
Methylglyoxal-Derived Advanced Glycation Endproducts in Multiple Sclerosis


Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Advanced Glycation Endproducts
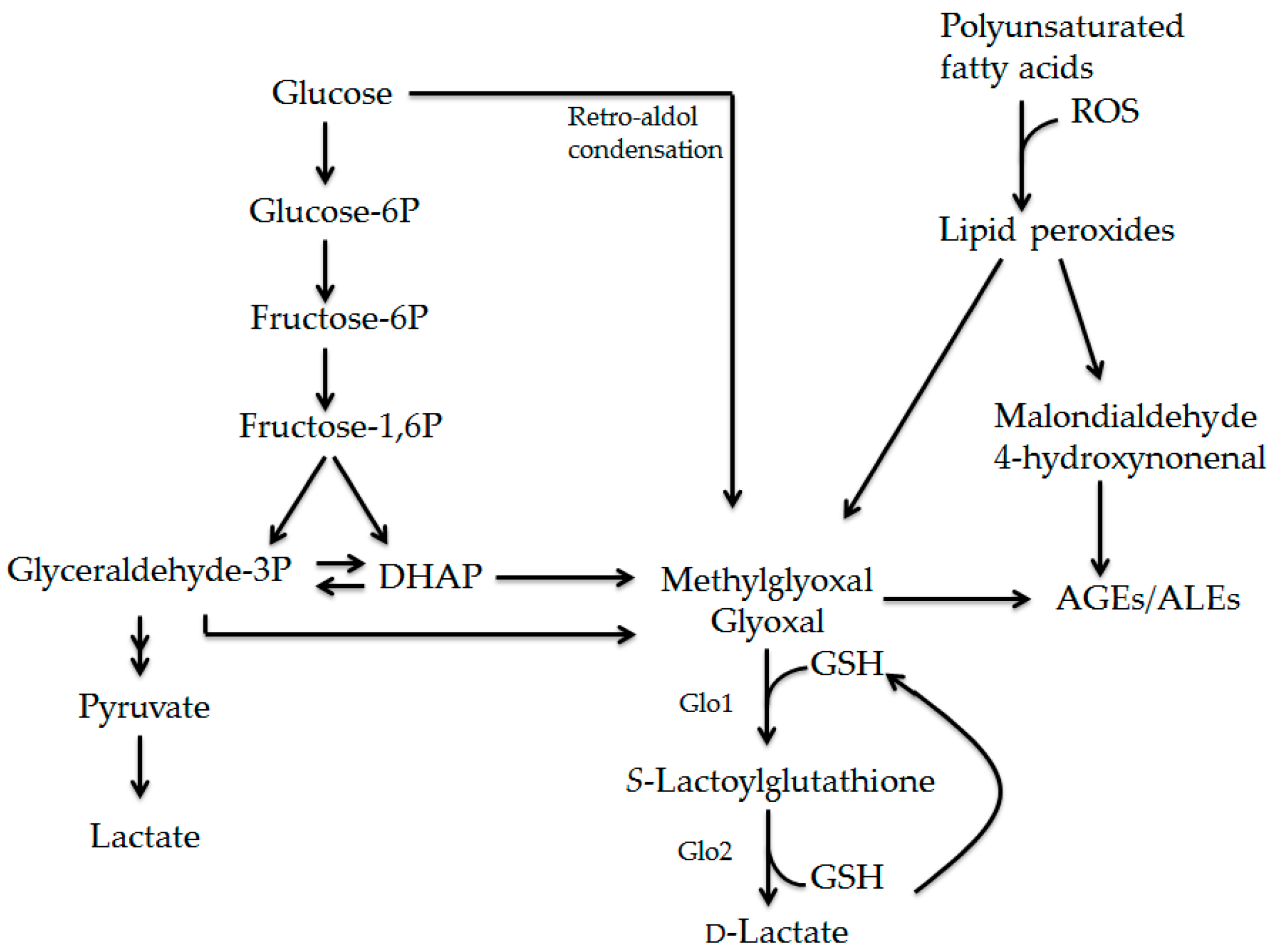
2.1. Formation of AGEs
2.2. Formation and Detoxification of Methylglyoxal
2.3. Biological Effects of Methylglyoxal and Advanced Glycation Endproducts
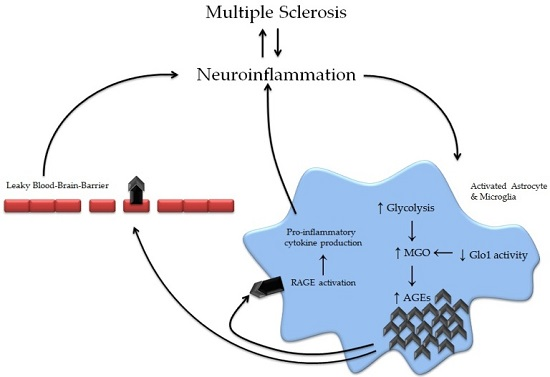
3. Advanced Glycation Endproducts in Multiple Sclerosis
3.1. Alterations in Advanced Glycation Endproduct Levels in Multiple Sclerosis
3.2. The Effects of Advanced Glycation Endproducts on Key Cells in MS Development
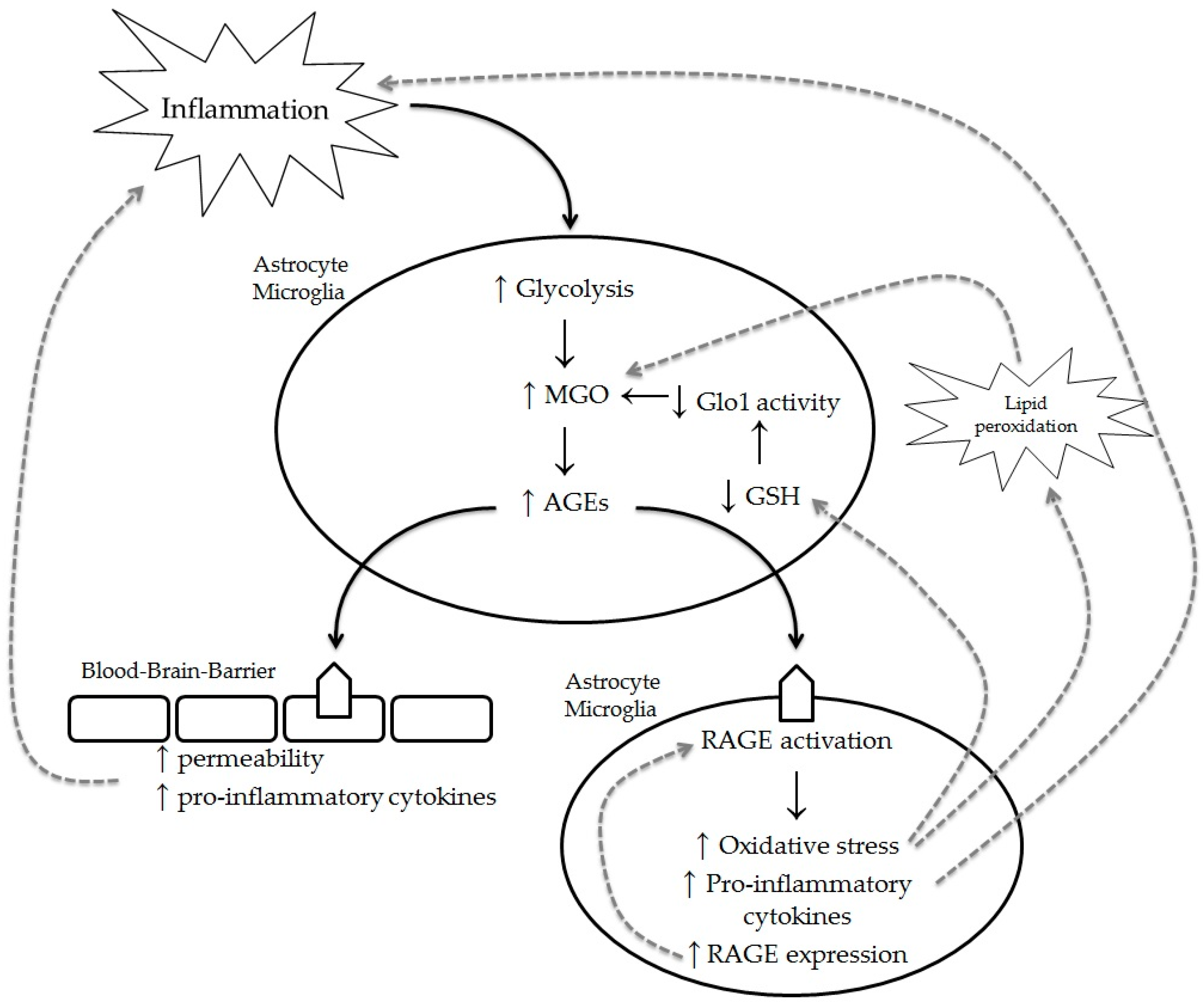
3.3. The Glyoxalase System in Multiple Sclerosis
3.4. Increased Glycolysis as an Underlying Mechanism for the Formation of Methylglyoxal-Derived Advanced Glycation Endproducts in Multiple Sclerosis
3.5. Increased Lipid Peroxidation as an Underlying Mechanism for the Formation of Methylglyoxal-Derived Advanced Glycation Endproducts in Multiple Sclerosis
3.6. Receptors for Advanced Glycation Endproducts in Multiple Sclerosis
4. Conclusions and Future Prospective
Conflicts of Interest
Abbreviations
| AGER1 | Advanced Glycation Endproduct Receptor 1 |
| AGEs | Advanced Glycation Endproducts |
| ALEs | Advanced Lipoxidation Endproducts |
| BBB | Blood–brain barrier |
| CD | Cluster of Differentiation |
| CEL | Nε-(carboxyethyl)-lysine |
| CML | Nε-(carboxymethyl)-lysine |
| CNS | Central Nervous System |
| CSF | Cerebrospinal Fluid |
| DHAP | Dihydroxyacetone Phosphate |
| DMTs | Disease Modifying Treatments |
| EAE | Experimental Autoimmune Encephalomyelitis |
| EDSS | Expended Disability Status Score |
| GAP | Glyceraldehyde-3-phosphate |
| Glo-1 | Glyoxalase-1 |
| Glo-2 | Glyoxalase-2 |
| GLUT | Glucose Transporter |
| GO | Glyoxal |
| GSH | Glutathione |
| GWAS | Genome-wide Association Studies |
| HLA | Human Leukocyte Antigen |
| HMGB | High-Mobility Group B |
| HNE | 4-Hydroxynonenal |
| IL | Interleukin |
| MDA | Malondialdehyde |
| MG-H1 | Methylglyoxal-derived Hydroimidazolone 1 |
| MGO | Methylglyoxal |
| MS | Multiple Sclerosis |
| NADPH | Nicotinamide Adenine Dinucleotide Phosphate |
| NAWM | Normal Appearing White Matter |
| NF-κB | Nuclear Factor-κB |
| PBMCs | Peripheral Blood Mononuclear Cells |
| PP-MS | Primary Progressive Multiple Sclerosis |
| RAGE | Receptor for Advanced Glycation Endproducts |
| ROS | Reactive Oxygen Species |
| RR-MS | Relapsing Remitting Multiple Sclerosis |
| SP-MS | Secondary Progressive Multiple Sclerosis |
| sRAGE | Soluble Receptor of Advanced Glycation Endproducts |
| THP | Tetrahydropyrimidine |
| TNFα | Tumor Necrosis Factor alpha |
References
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Bar-Or, A.; Oliveira, E.M.; Anderson, D.E.; Hafler, D.A. Molecular pathogenesis of multiple sclerosis. J. Neuroimmunol. 1999, 100, 252–259. [Google Scholar] [CrossRef]
