
Endoplasmic Reticulum (ER) Stress and Endocrine Disorders

Abstract
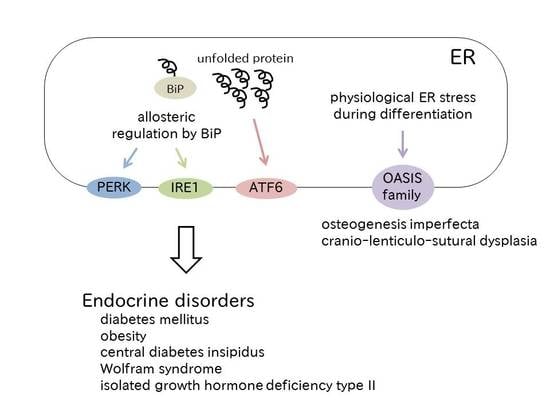
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Unfolded Protein Responses in Mammals
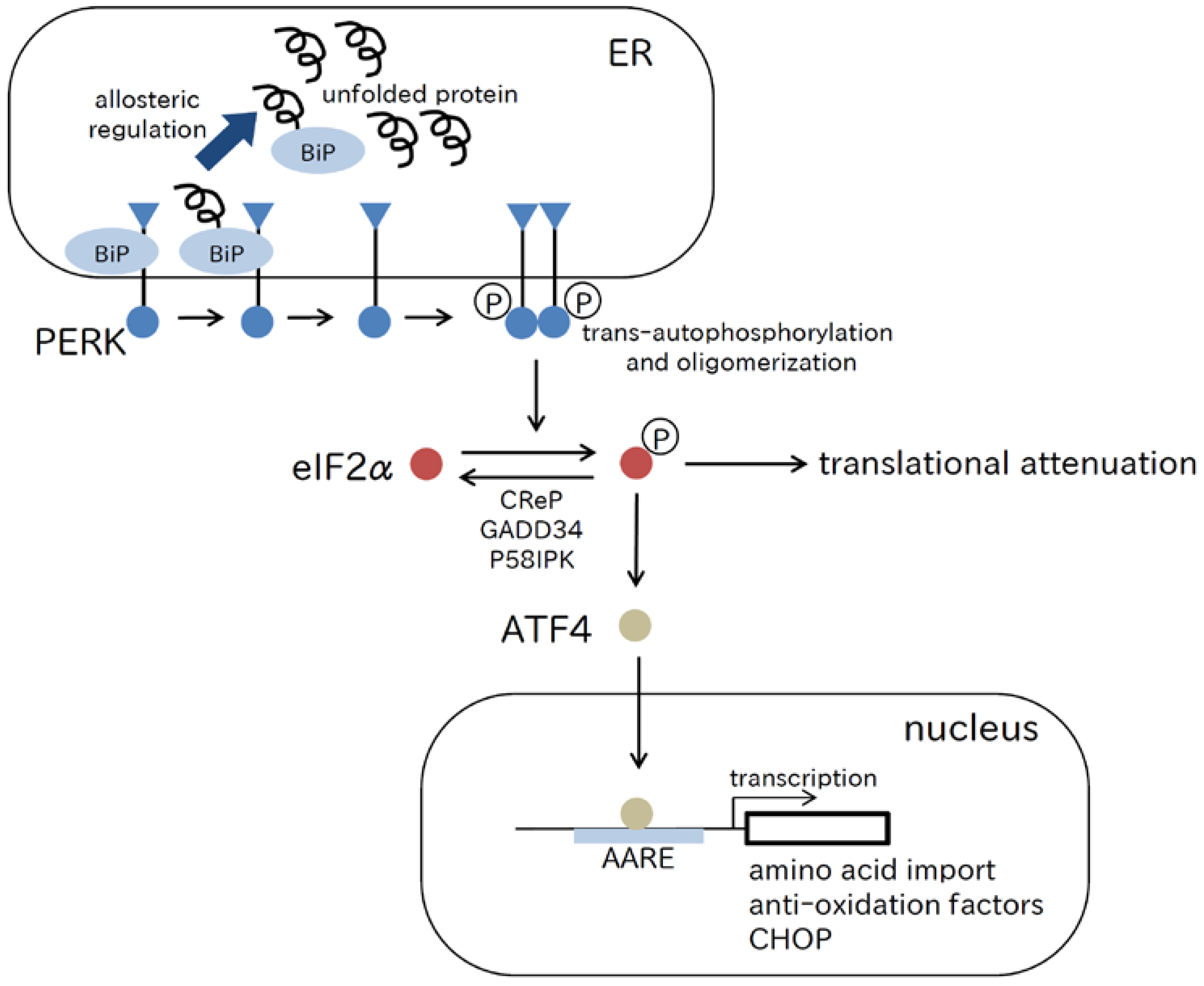
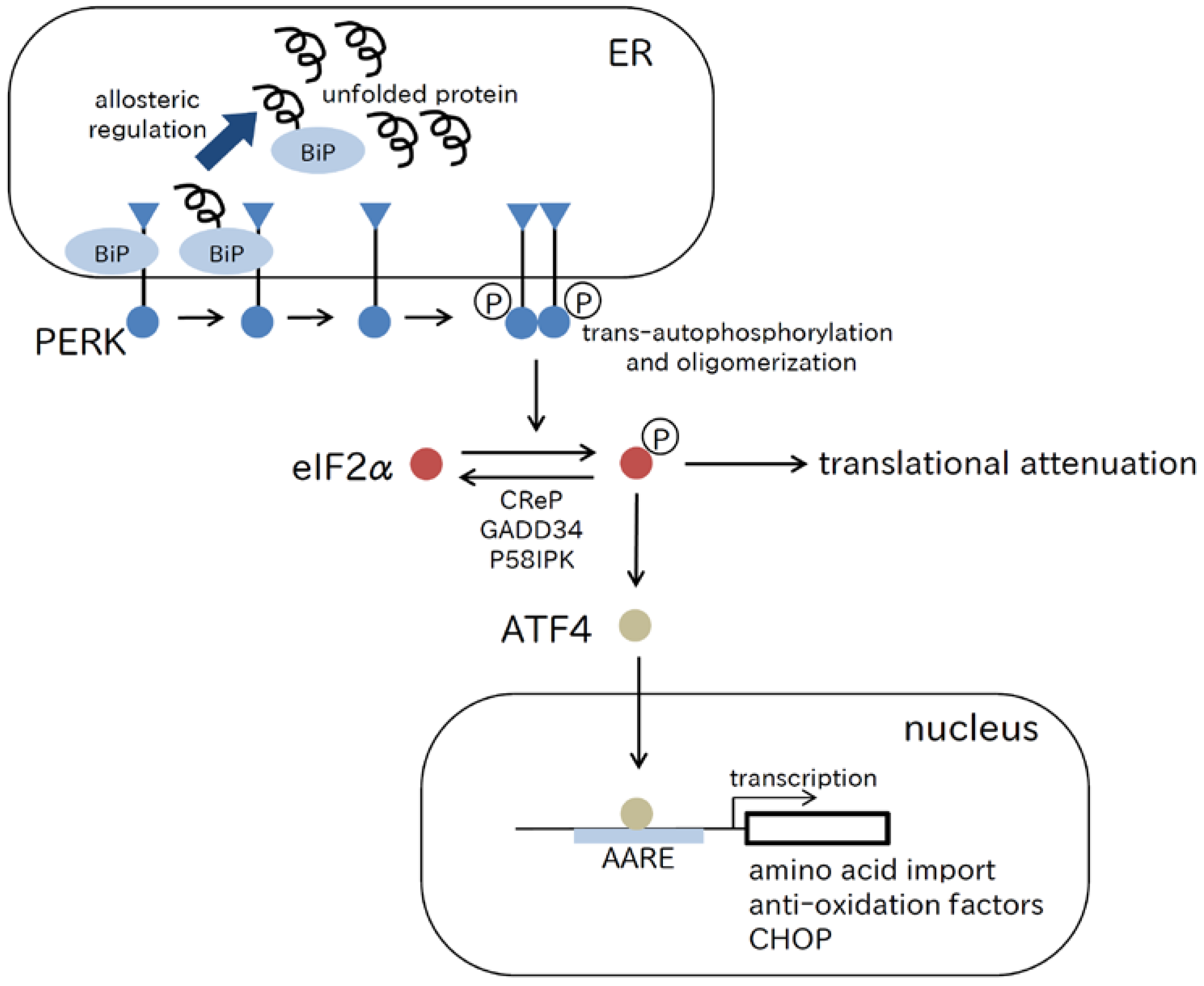
2.1. PERK Pathway
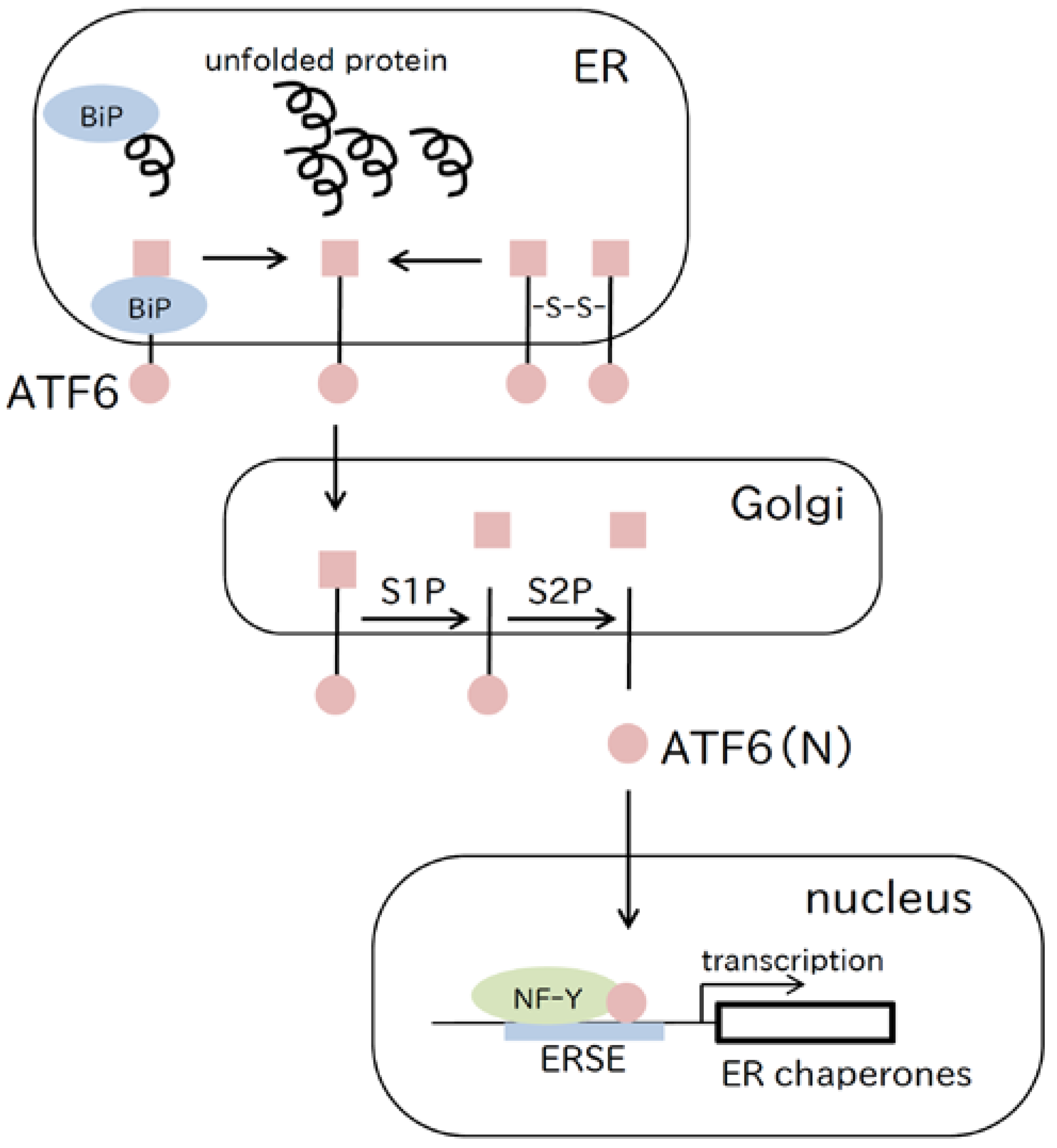
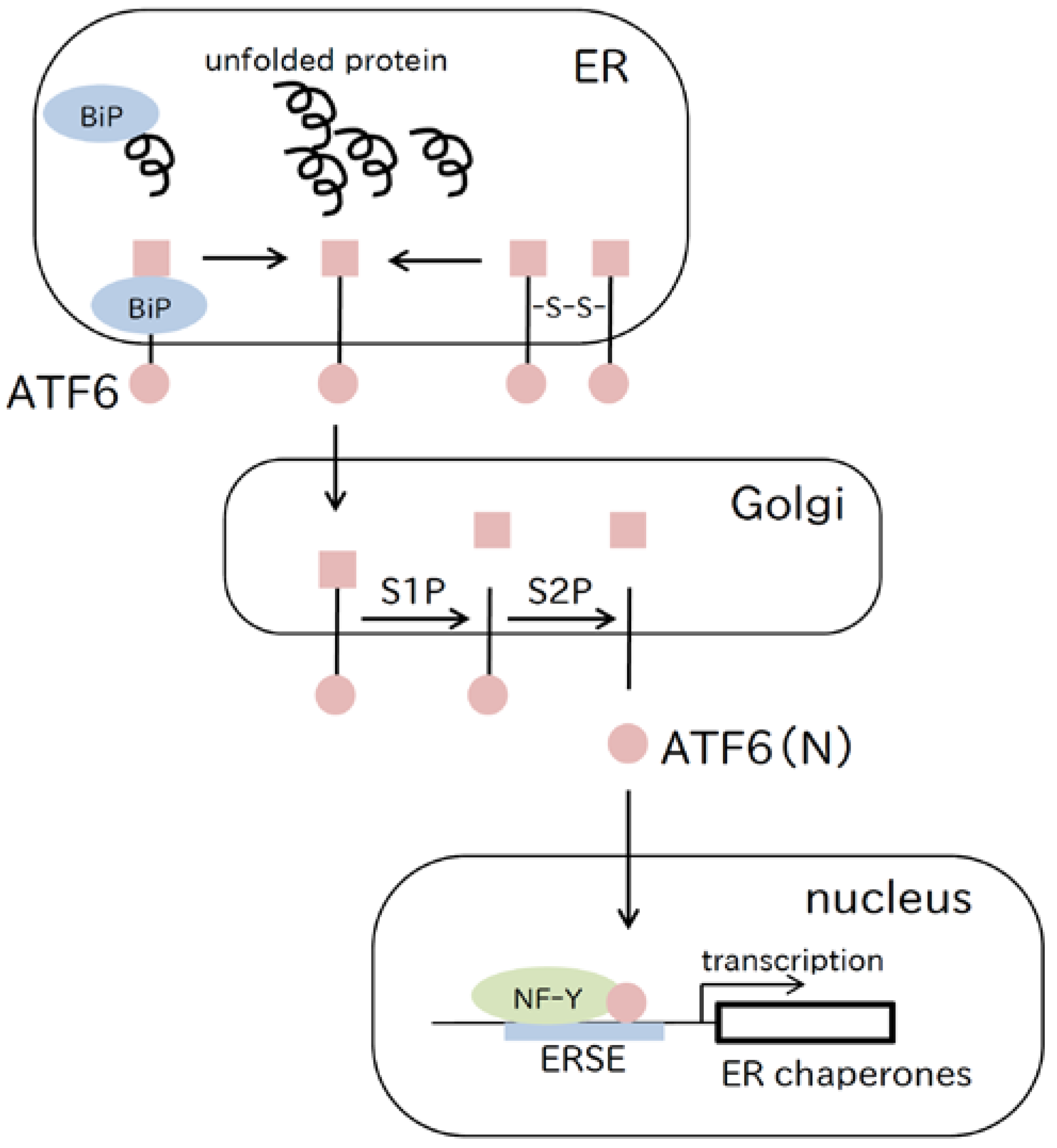
2.2. ATF6 Pathway
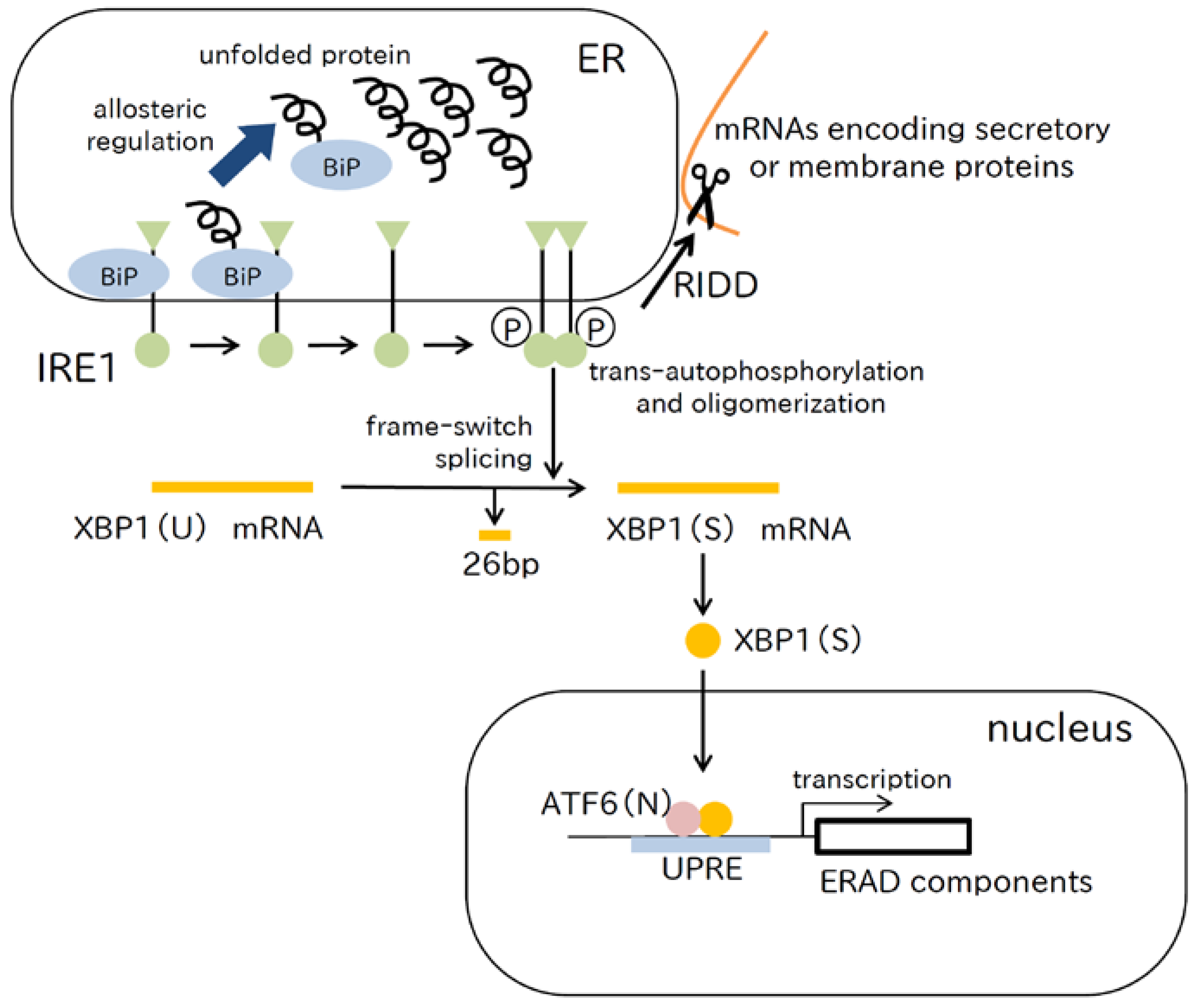
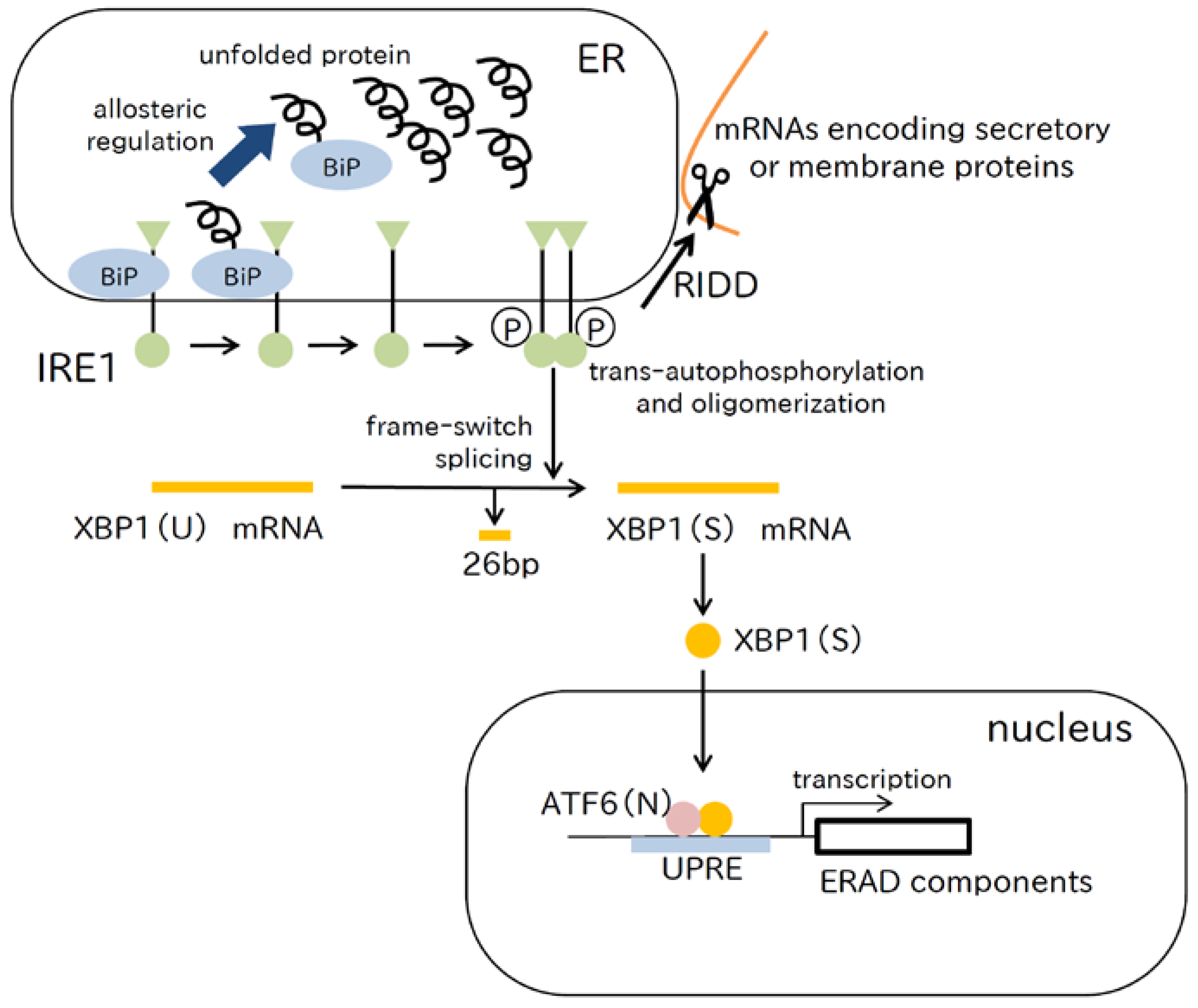
2.3. IRE1 Pathway
2.4. Phase Shift of the ER Stress Pathways
2.5. Apoptosis-Inducing Pathways
2.6. ER Stress-Independent Functions of the UPR
2.7. OASIS Family
3. ER Stress and Endocrine Disorders
3.1. Diabetes Mellitus
3.2. Obesity (Adiponectin and Leptin)
3.3. Central Diabetes Insipidus (CDI)
3.4. Wolfram Syndrome
3.5. Isolated Growth Hormone Deficiency Type II (IGHD2)
4. Treatment of ER Stress-Related Diseases
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ahner, A.; Brodsky, J.L. Checkpoints in ER-associated degradation: Excuse me, which way to the proteasome? Trends Cell Biol. 2004, 14, 474–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, H. ER stress and diseases. FEBS J. 2007, 274, 630–658. [Google Scholar] [CrossRef] [PubMed]
- Katayama, T.; Imaizumi, K.; Sato, N.; Miyoshi, K.; Kudo, T.; Hitomi, J.; Morihara, T.; Yoneda, T.; Gomi, F.; Mori, Y.; et al. Presenilin-1 mutations downregulate the signalling pathway of the unfolded-protein response. Nat. Cell Biol. 1999, 1, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Shimura, H.; Hattori, N.; Kubo, S.; Mizuno, Y.; Asakawa, S.; Minoshima, S.; Shimizu, N.; Iwai, K.; Chiba, T.; Tanaka, K.; et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 2000, 25, 302–305. [Google Scholar] [PubMed]
- Schaffar, G.; Breuer, P.; Boteva, R.; Behrends, C.; Tzvetkov, N.; Strippel, N.; Sakahira, H.; Siegers, K.; Hayer-Hartl, M.; Hartl, F.U. Cellular toxicity of polyglutamine expansion proteins: Mechanism of transcription factor deactivation. Mol. Cell 2004, 15, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Cui, J.; He, Q.; Chen, Z.; Arvan, P.; Liu, M. Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes. Mol. Asp. Med. 2015, 42, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Christis, C.; Fullaondo, A.; Schildknegt, D.; Mkrtchian, S.; Heck, A.J.; Braakman, I. Regulated increase in folding capacity prevents unfolded protein stress in the ER. J. Cell Sci. 2010, 123, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Haataja, L.; Manickam, N.; Soliman, A.; Tsai, B.; Liu, M.; Arvan, P. Disulfide mispairing during proinsulin folding in the endoplasmic reticulum. Diabetes 2016, 65, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Baglioni, S.; Corona, G.; Maggi, M.; Serio, M.; Peri, A. Identification of a novel mutation in the arginine vasopressin-neurophysin II gene affecting the sixth intrachain disulfide bridge of the neurophysin II moiety. Eur. J. Endocrinol. 2004, 151, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Arima, H.; Morishita, Y.; Hagiwara, D.; Hayashi, M.; Oiso, Y. Endoplasmic reticulum stress in vasopressin neurons of familial diabetes insipidus model mice: Aggregate formation and mRNA poly(A) tail shortening. Exp. Physiol. 2014, 99, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Li, J.; Ron, D.; Sha, B. The structure of the PERK kinase domain suggests the mechanism for its activation. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Carrara, M.; Prischi, F.; Nowak, P.R.; Kopp, M.C.; Ali, M.M. Noncanonical binding of BiP ATPase domain to Ire1 and Perk is dissociated by unfolded protein CH1 to initiate ER stress signaling. Elife 2015, 4, e03522. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Tsaytler, P.; Harding, H.P.; Ron, D.; Bertolotti, A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Das, I.; Krzyzosiak, A.; Schneider, K.; Wrabetz, L.; D’Antonio, M.; Barry, N.; Sigurdardottir, A.; Bertolotti, A. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science 2015, 348, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Liu, Z.; Lv, Y.; Zhao, N.; Guan, G.; Wang, J. Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death Dis. 2015, 6, e1822. [Google Scholar] [CrossRef] [PubMed]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournou, X.P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Li, F.; Shi, K.; Wu, P.; An, J.; Yang, Y.; Xu, C. Involvement of p38 in signal switching from autophagy to apoptosis via the Perk/eIF2α/ATF4 axis in selenite-treated NB4 cells. Cell Death Dis. 2014, 5, e1270. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Chen, X.; Shen, J.; Prywes, R. The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J. Biol. Chem. 2002, 277, 13045–13052. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Snapp, E.L.; Lippincott-Schwartz, J.; Prywes, R. Stable binding of ATF6 to BiP in the endoplasmic reticulum stress response. Mol. Cell. Biol. 2005, 25, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Schindler, A.J.; Schekman, R. In vitro reconstitution of ER-stress induced ATF6 transport in COPII vesicles. Proc. Natl. Acad. Sci. USA 2009, 106, 17775–17780. [Google Scholar] [CrossRef] [PubMed]
- Nadanaka, S.; Okada, T.; Yoshida, H.; Mori, K. Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol. Cell. Biol. 2007, 27, 1027–1043. [Google Scholar] [CrossRef] [PubMed]
