
In Vitro Characterization of the Two-Stage Non-Classical Reassembly Pathway of S-Layers


Abstract
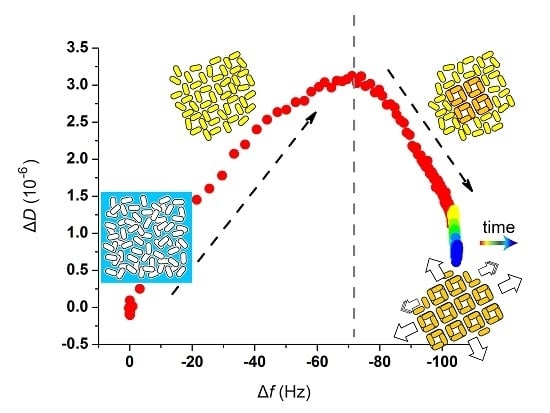
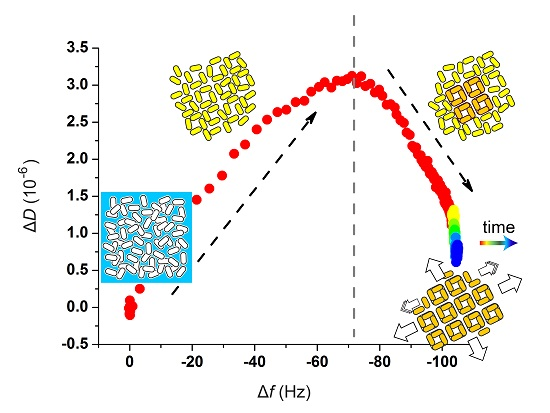
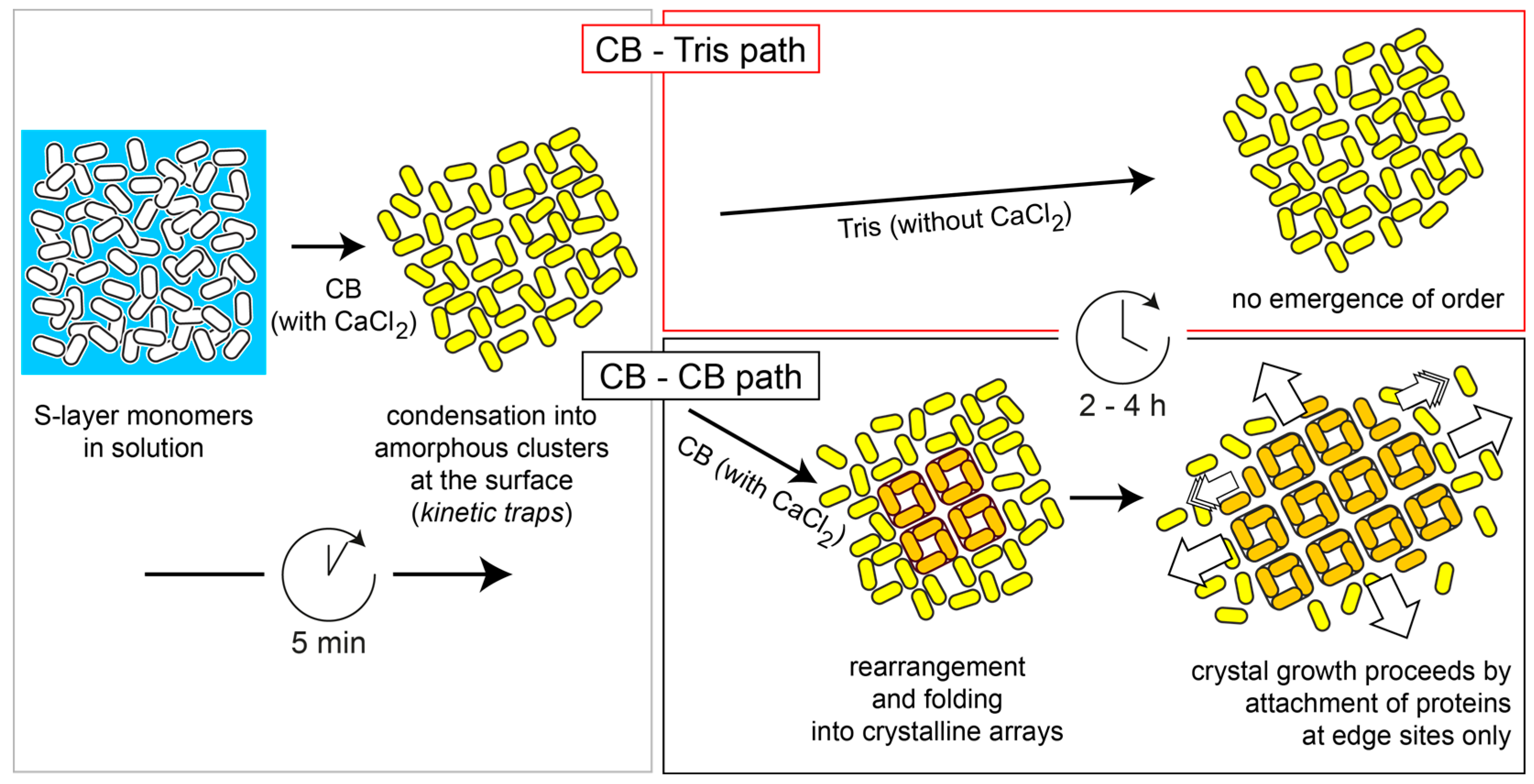
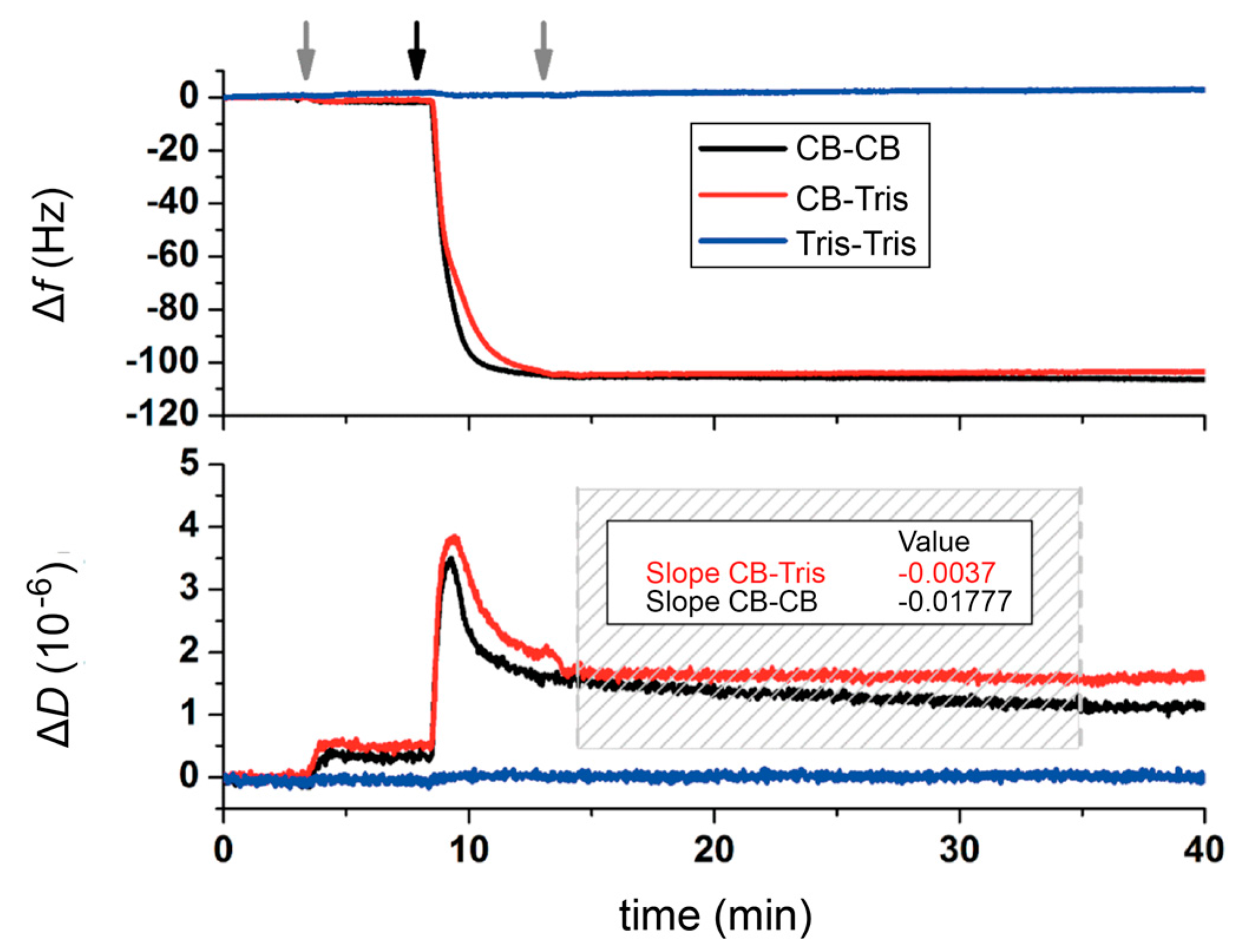
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Recombinant S-Layer Protein rSbpA
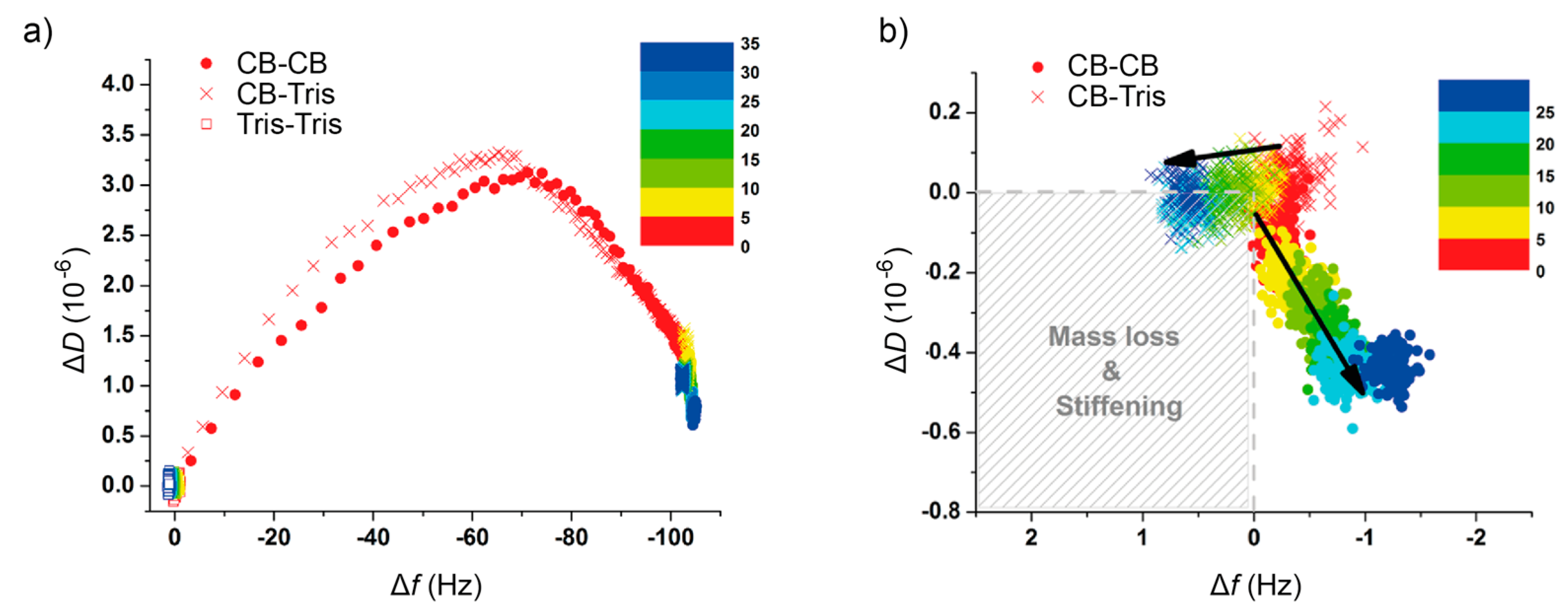
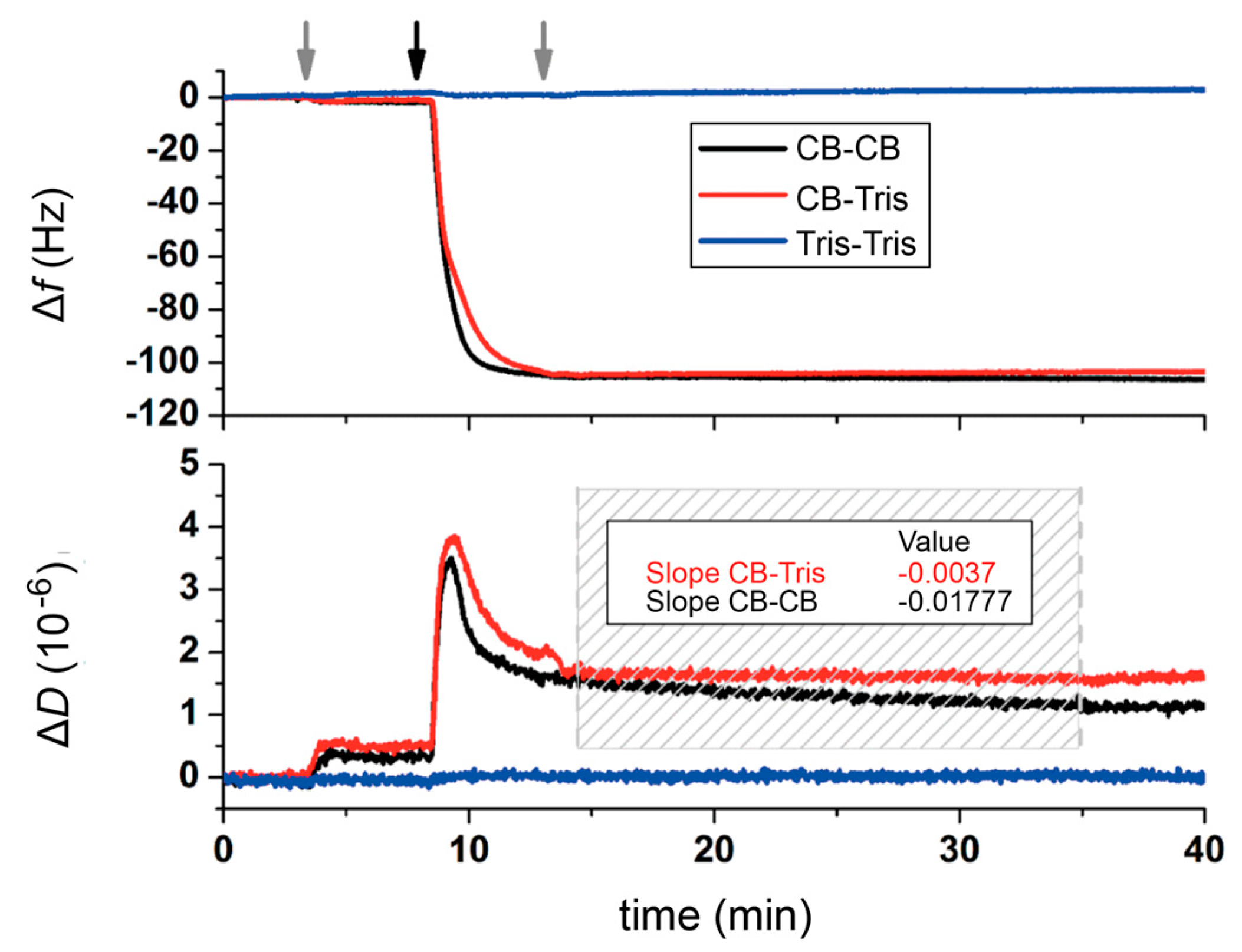
2.2. Quartz Crystal Microbalance with Dissipation (QCM-D) Studies
2.3. Atomic Force Microscopy (AFM)
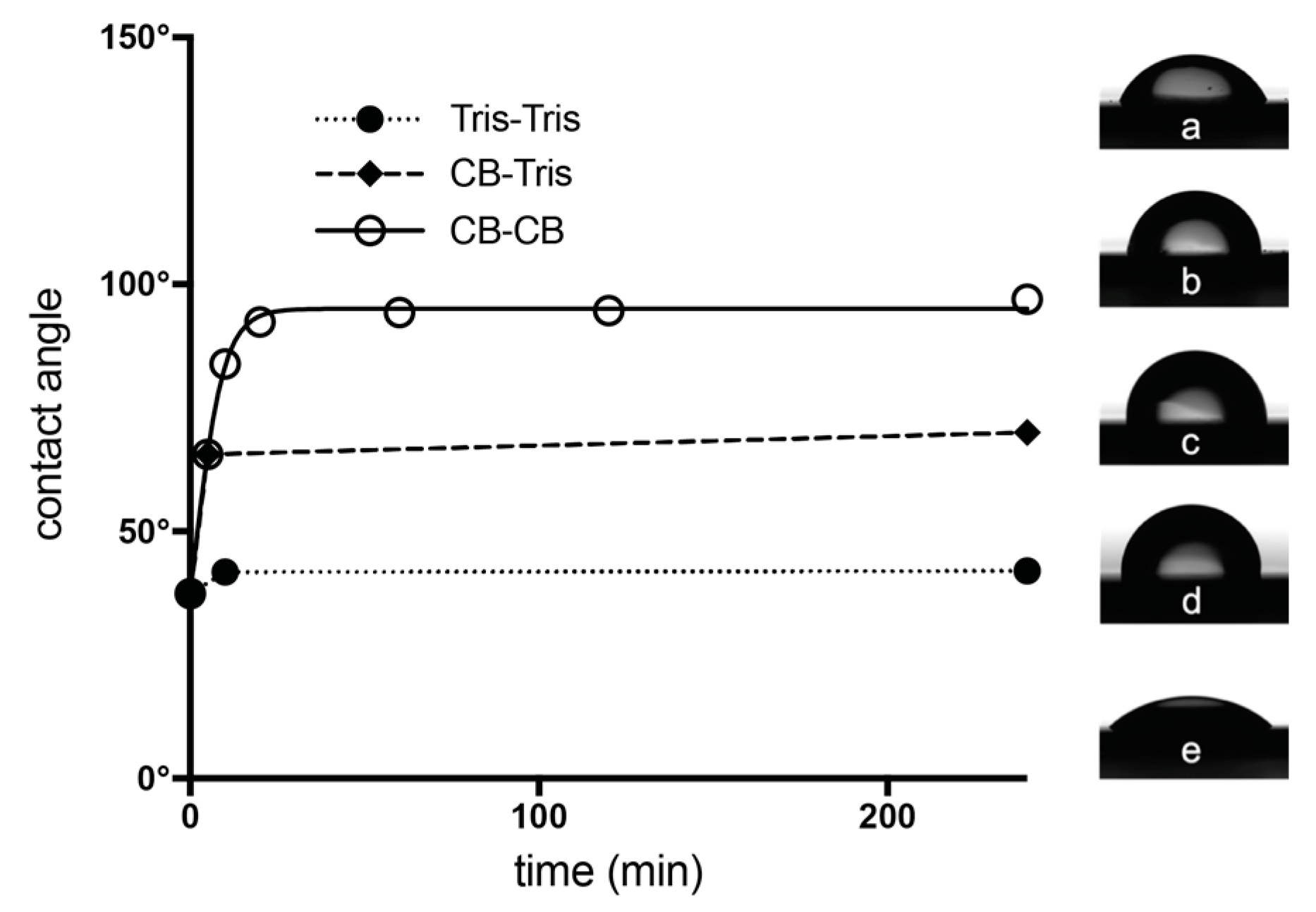
2.4. Contact Angle Measurements
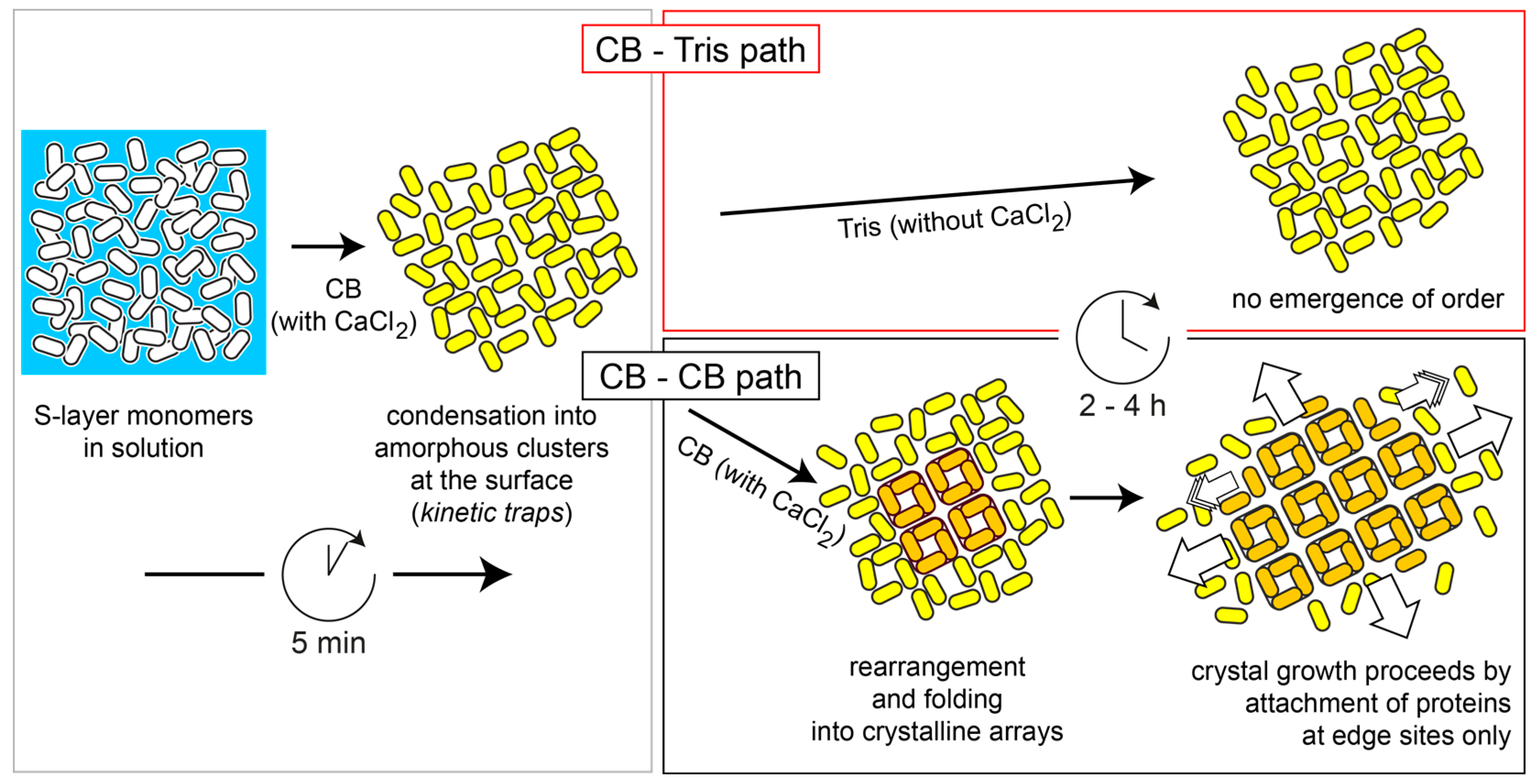
3. Discussion
4. Materials and Methods
4.1. Production of Monomeric S-Layer Protein Solutions
4.2. Quartz Crystal Microbalance with Dissipation (QCM-D) Monitoring
4.3. Recrystallization of S-Layer Proteins on Silicon Wafers
4.4. Atomic Force Microscopy
4.5. Contact Angle Measurements
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sleytr, U.B.; Schuster, B.; Egelseer, E.M.; Pum, D. S-layers: Principles and applications. FEMS Microbiol. Rev. 2014, 38, 823–864. [Google Scholar] [CrossRef] [PubMed]
- Pum, D.; Sleytr, U.B. Reassembly of S-layer proteins. Nanotechnology 2014, 25, 312001. [Google Scholar] [CrossRef] [PubMed]
- Pum, D.; Toca-Herrera, J.; Sleytr, U. S-layer protein self-assembly. Int. J. Mol. Sci. 2013, 14, 2484–2501. [Google Scholar] [CrossRef] [PubMed]
- Sleytr, U.B. Heterologous reattachment of regular arrays of glycoproteins on bacterial surfaces. Nature 1975, 257, 400–402. [Google Scholar] [CrossRef] [PubMed]