- Ellwardt, E.; Zipp, F. Molecular mechanisms linking neuroinflammation and neurodegeneration in MS. Exp. Neurol. 2014, 262, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Scalfari, A.; Neuhaus, A.; Daumer, M.; Muraro, P.A.; Ebers, G.C. Onset of secondary progressive phase and long-term evolution of multiple sclerosis. J. Neurol. Neurosurg. 2014, 85, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.S.; Lees, J.G.; Moalem-Taylor, G. The contribution of immune and glial cell types in experimental autoimmune encephalomyelitis and multiple sclerosis. Mult. Scler. Int. 2014, 2014, 285245. [Google Scholar] [CrossRef] [PubMed]
- Hoglund, R.A.; Maghazachi, A.A. Multiple sclerosis and the role of immune cells. World J. Exp. Med. 2014, 4, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, G.G.; Pacheco-Moises, F.P.; Macias-Islas, M.A.; Flores-Alvarado, L.J.; Mireles-Ramirez, M.A.; Gonzalez-Renovato, E.D.; Hernandez-Navarro, V.E.; Sanchez-Lopez, A.L.; Alatorre-Jimenez, M.A. Role of the blood–brain barrier in multiple sclerosis. Arch. Med. Res. 2014, 45, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, A.K.; Baarnhielm, M.; Olsson, T.; Alfredsson, L. Tobacco smoking, but not swedish snuff use, increases the risk of multiple sclerosis. Neurology 2009, 73, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.L.; Zhang, S.M.; O’Reilly, E.; Hernan, M.A.; Olek, M.J.; Willett, W.C.; Ascherio, A. Vitamin D intake and incidence of multiple sclerosis. Neurology 2004, 62, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Levin, L.I.; Munger, K.L.; O’Reilly, E.J.; Falk, K.I.; Ascherio, A. Primary infection with the epstein-barr virus and risk of multiple sclerosis. Ann. Neurol. 2010, 67, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Mallucci, G.; Peruzzotti-Jametti, L.; Bernstock, J.D.; Pluchino, S. The role of immune cells, glia and neurons in white and gray matter pathology in multiple sclerosis. Prog. Neurobiol. 2015, 127–128, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Simpson, S., Jr.; Blizzard, L.; Otahal, P.; van der Mei, I.; Taylor, B. Latitude is significantly associated with the prevalence of multiple sclerosis: A meta-analysis. J. Neurol. Neurosurg. 2011, 82, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Bogie, J.F.; Stinissen, P.; Hendriks, J.J. Macrophage subsets and microglia in multiple sclerosis. Acta Neuropathol. 2014, 128, 191–213. [Google Scholar] [CrossRef] [PubMed]
- Vainchtein, I.D.; Vinet, J.; Brouwer, N.; Brendecke, S.; Biagini, G.; Biber, K.; Boddeke, H.W.; Eggen, B.J. In acute experimental autoimmune encephalomyelitis, infiltrating macrophages are immune activated, whereas microglia remain immune suppressed. Glia 2014, 62, 1724–1735. [Google Scholar] [CrossRef] [PubMed]
- Nair, A.; Frederick, T.J.; Miller, S.D. Astrocytes in multiple sclerosis: A product of their environment. Cell. Mol. Life Sci. 2008, 65, 2702–2720. [Google Scholar] [CrossRef] [PubMed]
- Hemmer, B.; Kerschensteiner, M.; Korn, T. Role of the innate and adaptive immune responses in the course of multiple sclerosis. Lancet Neurol. 2015, 14, 406–419. [Google Scholar] [CrossRef]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Stitt, A.W.; Li, Y.M.; Gardiner, T.A.; Bucala, R.; Archer, D.B.; Vlassara, H. Advanced glycation end products (AGEs) co-localize with age receptors in the retinal vasculature of diabetic and of age-infused rats. Am. J. Pathol. 1997, 150, 523–531. [Google Scholar] [PubMed]