- Nadanaka, S.; Yoshida, H.; Mori, K. Reduction of disulfide bridges in the lumenal domain of ATF6 in response to glucose starvation. Cell Struct. Funct. 2006, 31, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Nadanaka, S.; Okada, T.; Okawa, K.; Mori, K. Luminal domain of ATF6 alone is sufficient for sensing endoplasmic reticulum stress and subsequent transport to the Golgi apparatus. Cell Struct. Funct. 2011, 36, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Haze, K.; Yanagi, H.; Yura, T.; Mori, K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 1998, 273, 33741–33749. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Yamamoto, K.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct. Funct. 2008, 33, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Bommiasamy, H.; Back, S.H.; Fagone, P.; Lee, K.; Meshinchi, S.; Vink, E.; Sriburi, R.; Frank, M.; Jackowski, S.; Kaufman, R.J.; et al. ATF6α induces XBP1-independent expansion of the endoplasmic reticulum. J. Cell Sci. 2009, 122, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Belmont, P.J.; Chen, W.J.; San Pedro, M.N.; Thuerauf, D.J.; Gellings Lowe, N.; Gude, N.; Hilton, B.; Wolkowicz, R.; Sussman, M.A.; Glembotski, C.C. Roles for endoplasmic reticulum-associated degradation and the novel endoplasmic reticulum stress response gene Derlin-3 in the ischemic heart. Circ. Res. 2010, 106, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef] [PubMed]
- Haze, K.; Okada, T.; Yoshida, H.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. Identification of the G13 (cAMP-response-element-binding protein-related protein) gene product related to activating transcription factor 6 as a transcriptional activator of the mammalian unfolded protein response. Biochem. J. 2001, 355, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.; Kaufman, R.J. ATF6α optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 2007, 13, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, A.; Kamide, T.; Hashida, K.; Ta, H.M.; Inahata, Y.; Takarada-Iemata, M.; Hattori, T.; Mori, K.; Takahashi, R.; Matsuyama, T.; et al. Deletion of ATF6α impairs astroglial activation and enhances neuronal death following brain ischemia in mice. J. Neurochem. 2015, 132, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Okada, T.; Ishikawa-Fujiwara, T.; Todo, T.; Kamei, Y.; Shigenobu, S.; Tanaka, M.; Saito, T.L.; Yoshimura, J.; Morishita, S.; et al. ATF6α/β-mediated adjustment of ER chaperone levels is essential for development of the notochord in medaka fish. Mol. Biol. Cell 2013, 24, 1387–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, T.; Taniguchi, Y.; Okada, T.; Takeda, S.; Mori, K. Vertebrate unfolded protein response: Mammalian signaling pathways are conserved in medaka fish. Cell Struct. Funct. 2011, 36, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, C.M.; Walter, P. Ceapins inhibit ATF6α signaling by selectively preventing transport of ATF6α to the Golgi apparatus during ER stress. Elife 2016, 5, e11880. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, C.M.; Garri, C.; Cain, E.L.; Ang, K.K.; Wilson, C.G.; Chen, S.; Hearn, B.R.; Jaishankar, P.; Aranda-Diaz, A.; Arkin, M.R.; et al. Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6α branch. Elife 2016, 5, e11878. [Google Scholar] [CrossRef] [PubMed]
- Mori, K. Signalling pathways in the unfolded protein response: Development from yeast to mammals. J. Biochem. 2009, 146, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Kimata, Y.; Ishiwata-Kimata, Y.; Ito, T.; Hirata, A.; Suzuki, T.; Oikawa, D.; Takeuchi, M.; Kohno, K. Two regulatory steps of ER-stress sensor IRE1 involving its cluster formation and interaction with unfolded proteins. J. Cell Biol. 2007, 179, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Gardner, B.M.; Walter, P. Unfolded proteins are IRE1-activating ligands that directly induce the unfolded protein response. Science 2011, 333, 1891–1894. [Google Scholar] [CrossRef] [PubMed]
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harbor Perspect. Biol. 2013, 5, a013169. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Yanagitani, K.; Imagawa, Y.; Iwawaki, T.; Hosoda, A.; Saito, M.; Kimata, Y.; Kohno, K. Cotranslational targeting of XBP1 protein to the membrane promotes cytoplasmic splicing of its own mRNA. Mol. Cell 2009, 34, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Yanagitani, K.; Kimata, Y.; Kadokura, H.; Kohno, K. Translational pausing ensures membrane targeting and cytoplasmic splicing of XBP1U mRNA. Science 2011, 331, 586–589. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Oku, M.; Suzuki, M.; Mori, K. pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J. Cell Biol. 2006, 172, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Uemura, A.; Taniguchi, M.; Matsuo, Y.; Oku, M.; Wakabayashi, S.; Yoshida, H. UBC9 regulates the stability of XBP1, a key transcription factor controlling the ER stress response. Cell Struct. Funct. 2013, 38, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated IRE1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, D.; Tokuda, M.; Hosoda, A.; Iwawaki, T. Identification of a consensus element recognized and cleaved by IRE1α. Nucleic Acids Res. 2010, 38, 6265–6273. [Google Scholar] [CrossRef] [PubMed]
- Upton, J.P.; Wang, L.; Han, D.; Wang, E.S.; Huskey, N.E.; Lim, L.; Truitt, M.; McManus, M.T.; Ruggero, D.; Goga, A.; et al. IRE1α cleaves select microRNAs during ER stress to derepress translation of proapoptotic Caspase-2. Science 2012, 338, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting ridd of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Kanekura, K.; Ma, X.; Murphy, J.T.; Zhu, L.J.; Diwan, A.; Urano, F. IRE1 prevents endoplasmic reticulum membrane permeabilization and cell death under pathological conditions. Sci. Signal. 2015, 8, ra62. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Hosokawa, N.; Kaufman, R.J.; Nagata, K.; Mori, K. A time-dependent phase shift in the mammalian unfolded protein response. Dev. Cell 2003, 4, 265–271. [Google Scholar] [CrossRef]