- Ilk, N.; Egelseer, E.M.; Sleytr, U.B. S-layer fusion proteins—Construction principles and applications. Curr. Opin. Biotechnol. 2011, 22, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Sleytr, U.B.; Glauert, A.M. Analysis of regular arrays of subunits on bacterial surfaces: Evidence for a dynamic process of assembly. J. Ultrastruct. Res. 1975, 50, 103–116. [Google Scholar] [CrossRef]
- Sleytr, U.B. Regular arrays of macromolecules on bacterial cell walls: Structure, chemistry, assembly, and function. Int. Rev. Cytol. 1978, 53, 1–62. [Google Scholar] [PubMed]
- Sleytr, U.B.; Messner, P.; Pum, D.; Sára, M. Crystalline bacterial cell surface layers (S-layers): From supramolecular cell structure to biomimetics and nanotechnology. Angew. Chem. Int. Ed. 1999, 38, 1034–1054. [Google Scholar] [CrossRef]
- Fagan, R.P.; Fairweather, N.F. Biogenesis and functions of bacterial S-layers. Nat. Rev. Microbiol. 2014, 12, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Breitwieser, A.; Gruber, K.; Sleytr, U.B. Evidence for an S-layer protein pool in the peptidoglycan of Bacillus stearothermophilus. J. Bacteriol. 1992, 174, 8008–8015. [Google Scholar] [CrossRef] [PubMed]
- Sleytr, U.B.; Messner, P. Self-assembly of crystalline bacterial cell surface layers (S-layers). In Electron Microscopy of Subcellular Dynamics; Plattner, H., Ed.; CRC Press: Boca Raton, FL, USA, 1989; pp. 13–31. [Google Scholar]
