
Advanced Glycation End Products Inhibit the Proliferation of Human Umbilical Vein Endothelial Cells by Inhibiting Cathepsin D

Abstract
:1. Introduction
2. Results
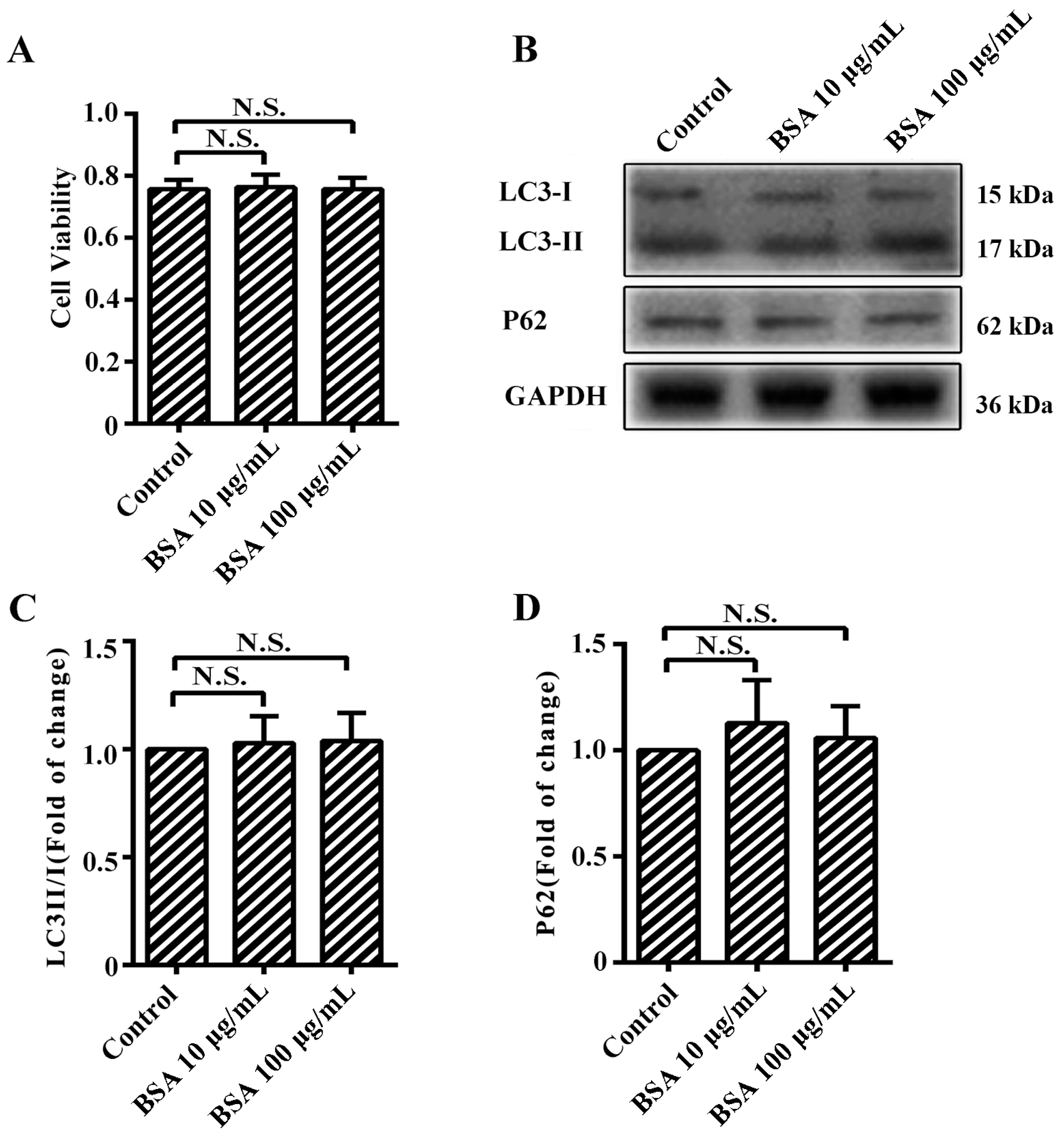
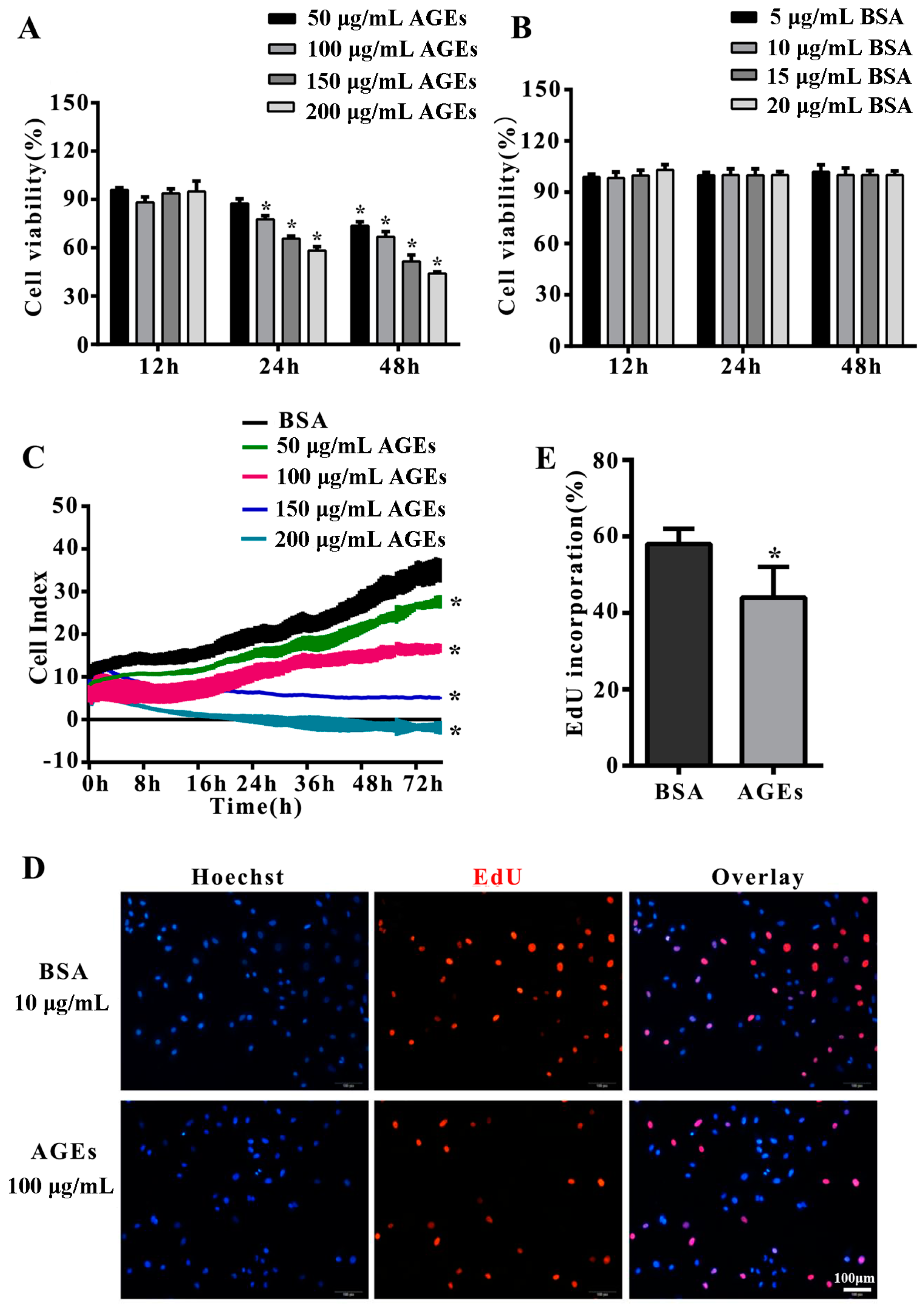