- Oyadomari, S.; Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004, 11, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Tsuru, A.; Imai, Y.; Saito, M.; Kohno, K. Novel mechanism of enhancing IRE1α-XBP1 signalling via the PERK-ATF4 pathway. Sci. Rep. 2016, 6, 24217. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Okada, T.; Haze, K.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 2000, 20, 6755–6767. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, I.; Weiwad, M.; Prell, E.; Ferrari, D.M. ERP29 deficiency affects sensitivity to apoptosis via impairment of the ATF6-CHOP pathway of stress response. Apoptosis 2014, 19, 801–815. [Google Scholar] [CrossRef] [PubMed]
- Frand, A.R.; Kaiser, C.A. The ERO1 gene of yeast is required for oxidation of protein dithiols in the endoplasmic reticulum. Mol. Cell 1998, 1, 161–170. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Wang, H.G. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem. 2004, 279, 45495–45502. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef] [PubMed]
- Mauro, C.; Crescenzi, E.; De Mattia, R.; Pacifico, F.; Mellone, S.; Salzano, S.; de Luca, C.; D’Adamio, L.; Palumbo, G.; Formisano, S.; et al. Central role of the scaffold protein tumor necrosis factor receptor-associated factor 2 in regulating endoplasmic reticulum stress-induced apoptosis. J. Biol. Chem. 2006, 281, 2631–2638. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, J.; Katayama, T.; Eguchi, Y.; Kudo, T.; Taniguchi, M.; Koyama, Y.; Manabe, T.; Yamagishi, S.; Bando, Y.; Imaizumi, K.; et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Aβ-induced cell death. J. Cell Biol. 2004, 165, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Binet, F.; Chiasson, S.; Girard, D. Evidence that endoplasmic reticulum (ER) stress and caspase-4 activation occur in human neutrophils. Biochem. Biophys. Res. Commun. 2010, 391, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.A.; Zhang, Z.; Svetlov, S.I.; Hayes, R.L.; Wang, K.K.; Larner, S.F. Calpain and caspase processing of caspase-12 contribute to the ER stress-induced cell death pathway in differentiated PC12 cells. Apoptosis 2010, 15, 1480–1493. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Yuan, J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J. Cell Biol. 2000, 150, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Chen, X.; Lee, A.H.; Glimcher, L.H. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat. Immunol. 2010, 11, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.W.; Kutzler, L.; Kimball, S.R.; Tabas, I. Toll-like receptor activation suppresses ER stress factor CHOP and translation inhibition through activation of eIF2B. Nat. Cell Biol. 2012, 14, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Reimold, A.M.; Iwakoshi, N.N.; Manis, J.; Vallabhajosyula, P.; Szomolanyi-Tsuda, E.; Gravallese, E.M.; Friend, D.; Grusby, M.J.; Alt, F.; Glimcher, L.H. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001, 412, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.C.; Dougan, S.K.; McGehee, A.M.; Love, J.C.; Ploegh, H.L. XBP-1 regulates signal transduction, transcription factors and bone marrow colonization in B cells. EMBO J. 2009, 28, 1624–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipson, K.L.; Fonseca, S.G.; Ishigaki, S.; Nguyen, L.X.; Foss, E.; Bortell, R.; Rossini, A.A.; Urano, F. Regulation of insulin biosynthesis in pancreatic β-cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab. 2006, 4, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Karali, E.; Bellou, S.; Stellas, D.; Klinakis, A.; Murphy, C.; Fotsis, T. VEGF signals through ATF6 and PERK to promote endothelial cell survival and angiogenesis in the absence of ER stress. Mol. Cell 2014, 54, 559–572. [Google Scholar] [CrossRef] [PubMed]
- DenBoer, L.M.; Hardy-Smith, P.W.; Hogan, M.R.; Cockram, G.P.; Audas, T.E.; Lu, R. Luman is capable of binding and activating transcription from the unfolded protein response element. Biochem. Biophys. Res. Commun. 2005, 331, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Murakami, T.; Tatsumi, K.; Ogata, M.; Kanemoto, S.; Otori, K.; Iseki, K.; Wanaka, A.; Imaizumi, K. OASIS, a CREB/ATF-family member, modulates UPR signalling in astrocytes. Nat. Cell Biol. 2005, 7, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Saito, A.; Hino, S.; Murakami, T.; Ogata, M.; Kanemoto, S.; Nara, S.; Yamashita, A.; Yoshinaga, K.; Hara, H.; et al. BBF2H7, a novel transmembrane bZip transcription factor, is a new type of endoplasmic reticulum stress transducer. Mol. Cell. Biol. 2007, 27, 1716–1729. [Google Scholar] [CrossRef] [PubMed]
- Omori, Y.; Imai, J.; Watanabe, M.; Komatsu, T.; Suzuki, Y.; Kataoka, K.; Watanabe, S.; Tanigami, A.; Sugano, S. CREB-H: A novel mammalian transcription factor belonging to the CREB/ATF family and functioning via the BOX-B element with a liver-specific expression. Nucleic Acids Res. 2001, 29, 2154–2162. [Google Scholar] [CrossRef] [PubMed]
- Nagamori, I.; Yabuta, N.; Fujii, T.; Tanaka, H.; Yomogida, K.; Nishimune, Y.; Nojima, H. Tisp40, a spermatid specific bZip transcription factor, functions by binding to the unfolded protein response element via the Rip pathway. Genes Cells 2005, 10, 575–594. [Google Scholar] [CrossRef] [PubMed]