- Jaenicke, R.; Welsch, R.; Sára, M.; Sleytr, U.B. Stability and self-assembly of the S-layer protein of the cell wall of Bacillus stearothermophilus. Biol. Chem. Hoppe Seyler 1985, 366, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, L.M.; Strickland, A.; Mark, S.S.; Bergkvist, M.; Sierra-Sastre, Y.; Batt, C.A. Entropically driven self-assembly of Lysinibacillus sphaericus S-layer proteins analyzed under various environmental conditions. Macromol. Biosci. 2010, 10, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Pum, D.; Weinhandl, M.; Hödl, C.; Sleytr, U.B. Large-scale recrystallization of the S-layer of Bacillus coagulans E38-66 at the air/water interface and on lipid films. J. Bacteriol. 1993, 175, 2762–2766. [Google Scholar] [CrossRef] [PubMed]
- Pum, D.; Sleytr, U.B. Large-scale reconstruction of crystalline bacterial surface layer proteins at the air-water interface and on lipids. Thin Solid Films 1994, 244, 882–886. [Google Scholar] [CrossRef]
- Pum, D.; Sleytr, U.B. Anisotropic crystal growth of the S-layer of Bacillus sphaericus CCM 2177 at the air/water interface. Colloids Surfaces A Physicochem. Eng. Asp. 1995, 102, 99–104. [Google Scholar] [CrossRef]
- Pum, D.; Sleytr, U.B. Monomolecular reassembly of a crystalline bacterial cell surface layer (S layer) on untreated and modified silicon surfaces. Supramol. Sci. 1995, 2, 193–197. [Google Scholar] [CrossRef]