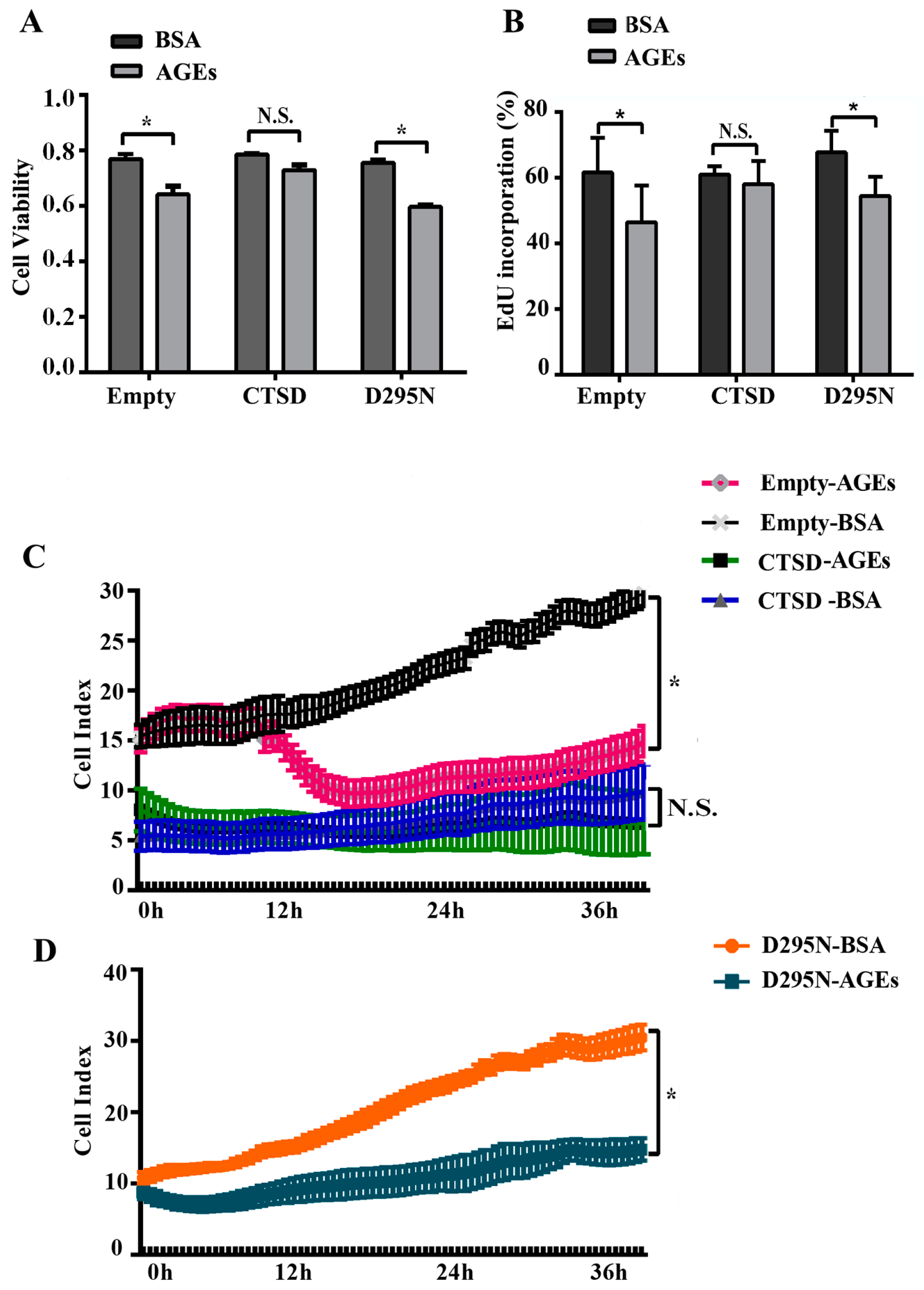
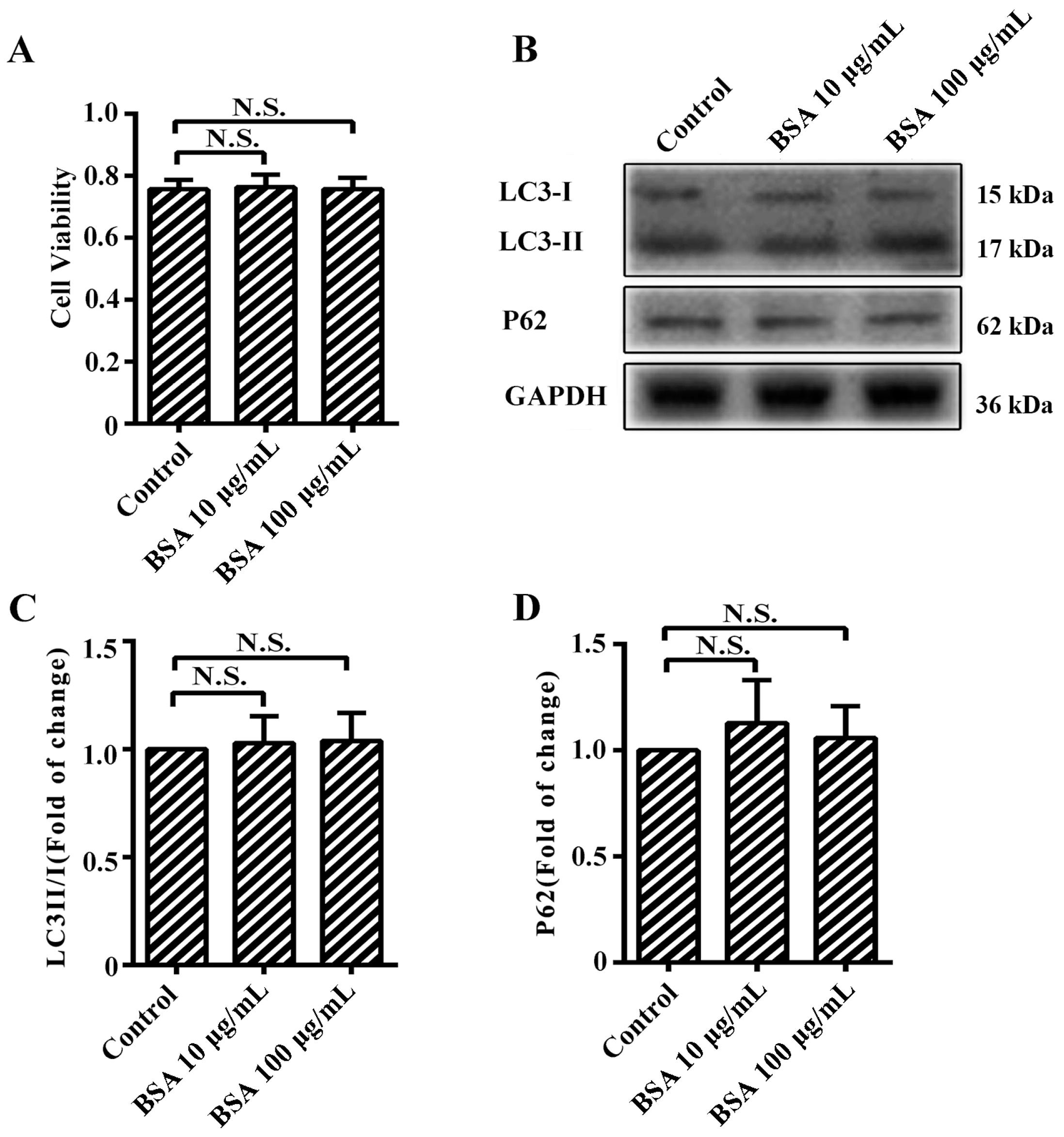