- Van Eupen, M.G.; Schram, M.T.; Colhoun, H.M.; Hanssen, N.M.; Niessen, H.W.; Tarnow, L.; Parving, H.H.; Rossing, P.; Stehouwer, C.D.; Schalkwijk, C.G. The methylglyoxal-derived AGE tetrahydropyrimidine is increased in plasma of individuals with type 1 diabetes mellitus and in atherosclerotic lesions and is associated with sVCAM-1. Diabetologia 2013, 56, 1845–1855. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, N.M.; Wouters, K.; Huijberts, M.S.; Gijbels, M.J.; Sluimer, J.C.; Scheijen, J.L.; Heeneman, S.; Biessen, E.A.; Daemen, M.J.; Brownlee, M.; et al. Higher levels of advanced glycation endproducts in human carotid atherosclerotic plaques are associated with a rupture-prone phenotype. Eur. Heart J. 2014, 35, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Gaens, K.H.J.; Goossens, G.H.; Niessen, P.M.; van Greevenbroek, M.M.; van der Kallen, C.J.H.; Niessen, H.W.; Rensen, S.S.; Buurman, W.A.; Greve, J.W.M.; Blaak, E.E.; et al. Nε-(carboxymethyl)lysine-receptor for advanced glycation end product axis is a key modulator of obesity-induced dysregulation of adipokine expression and insulin resistance. Arterioscl. Throm. Vas. 2014, 34, 1199–1208. [Google Scholar] [CrossRef] [PubMed]
- Gaens, K.H.; Niessen, P.M.; Rensen, S.S.; Buurman, W.A.; Greve, J.W.; Driessen, A.; Wolfs, M.G.; Hofker, M.H.; Bloemen, J.G.; Dejong, C.H.; et al. Endogenous formation of Nε-(carboxymethyl)lysine is increased in fatty livers and induces inflammatory markers in an in vitro model of hepatic steatosis. J. Hepatol. 2012, 56, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Ahmed, U.; Thornalley, P.J.; Hager, K.; Fleischer, G.; Munch, G. Protein glycation, oxidation and nitration adduct residues and free adducts of cerebrospinal fluid in Alzheimer’s disease and link to cognitive impairment. J. Neurochem. 2005, 92, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Dalfo, E.; Portero-Otin, M.; Ayala, V.; Martinez, A.; Pamplona, R.; Ferrer, I. Evidence of oxidative stress in the neocortex in incidental lewy body disease. J. Neuropathol. Exp. Neurol. 2005, 64, 816–830. [Google Scholar] [CrossRef] [PubMed]
- Ledesma, M.D.; Bonay, P.; Avila, J. Tau protein from Alzheimer’s disease patients is glycated at its tubulin-binding domain. J. Neurochem. 1995, 65, 1658–1664. [Google Scholar] [CrossRef] [PubMed]
- Vitek, M.P.; Bhattacharya, K.; Glendening, J.M.; Stopa, E.; Vlassara, H.; Bucala, R.; Manogue, K.; Cerami, A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 4766–4770. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, Z.; Hennies, C.; Sternberg, D.; Wang, P.; Kinkel, P.; Hojnacki, D.; Weinstock-Guttmann, B.; Munschauer, F. Diagnostic potential of plasma carboxymethyllysine and carboxyethyllysine in multiple sclerosis. J. Neuroinflam. 2010, 7, 72. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, Z.; Ostrow, P.; Vaughan, M.; Chichelli, T.; Munschauer, F. Age-rage in multiple sclerosis brain. Immunol. Investig. 