- Györvary, E.S.; Stein, O.; Pum, D.; Sleytr, U.B. Self-assembly and recrystallization of bacterial S-layer proteins at silicon supports imaged in real time by atomic force microscopy. J. Microsc. 2003, 212, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Flores, S.; Kasry, A.; Butt, H.J.; Vavilala, C.; Schmittel, M.; Pum, D.; Sleytr, U.B.; Toca-Herrera, J.L. From native to non-native two-dimensional protein lattices through underlying hydrophilic/hydrophobic nanoprotrusions. Angew. Chem. Int. Ed. 2008, 47, 4707–4710. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.E.; Moreno-Flores, S.; Pum, D.; Sleytr, U.B.; Toca-Herrera, J.L. Surface dependence of protein nanocrystal formation. Small 2010, 6, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Shin, S.H.; Bertozzi, C.R.; de Yoreo, J.J. Self-catalyzed growth of S layers via an amorphous-to-crystalline transition limited by folding kinetics. Proc. Natl. Acad. Sci. USA 2010, 107, 16536–16541. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.H.; Chung, S.; Sanii, B.; Comolli, L.R.; Bertozzi, C.R.; de Yoreo, J.J. Direct observation of kinetic traps associated with structural transformations leading to multiple pathways of S-layer assembly. Proc. Natl. Acad. Sci. USA 2012, 109, 12968–12973. [Google Scholar] [CrossRef] [PubMed]
- De Yoreo, J.J.; Chung, S.; Friddle, R.W. In situ atomic force microscopy as a tool for investigating interactions and assembly dynamics in biomolecular and biomineral systems. Adv. Funct. Mater. 2013, 23, 2525–2538. [Google Scholar] [CrossRef]