- Nagamori, I.; Yomogida, K.; Ikawa, M.; Okabe, M.; Yabuta, N.; Nojima, H. The testes-specific bZip type transcription factor Tisp40 plays a role in ER stress responses and chromatin packaging during spermiogenesis. Genes Cells 2006, 11, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.M.; Andrew, D.J. Transcriptional regulation of secretory capacity by bZip transcription factors. Front. Biol. 2015, 10, 28–51. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, S.; Fasanella, G.; Carreira, S.; Llarena, M.; Fox, R.; Barreca, C.; Andrew, D.; O’Hare, P. An orchestrated program regulating secretory pathway genes and cargos by the transmembrane transcription factor CREB-H. Traffic 2013, 14, 382–398. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Hino, S.; Murakami, T.; Kanemoto, S.; Kondo, S.; Saitoh, M.; Nishimura, R.; Yoneda, T.; Furuichi, T.; Ikegawa, S.; et al. Regulation of endoplasmic reticulum stress response by a BBF2H7-mediated Sec23a pathway is essential for chondrogenesis. Nat. Cell Biol. 2009, 11, 1197–1204. [Google Scholar] [CrossRef] [PubMed]
- Hino, K.; Saito, A.; Kido, M.; Kanemoto, S.; Asada, R.; Takai, T.; Cui, M.; Cui, X.; Imaizumi, K. Master regulator for chondrogenesis, Sox9, regulates transcriptional activation of the endoplasmic reticulum stress transducer BBF2H7/CREB3L2 in chondrocytes. J. Biol. Chem. 2014, 289, 13810–13820. [Google Scholar] [CrossRef] [PubMed]
- Boyadjiev, S.A.; Fromme, J.C.; Ben, J.; Chong, S.S.; Nauta, C.; Hur, D.J.; Zhang, G.; Hamamoto, S.; Schekman, R.; Ravazzola, M.; et al. Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat. Genet. 2006, 38, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Fromme, J.C.; Ravazzola, M.; Hamamoto, S.; Al-Balwi, M.; Eyaid, W.; Boyadjiev, S.A.; Cosson, P.; Schekman, R.; Orci, L. The genetic basis of a craniofacial disease provides insight into COPII coat assembly. Dev. Cell 2007, 13, 623–634. [Google Scholar] [CrossRef] [PubMed]
- Boyadjiev, S.A.; Kim, S.D.; Hata, A.; Haldeman-Englert, C.; Zackai, E.H.; Naydenov, C.; Hamamoto, S.; Schekman, R.W.; Kim, J. Cranio-lenticulo-sutural dysplasia associated with defects in collagen secretion. Clin. Genet. 2011, 80, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Saito, A.; Hino, S.; Kondo, S.; Kanemoto, S.; Chihara, K.; Sekiya, H.; Tsumagari, K.; Ochiai, K.; Yoshinaga, K.; et al. Signalling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nat. Cell Biol. 2009, 11, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Chihara, K.; Saito, A.; Murakami, T.; Hino, S.; Aoki, Y.; Sekiya, H.; Aikawa, Y.; Wanaka, A.; Imaizumi, K. Increased vulnerability of hippocampal pyramidal neurons to the toxicity of kainic acid in OASIS-deficient mice. J. Neurochem. 2009, 110, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Symoens, S.; Malfait, F.; D’Hondt, S.; Callewaert, B.; Dheedene, A.; Steyaert, W.; Bachinger, H.P.; De Paepe, A.; Kayserili, H.; Coucke, P.J. Deficiency for the ER-stress transducer OASIS causes severe recessive osteogenesis imperfecta in humans. Orphanet J. Rare Dis. 2013, 8, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forlino, A.; Cabral, W.A.; Barnes, A.M.; Marini, J.C. New perspectives on osteogenesis imperfecta. Nat. Rev. Endocrinol. 2011, 7, 540–557. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Hino, S.I.; Saito, A.; Kanemoto, S.; Kawasaki, N.; Asada, R.; Izumi, S.; Iwamoto, H.; Oki, M.; Miyagi, H.; et al. Activation of OASIS family, ER stress transducers, is dependent on its stabilization. Cell Death Differ. 2012, 19, 1939–1949. [Google Scholar] [CrossRef] [PubMed]
- Asada, R.; Saito, A.; Kawasaki, N.; Kanemoto, S.; Iwamoto, H.; Oki, M.; Miyagi, H.; Izumi, S.; Imaizumi, K. The endoplasmic reticulum stress transducer OASIS is involved in the terminal differentiation of goblet cells in the large intestine. J. Biol. Chem. 2012, 287, 8144–8153. [Google Scholar] [CrossRef] [PubMed]
- Kondo, S.; Saito, A.; Asada, R.; Kanemoto, S.; Imaizumi, K. Physiological unfolded protein response regulated by OASIS family members, transmembrane bZip transcription factors. IUBMB Life 2011, 63, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Asada, R.; Kanemoto, S.; Kondo, S.; Saito, A.; Imaizumi, K. The signalling from endoplasmic reticulum-resident bZip transcription factors involved in diverse cellular physiology. J. Biochem. 2011, 149, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.B.; O’Donnell, A.C.; Stamateris, R.E.; Ha, B.; McCloskey, K.M.; Reynolds, P.R.; Arvan, P.; Alonso, L.C. Insulin demand regulates β cell number via the unfolded protein response. J. Clin. Investig. 2015, 125, 3831–3846. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zeng, H.; Zhang, Y.; Jungries, R.; Chung, P.; Plesken, H.; Sabatini, D.D.; Ron, D. Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Mol. Cell 2001, 7, 1153–1163. [Google Scholar] [CrossRef]
- Oyadomari, S.; Koizumi, A.; Takeda, K.; Gotoh, T.; Akira, S.; Araki, E.; Mori, M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Investig. 2002, 109, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Delepine, M.; Nicolino, M.; Barrett, T.; Golamaully, M.; Lathrop, G.M.; Julier, C. EIF2AK3, encoding translation initiation factor 2-α kinase 3, is mutated in patients with Wolcott–Rallison syndrome. Nat. Genet. 2000, 25, 406–409. [Google Scholar] [PubMed]