2011, 40, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Gaens, K.H.; Stehouwer, C.D.; Schalkwijk, C.G. Advanced glycation endproducts and its receptor for advanced glycation endproducts in obesity. Curr. Opin. Lipidol. 2013, 24, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Allaman, I.; Belanger, M.; Magistretti, P.J. Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 2015, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Maessen, D.E.; Stehouwer, C.D.; Schalkwijk, C.G. The role of methylglyoxal and the glyoxalase system in diabetes and other age-related diseases. Clin. Sci. 2015, 128, 839–861. [Google Scholar] [CrossRef] [PubMed]
- Lange, J.N.; Wood, K.D.; Knight, J.; Assimos, D.G.; Holmes, R.P. Glyoxal formation and its role in endogenous oxalate synthesis. Adv. Urol. 2012, 2012, 819202. [Google Scholar] [CrossRef] [PubMed]
- Pamplona, R. Advanced lipoxidation end-products. Chem. Biol. Interact. 2011, 192, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Vistoli, G.; de Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): An overview of their mechanisms of formation. Free Radic. Res. 2013, 47, 3–27. [Google Scholar] [CrossRef] [PubMed]
- Matafome, P.; Sena, C.; Seica, R. Methylglyoxal, obesity, and diabetes. Endocrine 2013, 43, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Di Loreto, S.; Caracciolo, V.; Colafarina, S.; Sebastiani, P.; Gasbarri, A.; Amicarelli, F. Methylglyoxal induces oxidative stress-dependent cell injury and up-regulation of interleukin-1β and nerve growth factor in cultured hippocampal neuronal cells. Brain Res. 2004, 1006, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Figarola, J.L.; Singhal, J.; Rahbar, S.; Awasthi, S.; Singhal, S.S. LR-90 prevents methylglyoxal-induced oxidative stress and apoptosis in human endothelial cells. Apoptosis 2014, 19, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, M.W.; Hedegaard, R.V.; Andersen, J.M.; de Courten, B.; Bugel, S.; Nielsen, J.; Skibsted, L.H.; Dragsted, L.O. Advanced glycation endproducts in food and their effects on health. Food Chem.Toxicol. 2013, 60, 10–37. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete-Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.F.; Ramasamy, R.; Schmidt, A.M. The rage axis: A fundamental mechanism signaling danger to the vulnerable vasculature. Circ. Res. 2010, 106, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Dukic-Stefanovic, S.; Gasic-Milenkovic, J.; Deuther-Conrad, W.; Munch, G. Signal transduction pathways in mouse microglia N-11 cells activated by advanced glycation endproducts (AGEs). J. Neurochem. 2003, 87, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.L.; Li, Z.; Yuan, M.; Yu, A.C.; Zhu, X.; Tso, M.O. Sinomenine inhibits activation of rat retinal microglia induced by advanced glycation end products. Int. Immunopharmacol. 2007, 7, 1552–1558. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, K.; Liu, K.; Zhou, Y.; Zhang, T.; Wang, B.; Mi, M. DHA inhibited AGEs-induced retinal microglia activation via suppression of the PPARγ/NFκB pathway and reduction of signal transducers in the AGEs/RAGE axis recruitment into lipid rafts. Neurochem. Res. 2015, 40, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.B.; Uy, B.; Perera, A.; Nicholson, L.F. AGEs-RAGE mediated up-regulation of connexin43 in activated human microglial CHME-5 cells. Neurochem. Int. 2012, 60, 640–651. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, D.D.; Liang, Y.Y.; Wang, D.S.; Cai, N.S. Activation of astrocytes by advanced glycation end products: Cytokines induction and nitric oxide release. Acta Pharmacol. Sin. 2002, 23, 974–980. [Google Scholar] [PubMed]
- Begley, D.J.; Brightman, M.W. Structural and functional aspects of the blood–brain barrier. Prog. Drug Res. 2003, 61, 39–78. [Google Scholar] [PubMed]
- Hussain, M.; Bork, K.; Gnanapragassam, V.S.; Bennmann, D.; Jacobs, K.; Navarette-Santos, A.; Hofmann, B.; Simm, A.; Danker, K.; Horstkorte, R. Novel insights in the dysfunction of human blood–brain barrier after glycation. Mech. Ageing Dev. 2016, 155, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, F.; Sano, Y.; Tominaga, O.; Maeda, T.; Abe, M.A.; Kanda, T. Advanced glycation end-products disrupt the blood–brain barrier by stimulating the release of transforming growth factor-β by pericytes and vascular endothelial growth factor and matrix metalloproteinase-2 by endothelial cells in vitro. Neurobiol. Aging 2013, 34, 1902–1912. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, H.; Osanai, M.; Chiba, H.; Nishikiori, N.; Kojima, T.; Ohtsuka, K.; Sawada, N. Glyceraldehyde-derived advanced glycation end-products preferentially induce VEGF expression and reduce GDNF expression in human astrocytes. Biochem. Biophys. Res. Commun. 2005, 330, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.N.; Lim, J.L.; Nijland, P.G.; Witte, M.E.; van Horssen, J. Glutathione in multiple sclerosis: More than just an antioxidant? Mult. Scler. 2014, 20, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Scapagnini, G.; Ravagna, A.; Bella, R.; Foresti, R.; Bates, T.E.; Giuffrida-Stella, A.M.; Pennisi, G. Nitric oxide synthase is present in the cerebrospinal fluid of patients with active multiple sclerosis and is associated with increases in cerebrospinal fluid protein nitrotyrosine and S-nitrosothiols and with changes in glutathione levels. J. Neurosci. Res. 2002, 70, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.Y.; Lee, S.P.; Denney, D.R.; Lynch, S.G. Lower levels of glutathione in the brains of secondary progressive multiple sclerosis patients measured by 1H magnetic resonance chemical shift imaging at 3 T. Mult. Scler. 2011, 17, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.; Ratiney, H.; Hammond-Rosenbluth, K.E.; Pelletier, D.; Nelson, S.J. MR spectroscopic imaging of glutathione in the white and gray matter at 7 T with an application to multiple sclerosis. Magn. Reson. Imaging 2010, 28, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Junaid, M.A.; Kowal, D.; Barua, M.; Pullarkat, P.S.; Sklower Brooks, S.; Pullarkat, R.K. Proteomic studies identified a single nucleotide polymorphism in glyoxalase i as autism susceptibility factor. Am. J. Med. Genet. A 2004, 131, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Sidoti, A.; Antognelli, C.; Rinaldi, C.; D’Angelo, R.; Dattola, V.; Girlanda, P.; Talesa, V.; Amato, A. Glyoxalase I A111E, paraoxonase 1 Q192R and L55M polymorphisms: Susceptibility factors of multiple sclerosis? Mult. Scler. 