- Comolli, L.R.; Siegerist, C.E.; Shin, S.H.; Bertozzi, C.; Regan, W.; Zettl, A.; de Yoreo, J. Conformational transitions at an S-layer growing boundary resolved by cryo-TEM. Angew. Chem. Int. Ed. Engl. 2013, 52, 4829–4832. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Ilk, N.; Völlenkle, C.; Egelseer, E.M.; Breitwieser, A.; Sleytr, U.B.; Sára, M. Molecular characterization of the S-layer gene, sbpA, of Bacillus sphaericus CCM 2177 and production of a functional S-layer fusion protein with the ability to recrystallize in a defined orientation while presenting the fused allergen. Appl. Environ. Microbiol. 2002, 68, 3251–3260. [Google Scholar] [CrossRef] [PubMed]
- Rad, B.; Haxton, T.K.; Shon, A.; Shin, S.H.; Whitelam, S.; Ajo-Franklin, C.M. Ion-specific control of the self-assembly dynamics of a nanostructured protein lattice. ACS Nano 2015, 9, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Falke, S.; Drobot, B.; Oberthuer, D.; Kikhney, A.; Guenther, T.; Fahmy, K.; Svergun, D.; Betzel, C.; Raff, J. Analysis of self-assembly of S-layer protein slp-B53 from Lysinibacillus sphaericus. Eur. Biophys. J. 2017, 46, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Baranova, E.; Fronzes, R.; Garcia-Pino, A.; van Gerven, N.; Papapostolou, D.; Pehau-Arnaudet, G.; Pardon, E.; Steyaert, J.; Howorka, S.; Remaut, H. SbsB structure and lattice reconstruction unveil Ca2+ triggered S-layer assembly. Nature 2012, 487, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Haxton, T.K.; Whitelam, S. Design rules for the self-assembly of a protein crystal. Soft Matter 2012, 8, 3558–3562. [Google Scholar] [CrossRef]
- Whitelam, S. Control of Pathways and Yields of Protein Crystallization through the interplay of nonspecific and specific attractions. Phys. Rev. Lett. 2010, 105, 088102. [Google Scholar] [CrossRef] [PubMed]