- Usui, M.; Yamaguchi, S.; Tanji, Y.; Tominaga, R.; Ishigaki, Y.; Fukumoto, M.; Katagiri, H.; Mori, K.; Oka, Y.; Ishihara, H. ATF6α-null mice are glucose intolerant due to pancreatic β-cell failure on a high-fat diet but partially resistant to diet-induced insulin resistance. Metab. Clin. Exp. 2012, 61, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Saito, M.; Kohno, K. Pathogenic mechanism of diabetes development due to dysfunction of unfolded protein response. Yakugaku Zasshi J. Pharm. Soc. Jpn. 2016, 136, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Heidtman, K.; Hotamisligil, G.S.; Glimcher, L.H. Dual and opposing roles of the unfolded protein response regulated by IRE1α and XBP1 in proinsulin processing and insulin secretion. Proc. Natl. Acad. Sci. USA 2011, 108, 8885–8890. [Google Scholar] [CrossRef] [PubMed]
- Akai, R.; Hosoda, A.; Yoshino, M.; Iwawaki, T. Constitutive role of GADD34 and CREP in cancellation of phospho-eIF2α-dependent translational attenuation and insulin biosynthesis in pancreatic β-cells. Genes Cells 2015, 20, 871–886. [Google Scholar] [CrossRef] [PubMed]
- Lipson, K.L.; Ghosh, R.; Urano, F. The role of IRE1α in the degradation of insulin mRNA in pancreatic β-cells. PLoS ONE 2008, 3, e1648. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka, M.; Kayo, T.; Ikeda, T.; Koizumi, A. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes 1997, 46, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Araki, E.; Oyadomari, S.; Mori, M. Impact of endoplasmic reticulum stress pathway on pancreatic β-cells and diabetes mellitus. Exp. Biol. Med. 2003, 228, 1213–1217. [Google Scholar]
- Nozaki, J.; Kubota, H.; Yoshida, H.; Naitoh, M.; Goji, J.; Yoshinaga, T.; Mori, K.; Koizumi, A.; Nagata, K. The endoplasmic reticulum stress response is stimulated through the continuous activation of transcription factors ATF6 and XBP1 in Ins2+/Akita pancreatic β-cells. Genes Cells 2004, 9, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Kido, Y.; Asahara, S.; Kaisho, T.; Tanaka, T.; Hashimoto, N.; Shigeyama, Y.; Takeda, A.; Inoue, T.; Shibutani, Y.; et al. Ablation of C/EBPβ alleviates ER stress and pancreatic β-cell failure through the GRP78 chaperone in mice. J. Clin. Investig. 2010, 120, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Stoy, J.; Edghill, E.L.; Flanagan, S.E.; Ye, H.; Paz, V.P.; Pluzhnikov, A.; Below, J.E.; Hayes, M.G.; Cox, N.J.; Lipkind, G.M.; et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc. Natl. Acad. Sci. USA 2007, 104, 15040–15044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheuner, D.; Song, B.; McEwen, E.; Liu, C.; Laybutt, R.; Gillespie, P.; Saunders, T.; Bonner-Weir, S.; Kaufman, R.J. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 2001, 7, 1165–1176. [Google Scholar] [CrossRef]
- Oyadomari, S.; Yun, C.; Fisher, E.A.; Kreglinger, N.; Kreibich, G.; Oyadomari, M.; Harding, H.P.; Goodman, A.G.; Harant, H.; Garrison, J.L.; et al. Cotranslocational degradation protects the stressed endoplasmic reticulum from protein overload. Cell 2006, 126, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, N.; Asada, R.; Saito, A.; Kanemoto, S.; Imaizumi, K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci. Rep. 2012, 2, 799. [Google Scholar] [CrossRef] [PubMed]
- Jiao, P.; Ma, J.; Feng, B.; Zhang, H.; Diehl, J.A.; Chin, Y.E.; Yan, W.; Xu, H. FFA-induced adipocyte inflammation and insulin resistance: Involvement of ER stress and IKKβ pathways. Obesity 2011, 19, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, M.; Matsuda, M.; Maeda, N.; Funahashi, T.; Matsuzawa, Y.; Makishima, M.; Shimomura, I. Induction of adiponectin, a fat-derived antidiabetic and antiatherogenic factor, by nuclear receptors. Diabetes 2003, 52, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Maeda, N.; Takahashi, M.; Funahashi, T.; Kihara, S.; Nishizawa, H.; Kishida, K.; Nagaretani, H.; Matsuda, M.; Komuro, R.; Ouchi, N.; et al. PPARγ ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes 2001, 50, 2094–2099. [Google Scholar] [CrossRef] [PubMed]
- Hosogai, N.; Fukuhara, A.; Oshima, K.; Miyata, Y.; Tanaka, S.; Segawa, K.; Furukawa, S.; Tochino, Y.; Komuro, R.; Matsuda, M.; et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 2007, 56, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.B.; Kong, M.; Kim, T.M.; Suh, Y.H.; Kim, W.H.; Lim, J.H.; Song, J.H.; Jung, M.H. NFATC4 and ATF3 negatively regulate adiponectin gene expression in 3T3-L1 adipocytes. Diabetes 2006, 55, 1342–1352. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, F. Autophagy: Roles in obesity-induced ER stress and adiponectin downregulation in adipocytes. Autophagy 2010, 6, 1196–1197. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Liu, M.; Zhang, J.; Chen, H.; Dong, L.Q.; Liu, F. DsbA-L alleviates endoplasmic reticulum stress-induced adiponectin downregulation. Diabetes 2010, 59, 2809–2816. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Chen, H.; Wei, L.; Hu, D.; Dong, K.; Jia, W.; Dong, L.Q.; Liu, F. Endoplasmic reticulum (ER) localization is critical for DsbA-l protein to suppress ER stress and adiponectin down-regulation in adipocytes. J. Biol. Chem. 2015, 290, 10143–10148. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhou, L.; Xu, A.; Lam, K.S.; Wetzel, M.D.; Xiang, R.; Zhang, J.; Xin, X.; Dong, L.Q.; Liu, F. A disulfide-bond a oxidoreductase-like protein (DsbA-L) regulates adiponectin multimerization. Proc. Natl. Acad. Sci. USA 2008, 105, 18302–18307. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Kang, Y.M.; Kim, C.H.; Jung, M.H. ATF3 negatively regulates adiponectin receptor 1 expression. Biochem. Biophys. Res. Commun. 2010, 400, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Koh, I.U.; Lim, J.H.; Joe, M.K.; Kim, W.H.; Jung, M.H.; Yoon, J.B.; Song, J. AdipoR2 is transcriptionally regulated by ER stress-inducible ATF3 in HepG2 human hepatocyte cells. FEBS J. 2010, 277, 2304–2317. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Zhang, X.; Huang, H.; Ding, N.; Zhang, S.; Hutchinson, S.Z. Adiponectin protects rat myocardium against chronic intermittent hypoxia-induced injury via inhibition of endoplasmic reticulum stress. PLoS ONE 2014, 9, e94545. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Gan, L.; Wu, T.; Feng, F.; Luo, D.; Gu, H.; Liu, S.; Sun, C. Adiponectin reduces ER stress-induced apoptosis through PPARα transcriptional regulation of ATF2 in mouse adipose. Cell Death Dis. 2016, 7, e2487. [Google Scholar] [CrossRef] [PubMed]