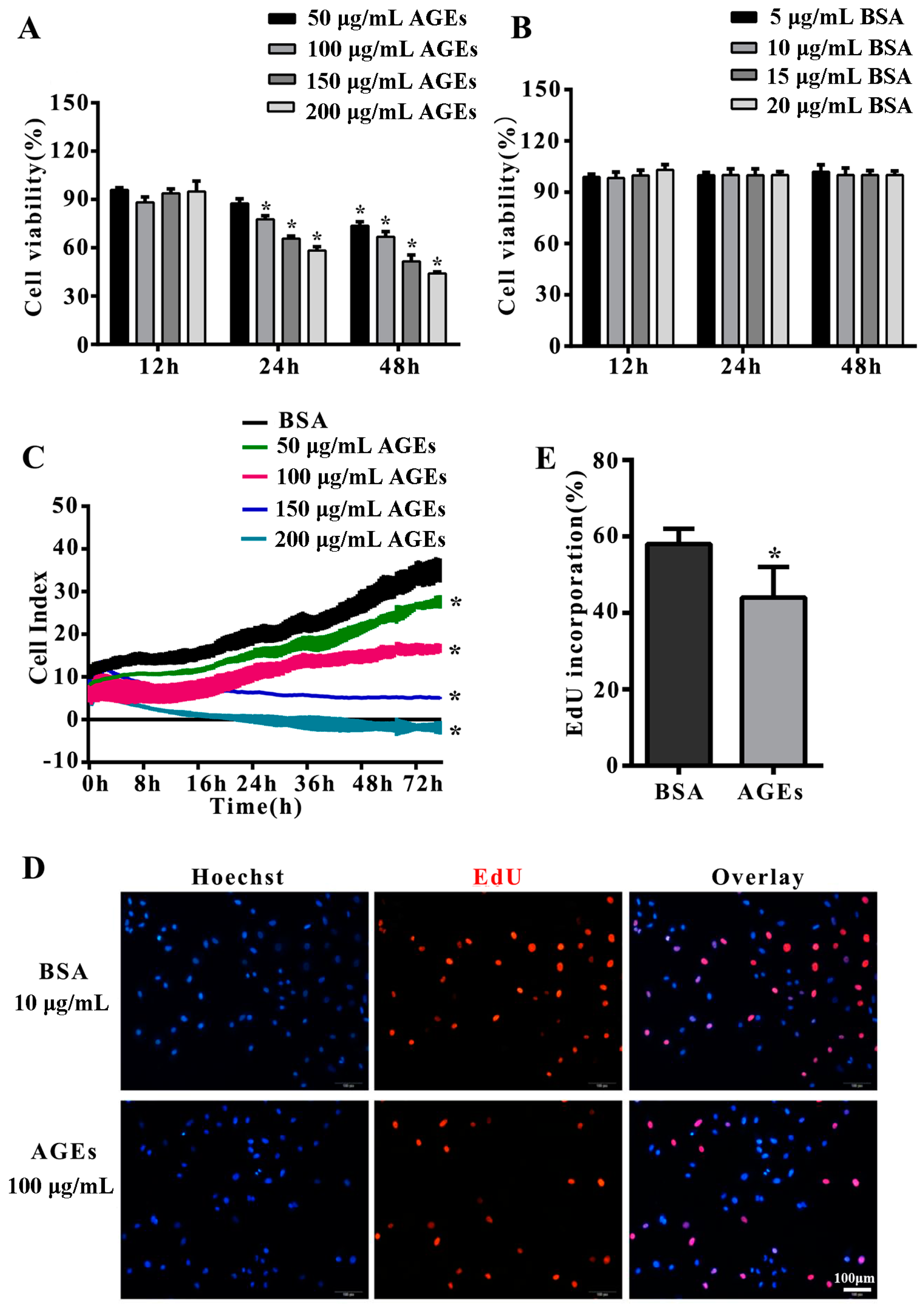
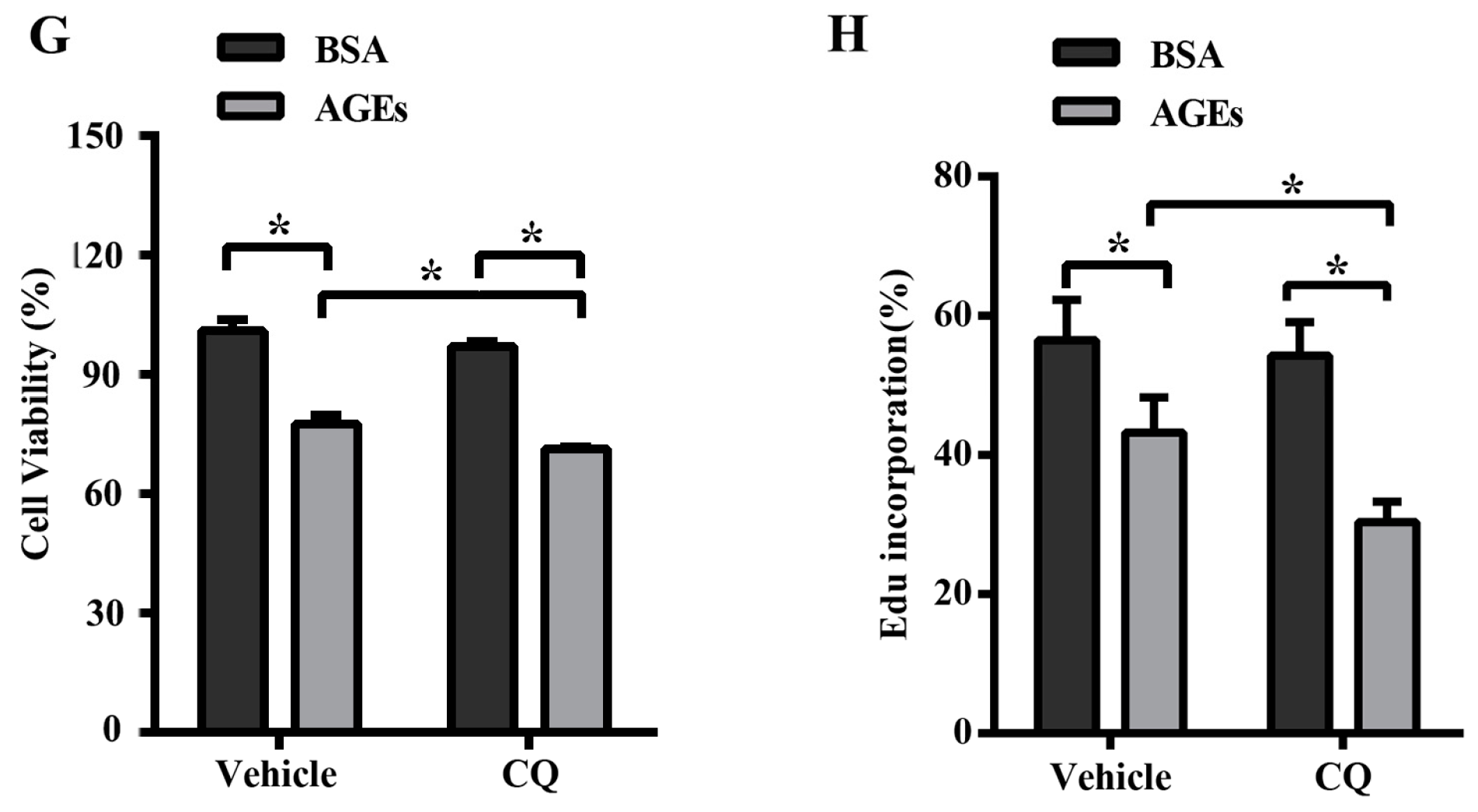