- Baneyx, F.; Matthaei, J.F. Self-assembled two-dimensional protein arrays in bionanotechnology: From S-layers to designed lattices. Curr. Opin. Biotechnol. 2014, 28, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Pavkov-Keller, T.; Howorka, S.; Keller, W. The structure of bacterial S-layer proteins. Prog. Mol. Biol. Transl. Sci. 2011, 103, 73–130. [Google Scholar] [PubMed]
- Huber, C.; Ilk, N.; Rünzler, D.; Egelseer, E.M.; Weigert, S.; Sleytr, U.B.; Sára, M. The three S-layer-like homology motifs of the S-layer protein SbpA of Bacillus sphaericus CCM 2177 are not sufficient for binding to the pyruvylated secondary cell wall polymer. Mol. Microbiol. 2005, 55, 197–205. [Google Scholar] [CrossRef] [PubMed]
- De Yoreo, J.J.; Chung, S.; Nielsen, M.H. The Dynamics and Energetics of Matrix Assembly and Mineralization. Calcif. Tissue Int. 2013, 93, 316–328. [Google Scholar] [CrossRef] [PubMed]
- De Yoreo, J. Crystal nucleation: More than one pathway. Nat. Mater. 2013, 12, 284–285. [Google Scholar] [CrossRef] [PubMed]
- Rothbauer, M.; Kupcu, S.; Sticker, D.; Sleytr, U.B.; Ertl, P. Exploitation of S-layer anisotropy: pH-Dependent nanolayer orientation for cellular micropatterning. ACS Nano 2013, 7, 8020–8030. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, F.C.; Sear, R.P. Materials science now you see them. Science 2008, 322, 1802–1803. [Google Scholar] [CrossRef] [PubMed]