- Halaas, J.L.; Gajiwala, K.S.; Maffei, M.; Cohen, S.L.; Chait, B.T.; Rabinowitz, D.; Lallone, R.L.; Burley, S.K.; Friedman, J.M. Weight-reducing effects of the plasma protein encoded by the obese gene. Science 1995, 269, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Marwarha, G.; Dasari, B.; Ghribi, O. Endoplasmic reticulum stress-induced CHOP activation mediates the down-regulation of leptin in human neuroblastoma SH-SY5Y cells treated with the oxysterol 27-hydroxycholesterol. Cell. Signal. 2012, 24, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, L.; Ergin, A.S.; Lu, A.; Chung, J.; Sarkar, S.; Nie, D.; Myers, M.G., Jr.; Ozcan, U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009, 9, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Schneeberger, M.; Dietrich, M.O.; Sebastian, D.; Imbernon, M.; Castano, C.; Garcia, A.; Esteban, Y.; Gonzalez-Franquesa, A.; Rodriguez, I.C.; Bortolozzi, A.; et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 2013, 155, 172–187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.H.; Iwakoshi, N.N.; Ozdelen, E.; Tuncman, G.; Gorgun, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Boden, G.; Duan, X.; Homko, C.; Molina, E.J.; Song, W.; Perez, O.; Cheung, P.; Merali, S. Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes 2008, 57, 2438–2444. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, K.; Miyazaki, M.; Matsuhisa, M.; Takano, K.; Nakatani, Y.; Hatazaki, M.; Tamatani, T.; Yamagata, K.; Miyagawa, J.; Kitao, Y.; et al. The endoplasmic reticulum chaperone improves insulin resistance in type 2 diabetes. Diabetes 2005, 54, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, Y.; Kaneto, H.; Kawamori, D.; Yoshiuchi, K.; Hatazaki, M.; Matsuoka, T.A.; Ozawa, K.; Ogawa, S.; Hori, M.; Yamasaki, Y.; et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J. Biol. Chem. 2005, 280, 847–851. [Google Scholar] [CrossRef] [PubMed]
- Azuma, Y.; Hagiwara, D.; Lu, W.; Morishita, Y.; Suga, H.; Goto, M.; Banno, R.; Sugimura, Y.; Oyadomari, S.; Mori, K.; et al. Activating transcription factor 6α is required for the vasopressin neuron system to maintain water balance under dehydration in male mice. Endocrinology 2014, 155, 4905–4914. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Arima, H.; Ozaki, N.; Morishita, Y.; Hiroi, M.; Nagasaki, H.; Kinoshita, N.; Ueda, M.; Shiota, A.; Oiso, Y. Progressive polyuria without vasopressin neuron loss in a mouse model for familial neurohypophysial diabetes insipidus. Am. J. Physiol. 2009, 296, R1641–R1649. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, D.; Arima, H.; Morishita, Y.; Wenjun, L.; Azuma, Y.; Ito, Y.; Suga, H.; Goto, M.; Banno, R.; Sugimura, Y.; et al. Arginine vasopressin neuronal loss results from autophagy-associated cell death in a mouse model for familial neurohypophysial diabetes insipidus. Cell Death Dis. 2014, 5, e1148. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Huerta, P.; Troncoso-Escudero, P.; Jerez, C.; Hetz, C.; Vidal, R.L. The intersection between growth factors, autophagy and ER stress: A new target to treat neurodegenerative diseases? Brain Res. 2016, 1649, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Miyazaki, S.; Matsuyama, S.; Takeda, M.; Kawano, M.; Nakagawa, H.; Nishimura, K.; Matsuo, S. Selection of autophagy or apoptosis in cells exposed to ER-stress depends on ATF4 expression pattern with or without CHOP expression. Biol. Open 2013, 2, 1084–1090. [Google Scholar] [CrossRef] [PubMed]
- Morishita, Y.; Arima, H.; Hiroi, M.; Hayashi, M.; Hagiwara, D.; Asai, N.; Ozaki, N.; Sugimura, Y.; Nagasaki, H.; Shiota, A.; et al. Poly(A) tail length of neurohypophysial hormones is shortened under endoplasmic reticulum stress. Endocrinology 2011, 152, 4846–4855. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Hoffmann, A.; Kaiser, E.K.; Grunwald, W.C., Jr.; Cool, D.R. Misfolding of mutated vasopressin causes ER-retention and activation of ER-stress markers in neuro-2A cells. Open Neuroendocrinol. J. 2011, 4, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, M.; Bordieri, L.; Greenwood, M.P.; Rosso Melo, M.; Colombari, D.S.; Colombari, E.; Paton, J.F.; Murphy, D. Transcription factor CREB3L1 regulates vasopressin gene expression in the rat hypothalamus. J. Neurosci. 2014, 34, 3810–3820. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Tanizawa, Y.; Wasson, J.; Behn, P.; Kalidas, K.; Bernal-Mizrachi, E.; Mueckler, M.; Marshall, H.; Donis-Keller, H.; Crock, P.; et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat. Genet. 1998, 20, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Kawano, J.; Takeda, K.; Yujiri, T.; Tanabe, K.; Anno, T.; Akiyama, M.; Nozaki, J.; Yoshinaga, T.; Koizumi, A.; et al. Endoplasmic reticulum stress induces WFS1 gene expression in pancreatic β-cells via transcriptional activation. Eur. J. Endocrinol. 2005, 153, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.G.; Fukuma, M.; Lipson, K.L.; Nguyen, L.X.; Allen, J.R.; Oka, Y.; Urano, F. WFS1 is a novel component of the unfolded protein response and maintains homeostasis of the endoplasmic reticulum in pancreatic β-cells. J. Biol. Chem. 2005, 280, 39609–39615. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.G.; Ishigaki, S.; Oslowski, C.M.; Lu, S.; Lipson, K.L.; Ghosh, R.; Hayashi, E.; Ishihara, H.; Oka, Y.; Permutt, M.A.; et al. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J. Clin. Investig. 2010, 120, 744–755. [Google Scholar] [CrossRef] [PubMed]