2007, 13, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Bittner, C.X.; Loaiza, A.; Ruminot, I.; Larenas, V.; Sotelo-Hitschfeld, T.; Gutierrez, R.; Cordova, A.; Valdebenito, R.; Frommer, W.B.; Barros, L.F. High resolution measurement of the glycolytic rate. Front. Neuroenerg. 2010, 2, 26. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Mendez, A.; Almeida, A.; Fernandez, E.; Maestre, C.; Moncada, S.; Bolanos, J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-CDH1. Nat. Cell Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Esaki, T.; Shimoji, K.; Cook, M.; Law, M.J.; Kaufman, E.; Sokoloff, L. Dichloroacetate effects on glucose and lactate oxidation by neurons and astroglia in vitro and on glucose utilization by brain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4879–4884. [Google Scholar] [CrossRef] [PubMed]
- Karnovsky, M.L. Metabolic basis of phagocytic activity. Physiol. Rev. 1962, 42, 143–168. [Google Scholar] [PubMed]
- Bogie, J.F.; Timmermans, S.; Huynh-Thu, V.A.; Irrthum, A.; Smeets, H.J.; Gustafsson, J.A.; Steffensen, K.R.; Mulder, M.; Stinissen, P.; Hellings, N.; et al. Myelin-derived lipids modulate macrophage activity by liver X receptor activation. PLoS ONE 2012, 7, e44998. [Google Scholar] [CrossRef] [PubMed]
- Belanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [PubMed]
- Nijland, P.G.; Michailidou, I.; Witte, M.E.; Mizee, M.R.; van der Pol, S.M.; van Het Hof, B.; Reijerkerk, A.; Pellerin, L.; van der Valk, P.; de Vries, H.E.; et al. Cellular distribution of glucose and monocarboxylate transporters in human brain white matter and multiple sclerosis lesions. Glia 2014, 62, 1125–1141. [Google Scholar] [CrossRef] [PubMed]
- Jurcovicova, J. Glucose transport in brain-effect of inflammation. Endocr. Regul. 2014, 48, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Stys, P.K. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. Lancet Neurol. 2009, 8, 280–291. [Google Scholar] [CrossRef]
- Schiepers, C.; van Hecke, P.; Vandenberghe, R.; van Oostende, S.; Dupont, P.; Demaerel, P.; Bormans, G.; Carton, H. Positron emission tomography, magnetic resonance imaging and proton NMR spectroscopy of white matter in multiple sclerosis. Mult. Scler. 1997, 3, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Schocke, M.F.; Berger, T.; Felber, S.R.; Wolf, C.; Deisenhammer, F.; Kremser, C.; Seppi, K.; Aichner, F.T. Serial contrast-enhanced magnetic resonance imaging and spectroscopic imaging of acute multiple sclerosis lesions under high-dose methylprednisolone therapy. NeuroImage 2003, 20, 1253–1263. [Google Scholar] [CrossRef]
- Morland, C.; Henjum, S.; Iversen, E.G.; Skrede, K.K.; Hassel, B. Evidence for a higher glycolytic than oxidative metabolic activity in white matter of rat brain. Neurochem. Int. 2007, 50, 703–709. [Google Scholar] [CrossRef] [PubMed]
- Funfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Saab, A.S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Mobius, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2012, 485, 517–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Horssen, J.; Witte, M.E.; Schreibelt, G.; de Vries, H.E. Radical changes in multiple sclerosis pathogenesis. Biochim. Biophys. Acta 2011, 1812, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Ljubisavljevic, S. Oxidative stress and neurobiology of demyelination. Mol. Neurobiol. 2014, 53, 744–758. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Haghighi, S.; Andersen, O.; Yao, Y.; Rosengren, L.; Blennow, K.; Pratico, D.; Zetterberg, H. Elevated cerebrospinal fluid F2-isoprostane levels indicating oxidative stress in healthy siblings of multiple sclerosis patients. Neurosci. Lett. 2007, 414, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Colton, C.A.; Gilbert, D.L. Microglia, an in vivo source of reactive oxygen species in the brain. Adv. Neurol. 1993, 59, 321–326. [Google Scholar] [PubMed]
- Guan, J.Z.; Guan, W.P.; Maeda, T.; Xie, G.Q.; Wan, G.Z.; Makino, N. Patients with multiple sclerosis show increased oxidative stress markers and somatic telomere length shortening. Mol. Cell. Biochem. 2015, 400, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Van Horssen, J.; Schreibelt, G.; Drexhage, J.; Hazes, T.; Dijkstra, C.D.; van der Valk, P.; de Vries, H.E. Severe oxidative damage in multiple sclerosis lesions coincides with enhanced antioxidant enzyme expression. Free Radic. Biol. Med. 2008, 45, 1729–1737. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Xie, K.; Wang, C.; Bi, J. Oxidative stress induced by lipid peroxidation is related with inflammation of demyelination and neurodegeneration in multiple sclerosis. Eur. Neurol. 2014, 72, 249–254. [Google Scholar] [PubMed]
- Andersson, A.; Covacu, R.; Sunnemark, D.; Danilov, A.I.; dal Bianco, A.; Khademi, M.; Wallstrom, E.; Lobell, A.; Brundin, L.; Lassmann, H.; et al. Pivotal advance: HMGB1 expression in active lesions of human and experimental multiple sclerosis. J. Leukoc. Biol. 2008, 84, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.S.; Wu, Z.Y.; Zhang, H.P.; Furtado, G.; Chen, X.; Yan, S.F.; Schmidt, A.M.; Brown, C.; Stern, A.; LaFaille, J.; et al. Suppression of experimental autoimmune encephalomyelitis by selective blockade of encephalitogenic T-cell infiltration of the central nervous system. Nat. Med. 2003, 9, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Keller, J.N. Evaluation of rage isoforms, ligands, and signaling in the brain. Biochim. Biophys. Acta 2005, 1746, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Liliensiek, B.; Weigand, M.A.; Bierhaus, A.; Nicklas, W.; Kasper, M.; Hofer, S.; Plachky, J.; Grone, H.J.; Kurschus, F.C.; Schmidt, A.M.; et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J. Clin. Investig. 