- De Yoreo, J.J.; Gilbert, P.U.P.A.; Sommerdijk, N.A.J.M.; Penn, R.L.; Whitelam, S.; Joester, D.; Zhang, H.Z.; Rimer, J.D.; Navrotsky, A.; Banfield, J.F.; et al. Crystallization by particle attachment in synthetic, biogenic, and geologic environments. Science 2015, 349. [Google Scholar] [CrossRef] [PubMed]
- Vekilov, P.G.; Chung, S.; Olafson, K.N. Shape change in crystallization of biological macromolecules. Mrs Bull. 2016, 41, 375–380. [Google Scholar] [CrossRef]
- Sleutel, M.; van Driessche, A.E.S. Role of clusters in nonclassical nucleation and growth of protein crystals. Proc. Natl. Acad. Sci. USA 2014, 111, E546–E553. [Google Scholar] [CrossRef] [PubMed]
- Sleutel, M.; Lutsko, J.; van Driessche, A.E.S.; Duran-Olivencia, M.A.; Maes, D. Observing classical nucleation theory at work by monitoring phase transitions with molecular precision. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Wolde, P.R.T.; Frenkel, D. Enhancement of protein crystal nucleation by critical density fluctuations. Science 1997, 277, 1975–1978. [Google Scholar] [CrossRef] [PubMed]
- Völlenkle, C.; Weigert, S.; Ilk, N.; Egelseer, E.; Weber, V.; Loth, F.; Falkenhagen, D.; Sleytr, U.B.; Sára, M. Construction of a functional S-layer fusion protein comprising an immunoglobulin G-binding domain for development of specific adsorbents for extracorporeal blood purification. Appl. Environ. Microbiol. 2004, 70, 1514–1521. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.; Liu, J.; Egelseer, E.M.; Moll, D.; Knoll, W.; Sleytr, U.B.; Sára, M. Heterotetramers formed by an S-layer-streptavidin fusion protein and core-streptavidin as nanoarrayed template for biochip development. Small 2006, 2, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Ucisik, M.H.; Kupcu, S.; Breitwieser, A.; Gelbmann, N.; Schuster, B.; Sleytr, U.B. S-layer fusion protein as a tool functionalizing emulsomes and CurcuEmulsomes for antibody binding and targeting. Colloids Surf. B Biointerfaces 2015, 128, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Hutter, J.L.; Bechhoefer, J. Calibration of atomic-force microscope tips. Rev. Sci. Instrum. 1993, 64. [Google Scholar] [CrossRef]
- Butt, H.J.; Jaschke, M. Calculation of thermal noise in atomic force microscopy. Nanotechnology 1995, 6. [Google Scholar] [CrossRef]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breitwieser, A.; Iturri, J.; Toca-Herrera, J.-L.; Sleytr, U.B.; Pum, D. In Vitro Characterization of the Two-Stage Non-Classical Reassembly Pathway of S-Layers. Int. J. Mol. Sci. 2017, 18, 400. https://doi.org/10.3390/ijms18020400
Breitwieser A, Iturri J, Toca-Herrera J-L, Sleytr UB, Pum D. In Vitro Characterization of the Two-Stage Non-Classical Reassembly Pathway of S-Layers. International Journal of Molecular Sciences. 2017; 18(2):400. https://doi.org/10.3390/ijms18020400
Chicago/Turabian StyleBreitwieser, Andreas, Jagoba Iturri, Jose-Luis Toca-Herrera, Uwe B. Sleytr, and Dietmar Pum. 2017. "In Vitro Characterization of the Two-Stage Non-Classical Reassembly Pathway of S-Layers" International Journal of Molecular Sciences 18, no. 2: 400. https://doi.org/10.3390/ijms18020400