- Bespalova, I.N.; Van Camp, G.; Bom, S.J.; Brown, D.J.; Cryns, K.; DeWan, A.T.; Erson, A.E.; Flothmann, K.; Kunst, H.P.; Kurnool, P.; et al. Mutations in the Wolfram syndrome 1 gene (WFS1) are a common cause of low frequency sensorineural hearing loss. Hum. Mol. Genet. 2001, 10, 2501–2508. [Google Scholar] [CrossRef] [PubMed]
- Swift, M.; Swift, R.G. Psychiatric disorders and mutations at the Wolfram syndrome locus. Biol. Psychiatry 2000, 47, 787–793. [Google Scholar] [CrossRef]
- Hansen, L.; Eiberg, H.; Barrett, T.; Bek, T.; Kjaersgaard, P.; Tranebjaerg, L.; Rosenberg, T. Mutation analysis of the WFS1 gene in seven Danish Wolfram syndrome families; four new mutations identified. Eur. J. Hum. Genet. 2005, 13, 1275–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urano, F. Wolfram syndrome: Diagnosis, management, and treatment. Curr. Diabetes Rep. 2016, 16, 6. [Google Scholar] [CrossRef] [PubMed]
- Riggs, A.C.; Bernal-Mizrachi, E.; Ohsugi, M.; Wasson, J.; Fatrai, S.; Welling, C.; Murray, J.; Schmidt, R.E.; Herrera, P.L.; Permutt, M.A. Mice conditionally lacking the wolfram gene in pancreatic islet β-cells exhibit diabetes as a result of enhanced endoplasmic reticulum stress and apoptosis. Diabetologia 2005, 48, 2313–2321. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Hua, H.; Foo, K.; Martinez, H.; Watanabe, K.; Zimmer, M.; Kahler, D.J.; Freeby, M.; Chung, W.; LeDuc, C.; et al. β-cell dysfunction due to increased ER stress in a stem cell model of Wolfram syndrome. Diabetes 2014, 63, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Alatzoglou, K.S.; Kular, D.; Dattani, M.T. Autosomal dominant growth hormone deficiency (type II). Pediatr. Endocrinol. Rev. 2015, 12, 347–355. [Google Scholar] [PubMed]
- Akinci, A.; Kanaka, C.; Eble, A.; Akar, N.; Vidinlisan, S.; Mullis, P.E. Isolated growth hormone (GH) deficiency type IA associated with a 45-kilobase gene deletion within the human GH gene cluster. J. Clin. Endocrinol. Metab. 1992, 75, 437–441. [Google Scholar] [PubMed]
- Cogan, J.D.; Phillips, J.A., 3rd; Schenkman, S.S.; Milner, R.D.; Sakati, N. Familial growth hormone deficiency: A model of dominant and recessive mutations affecting a monomeric protein. J. Clin. Endocrinol. Metab. 1994, 79, 1261–1265. [Google Scholar] [PubMed]
- Binder, G.; Ranke, M.B. Screening for growth hormone (GH) gene splice-site mutations in sporadic cases with severe isolated GH deficiency using ectopic transcript analysis. J. Clin. Endocrinol. Metab. 1995, 80, 1247–1252. [Google Scholar] [PubMed]
- Hayashi, Y.; Yamamoto, M.; Ohmori, S.; Kamijo, T.; Ogawa, M.; Seo, H. Inhibition of growth hormone (GH) secretion by a mutant GH-I gene product in neuroendocrine cells containing secretory granules: An implication for isolated GH deficiency inherited in an autosomal dominant manner. J. Clin. Endocrinol. Metab. 1999, 84, 2134–2139. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Wajnrajch, M.P.; Kim, S.S.; Plotnick, L.P.; Wang, J.; Gertner, J.M.; Leibel, R.L.; Dannies, P.S. Autosomal dominant growth hormone (GH) deficiency type II: The Del32-71-GH deletion mutant suppresses secretion of wild-type GH. Endocrinology 2000, 141, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Salemi, S.; Yousefi, S.; Eble, A.; Deladoey, J.; Mullis, P.E. Impact of Del32-71-GH (exon 3 skipped GH) on intracellular GH distribution, secretion and cell viability: A quantitative confocal microscopy analysis. Horm. Res. 2006, 65, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Kannenberg, K.; Wittekindt, N.E.; Tippmann, S.; Wolburg, H.; Ranke, M.B.; Binder, G. Mutant and misfolded human growth hormone is rapidly degraded through the proteasomal degradation pathway in a cellular model for isolated growth hormone deficiency type II. J. Neuroendocrinol. 2007, 19, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Iliev, D.I.; Wittekindt, N.E.; Ranke, M.B.; Binder, G. Structural analysis of human growth hormone with respect to the dominant expression of growth hormone (GH) mutations in isolated GH deficiency type II. Endocrinology 2005, 146, 1411–1417. [Google Scholar] [CrossRef] [PubMed]
- Graves, T.K.; Patel, S.; Dannies, P.S.; Hinkle, P.M. Misfolded growth hormone causes fragmentation of the Golgi apparatus and disrupts endoplasmic reticulum-to-Golgi traffic. J. Cell Sci. 2001, 114, 3685–3694. [Google Scholar] [PubMed]
- McGuinness, L.; Magoulas, C.; Sesay, A.K.; Mathers, K.; Carmignac, D.; Manneville, J.B.; Christian, H.; Phillips, J.A., III; Robinson, I.C. Autosomal dominant growth hormone deficiency disrupts secretory vesicles in vitro and in vivo in transgenic mice. Endocrinology 2003, 144, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Ariyasu, D.; Yoshida, H.; Yamada, M.; Hasegawa, Y. Endoplasmic reticulum stress and apoptosis contribute to the pathogenesis of dominantly inherited isolated GH deficiency due to GH1 gene splice site mutations. Endocrinology 2013, 154, 3228–3239. [Google Scholar] [CrossRef] [PubMed]