2004, 113, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, Z.; Chiotti, A.; Tario, J.; Chichelli, T.; Patel, N.; Chadha, K.; Yu, J.; Karmon, Y. Reduced expression of membrane-bound (m)RAGE is a biomarker of multiple sclerosis disease progression. Immunobiology 2016, 221, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, Z.; Weinstock-Guttman, B.; Hojnacki, D.; Zamboni, P.; Zivadinov, R.; Chadha, K.; Lieberman, A.; Kazim, L.; Drake, A.; Rocco, P.; et al. Soluble receptor for advanced glycation end products in multiple sclerosis: A potential marker of disease severity. Mult. Scler. 2008, 14, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.A.; Drury, S.; Hudson, B.I.; Gleason, M.R.; Qu, W.; Lu, Y.; Lalla, E.; Chitnis, S.; Monteiro, J.; Stickland, M.H.; et al. Rage and arthritis: The G82S polymorphism amplifies the inflammatory response. Genes Immun. 2002, 3, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Stickland, M.H.; Futers, T.S.; Grant, P.J. Effects of novel polymorphisms in the rage gene on transcriptional regulation and their association with diabetic retinopathy. Diabetes 2001, 50, 1505–1511. [Google Scholar] [CrossRef] [PubMed]
- Tiszlavicz, Z.; Gyulai, Z.; Bencsik, K.; Szolnoki, Z.; Kocsis, A.K.; Somogyvari, F.; Vecsei, L.; Mandi, Y. Rage gene polymorphisms in patients with multiple sclerosis. J. Mol. Neurosci. 2009, 39, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhao, B.; Dai, D.; Yao, S.; Liang, W.; Yao, L.; Yang, Z. A functional p.82G>S polymorphism in the RAGE gene is associated with multiple sclerosis in the chinese population. Mult. Scler. 2011, 17, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Ramdas, M.; Zhu, L.; Chen, X.; Striker, G.E.; Vlassara, H. Oral advanced glycation endproducts (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses age receptor-1 and sirtuin 1. Proc. Natl. Acad. Sci. USA 2012, 109, 15888–15893. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Jiang, J.X.; Zhang, G.X. Macrophages: A double-edged sword in experimental autoimmune encephalomyelitis. Immunol. Lett. 2014, 160, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Kotter, M.R.; Zhao, C.; van Rooijen, N.; Franklin, R.J. Macrophage-depletion induced impairment of experimental CNS remyelination is associated with a reduced oligodendrocyte progenitor cell response and altered growth factor expression. Neurobiol. Dis. 2005, 18, 166–175. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wetzels, S.; Wouters, K.; Schalkwijk, C.G.; Vanmierlo, T.; Hendriks, J.J.A. Methylglyoxal-Derived Advanced Glycation Endproducts in Multiple Sclerosis. Int. J. Mol. Sci. 2017, 18, 421. https://doi.org/10.3390/ijms18020421
Wetzels S, Wouters K, Schalkwijk CG, Vanmierlo T, Hendriks JJA. Methylglyoxal-Derived Advanced Glycation Endproducts in Multiple Sclerosis. International Journal of Molecular Sciences. 2017; 18(2):421. https://doi.org/10.3390/ijms18020421
Chicago/Turabian StyleWetzels, Suzan, Kristiaan Wouters, Casper G. Schalkwijk, Tim Vanmierlo, and Jerome J. A. Hendriks. 2017. "Methylglyoxal-Derived Advanced Glycation Endproducts in Multiple Sclerosis" International Journal of Molecular Sciences 18, no. 2: 421. https://doi.org/10.3390/ijms18020421