counteract the effect of an inhibitory 5′ splice site in a 3′ untranslated region. Mol. Cell. Biol. 1995, 15, 2962–2971. [Google Scholar] [CrossRef] [PubMed]
- Cumming, S.A.; Repellin, C.E.; McPhilips, M.; Redford, J.C.; Clements, J.B.; Graham, S.V. The human papillomavirus type 31 untranslated region contains a complex bipartite negative regulatory element. J. Virol. 2002, 76, 5993–6003. [Google Scholar] [CrossRef] [PubMed]
- Koffa, M.D.; Graham, S.V.; Takagaki, Y.; Manley, J.L.; Clements, J.B. The human papillomavirus type 16 negative regulatory RNA element interacts with three proteins that act at different posttranscriptional levels. Proc. Natl. Acad. Sci. USA 2000, 97, 4677–4682. [Google Scholar] [CrossRef] [PubMed]
- Cheunim, T.; Zhang, J.; Milligan, S.G.; McPhillips, M.G.; Graham, S.V. The alternative splicing factor hnRNP A1 is up-regulated during virus-infected epithelial cell differentiation and binds the human papillomavirus type 16 late regulatory element. Virus Res. 2008, 131, 189–198. [Google Scholar] [CrossRef] [PubMed]
- McPhillips, M.G.; Veerapraditsin, T.; Cumming, S.A.; Karali, D.; Milligan, S.G.; Boner, W.; Morgan, I.M.; Graham, S.V. SF2/ASF binds the human papillomavirus type 16 late RNA control element and is regulated during differentiation of virus-infected epithelial cells. J. Virol. 2004, 78, 10598–10605. [Google Scholar] [CrossRef] [PubMed]
- Cumming, S.A.; Chuen-Im, T.; Zhang, J.; Graham, S.V. The RNA stability regulator HuR regulates L1 protein expression in vivo in differentiating cervical epithelial cells. Virology 2009, 383, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Sokolowski, M.; Furneaux, H.; Schwartz, S. The inhibitory activity of the AU-rich RNA element in the human papillomavirus type 1 late 3′ untranslated region correlates with its affinity for the elav-like HuR protein. J. Virol. 1999, 73, 1080–1091. [Google Scholar] [PubMed]
- Sokolowski, M.; Zhao, C.; Tan, W.; Schwartz, S. AU-rich mRNA instability elements on human papillomavirus type 1 late mRNAs and c-fos mRNAs interact with the same cellular factors. Oncogene 1997, 15, 2303–2319. [Google Scholar] [CrossRef] [PubMed]
- Wiklund, L.; Sokolowski, M.; Carlsson, A.; Rush, M.; Schwartz, S. Inhibtion of translation by UAUUUAU and UAUUUUUAU motifs of the AU-rich RNA instability in the HPV-1 late 3′ untranslated region. J. Biol. Chem. 2002, 277, 40462–40471. [Google Scholar] [CrossRef] [PubMed]
- Felber, B.K.; Pavlakis, G.N. Molecular biology of HIV-1: Positive and negative regulatory elements important for virus expression. AIDS 1993, 7, S51–S62. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Schwartz, S. The Rev protein of human immunodeficiency virus type 1 counteracts the effect of an AU-rich negative element in the human papillomavirus type 1 late 3′ untranslated region. J. Virol. 1995, 69, 2932–2945. [Google Scholar] [PubMed]
- Tan, W.; Felber, B.K.; Zolotukhin, A.S.; Pavlakis, G.N.; Schwartz, S. Efficient expression of the human papillomavirus type 16 L1 protein in epithelial cells by using Rev and the Rev-responsive element of human immunodeficiency virus or the cis-acting transactivation element of simian retrovirus type 1. J. Virol. 1995, 69, 5607–5620. [Google Scholar] [PubMed]
- Clarke, M.A.; Wentzensen, N.; Mirabello, L.; Ghosh, A.; Wacholder, S.; Harari, A.; Lorincz, A.; Schiffman, M.; Burk, R.D. Human papillomavirus DNA methylation as a potential biomarker for cervical cancer. Cancer Epidemiol. Biomark. Prev. 2012, 21, 2125–2137. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Fattah, T.J.; Yu, H.; Nygren, J.; Mossberg, A.K.; Schwartz, S. Acetylation of intragenic histones on HPV16 correlates with enhanced HPV16 gene expression. Virology 2015, 482, 244–259. [Google Scholar] [CrossRef] [PubMed]
- Johannsen, E.; Lambert, P.F. Epigenetics of human papillomaviruses. Virology 2013, 445, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Paris, C.; Pentland, I.; Groves, I.; Roberts, D.C.; Powis, S.J.; Coleman, N.; Roberts, S.; Parish, J.L. CCCTC-binding factor recruitment to the early region of the human papillomavirus type 18 genome regulates viral oncogene expression. J. Virol. 2015, 89, 4770–4785. [Google Scholar] [CrossRef] [PubMed]
- Pentland, I.; Parish, J.L. Targeting CTCF to control virus gene expression: A common theme amongst diverse DNA viruses. Viruses 2015, 7, 3574–3585. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HPV16 Protein | Protein Function |
|---|---|
| E1 | Replication of viral DNA |
| E2 | Replication of viral DNA |
| E4 | Assists at egress of virus |
| E5 | Promitotic function |
| E6 | Antiapoptotic function |
| E7 | Promitotic function |
| L1 | Major capsid protein |
| L2 | Minor capsid protein |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.; Kajitani, N.; Schwartz, S. Splicing and Polyadenylation of Human Papillomavirus Type 16 mRNAs. Int. J. Mol. Sci. 2017, 18, 366. https://doi.org/10.3390/ijms18020366
Wu C, Kajitani N, Schwartz S. Splicing and Polyadenylation of Human Papillomavirus Type 16 mRNAs. International Journal of Molecular Sciences. 2017; 18(2):366. https://doi.org/10.3390/ijms18020366
Chicago/Turabian StyleWu, Chengjun, Naoko Kajitani, and Stefan Schwartz. 2017. "Splicing and Polyadenylation of Human Papillomavirus Type 16 mRNAs" International Journal of Molecular Sciences 18, no. 2: 366. https://doi.org/10.3390/ijms18020366