- Lochmatter, D.; Strom, M.; Eble, A.; Petkovic, V.; Fluck, C.E.; Bidlingmaier, M.; Robinson, I.C.; Mullis, P.E. Isolated GH deficiency type II: Knockdown of the harmful ∆3GH recovers wt-GH secretion in rat tumor pituitary cells. Endocrinology 2010, 151, 4400–4409. [Google Scholar] [CrossRef] [PubMed]
- Miletta, M.C.; Petkovic, V.; Eble, A.; Fluck, C.E.; Mullis, P.E. Rescue of isolated GH deficiency type II (IGHD II) via pharmacologic modulation of GH-1 splicing. Endocrinology 2016, 157, 3972–3982. [Google Scholar] [CrossRef] [PubMed]
- Coschigano, K.T.; Clemmons, D.; Bellush, L.L.; Kopchick, J.J. Assessment of growth parameters and life span of GHR/BP gene-disrupted mice. Endocrinology 2000, 141, 2608–2613. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.D.; Jousse, C.; Marciniak, S.J.; Zhang, Y.; Novoa, I.; Scheuner, D.; Kaufman, R.J.; Ron, D.; Harding, H.P. Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J. 2004, 23, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Kudo, T.; Kanemoto, S.; Hara, H.; Morimoto, N.; Morihara, T.; Kimura, R.; Tabira, T.; Imaizumi, K.; Takeda, M. A molecular chaperone inducer protects neurons from ER stress. Cell Death Differ. 2008, 15, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Hino, S.; Kondo, S.; Yoshinaga, K.; Saito, A.; Murakami, T.; Kanemoto, S.; Sekiya, H.; Chihara, K.; Aikawa, Y.; Hara, H.; et al. Regulation of ER molecular chaperone prevents bone loss in a murine model for osteoporosis. J. Bone Miner. Metab. 2010, 28, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Oida, Y.; Izuta, H.; Oyagi, A.; Shimazawa, M.; Kudo, T.; Imaizumi, K.; Hara, H. Induction of BiP, an ER-resident protein, prevents the neuronal death induced by transient forebrain ischemia in gerbil. Brain Res. 2008, 1208, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Oida, Y.; Hamanaka, J.; Hyakkoku, K.; Shimazawa, M.; Kudo, T.; Imaizumi, K.; Yasuda, T.; Hara, H. Post-treatment of a BiP inducer prevents cell death after middle cerebral artery occlusion in mice. Neurosci. Lett. 2010, 484, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; Lavail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Upton, J.P.; Hagen, A.; Callahan, J.; Oakes, S.A.; Papa, F.R. A kinase inhibitor activates the IRE1α RNase to confer cytoprotection against ER stress. Biochem. Biophys. Res. Commun. 2008, 365, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Papa, F.R.; Zhang, C.; Shokat, K.; Walter, P. Bypassing a kinase activity with an ATP-competitive drug. Science 2003, 302, 1533–1537. [Google Scholar] [CrossRef] [PubMed]
- Kozuka, C.; Yabiku, K.; Sunagawa, S.; Ueda, R.; Taira, S.; Ohshiro, H.; Ikema, T.; Yamakawa, K.; Higa, M.; Tanaka, H.; et al. Brown rice and its component, gamma-oryzanol, attenuate the preference for high-fat diet by decreasing hypothalamic endoplasmic reticulum stress in mice. Diabetes 2012, 61, 3084–3093. [Google Scholar] [CrossRef] [PubMed]
- Tuchman, M.; Lee, B.; Lichter-Konecki, U.; Summar, M.L.; Yudkoff, M.; Cederbaum, S.D.; Kerr, D.S.; Diaz, G.A.; Seashore, M.R.; Lee, H.S.; et al. Cross-sectional multicenter study of patients with urea cycle disorders in the United States. Mol. Genet. Metab. 2008, 94, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Basseri, S.; Lhotak, S.; Sharma, A.M.; Austin, R.C. The chemical chaperone 4-phenylbutyrate inhibits adipogenesis by modulating the unfolded protein response. J. Lipid res. 2009, 50, 2486–2501. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, N.; Fukata, Y.; Kase, D.; Miyazaki, T.; Jaegle, M.; Ohkawa, T.; Takahashi, N.; Iwanari, H.; Mochizuki, Y.; Hamakubo, T.; et al. Chemical corrector treatment ameliorates increased seizure susceptibility in a mouse model of familial epilepsy. Nat. Med. 2015, 21, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Khaoustov, V.I.; Chung, C.C.; Sohn, J.; Krishnan, B.; Lewis, D.E.; Yoffe, B. Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress-induced caspase-12 activation. Hepatology 2002, 36, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Yalcin, A.; Lee, G.Y.; Li, P.; Fan, J.; Arruda, A.P.; Pers, B.M.; Yilmaz, M.; Eguchi, K.; Hotamisligil, G.S. Phenotypic assays identify azoramide as a small-molecule modulator of the unfolded protein response with antidiabetic activity. Sci. Transl. Med. 2015, 7, 292ra298. [Google Scholar] [CrossRef] [PubMed]
- Minamiyama, M.; Katsuno, M.; Adachi, H.; Doi, H.; Kondo, N.; Iida, M.; Ishigaki, S.; Fujioka, Y.; Matsumoto, S.; Miyazaki, Y.; et al. Naratriptan mitigates CGRP1-associated motor neuron degeneration caused by an expanded polyglutamine repeat tract. Nat. Med. 2012, 18, 1531–1538. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ma, M.; Teng, J.; Che, X.; Zhang, W.; Feng, S.; Zhou, S.; Zhang, Y.; Wu, E.; Ding, X. Valproate attenuates 25-kDa C-terminal fragment of TDP-43-induced neuronal toxicity via suppressing endoplasmic reticulum stress and activating autophagy. Int. J. Biol. Sci. 2015, 11, 752–761. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ariyasu, D.; Yoshida, H.; Hasegawa, Y. Endoplasmic Reticulum (ER) Stress and Endocrine Disorders. Int. J. Mol. Sci. 2017, 18, 382. https://doi.org/10.3390/ijms18020382
Ariyasu D, Yoshida H, Hasegawa Y. Endoplasmic Reticulum (ER) Stress and Endocrine Disorders. International Journal of Molecular Sciences. 2017; 18(2):382. https://doi.org/10.3390/ijms18020382
Chicago/Turabian StyleAriyasu, Daisuke, Hiderou Yoshida, and Yukihiro Hasegawa. 2017. "Endoplasmic Reticulum (ER) Stress and Endocrine Disorders" International Journal of Molecular Sciences 18, no. 2: 382. https://doi.org/10.3390/ijms18020382