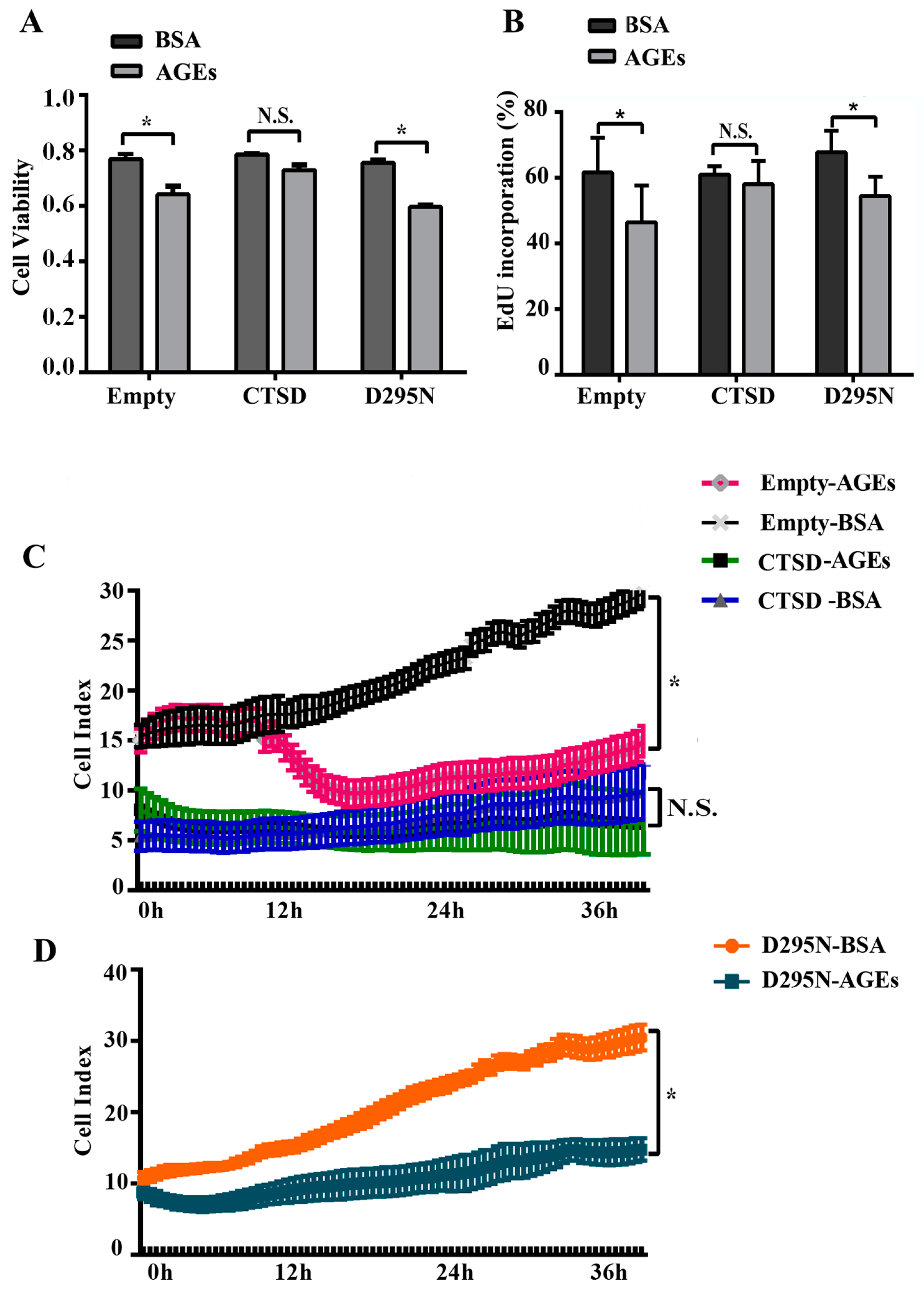
2.1. Advanced Glycation end-Products (AGEs) Inhibited the Proliferation of Human Umbilical Vein Endothelial Cells (HUVECs)
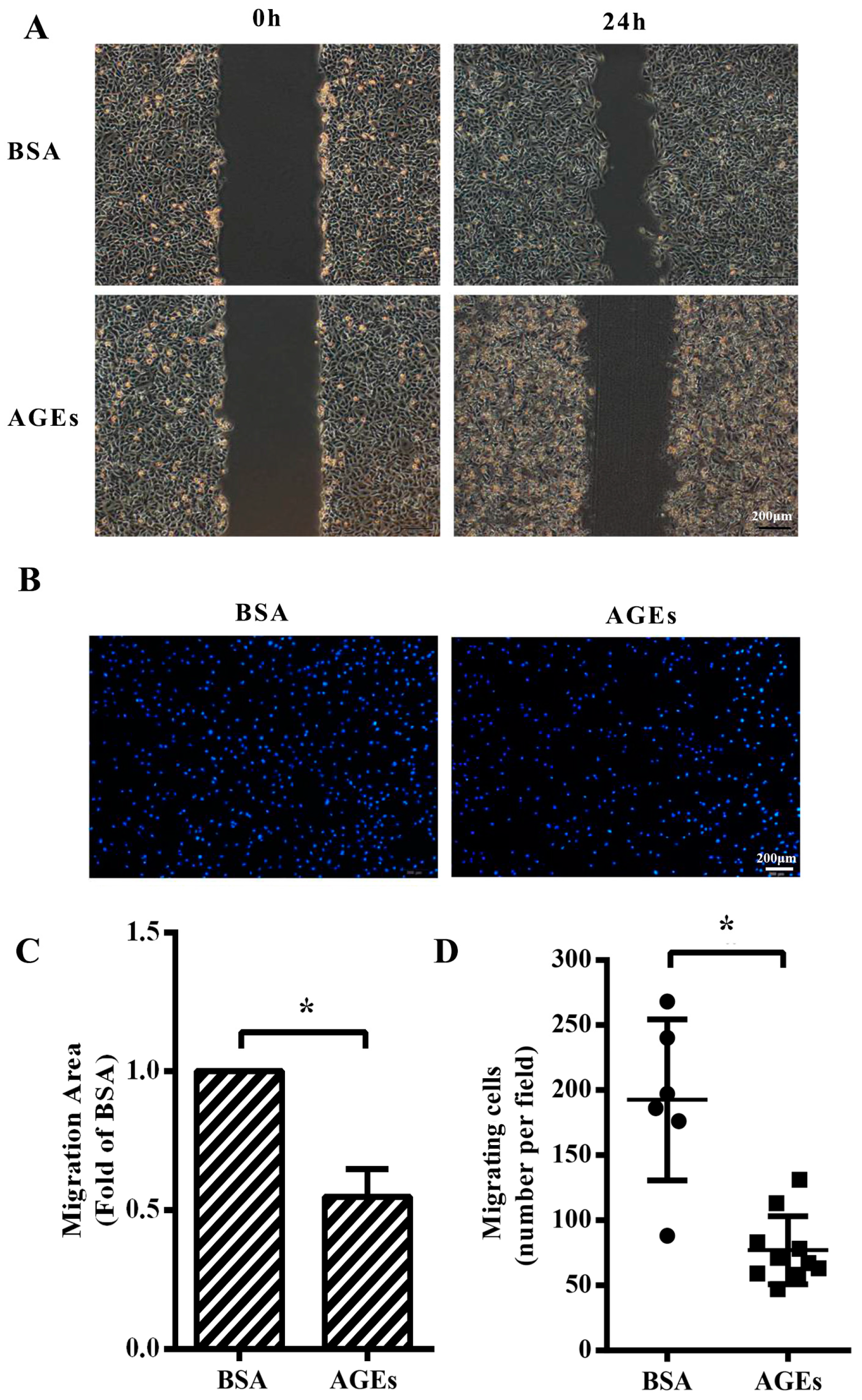
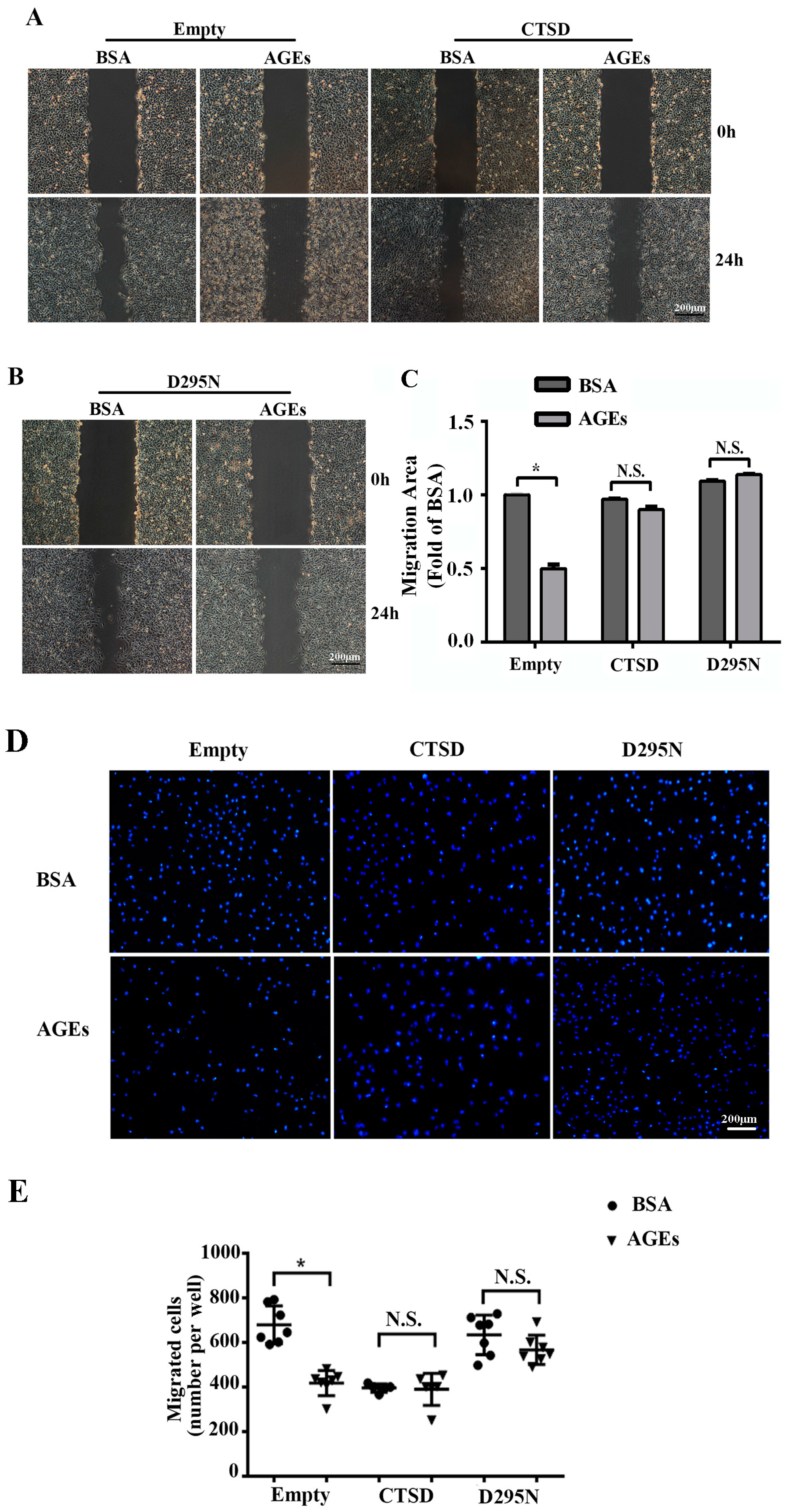
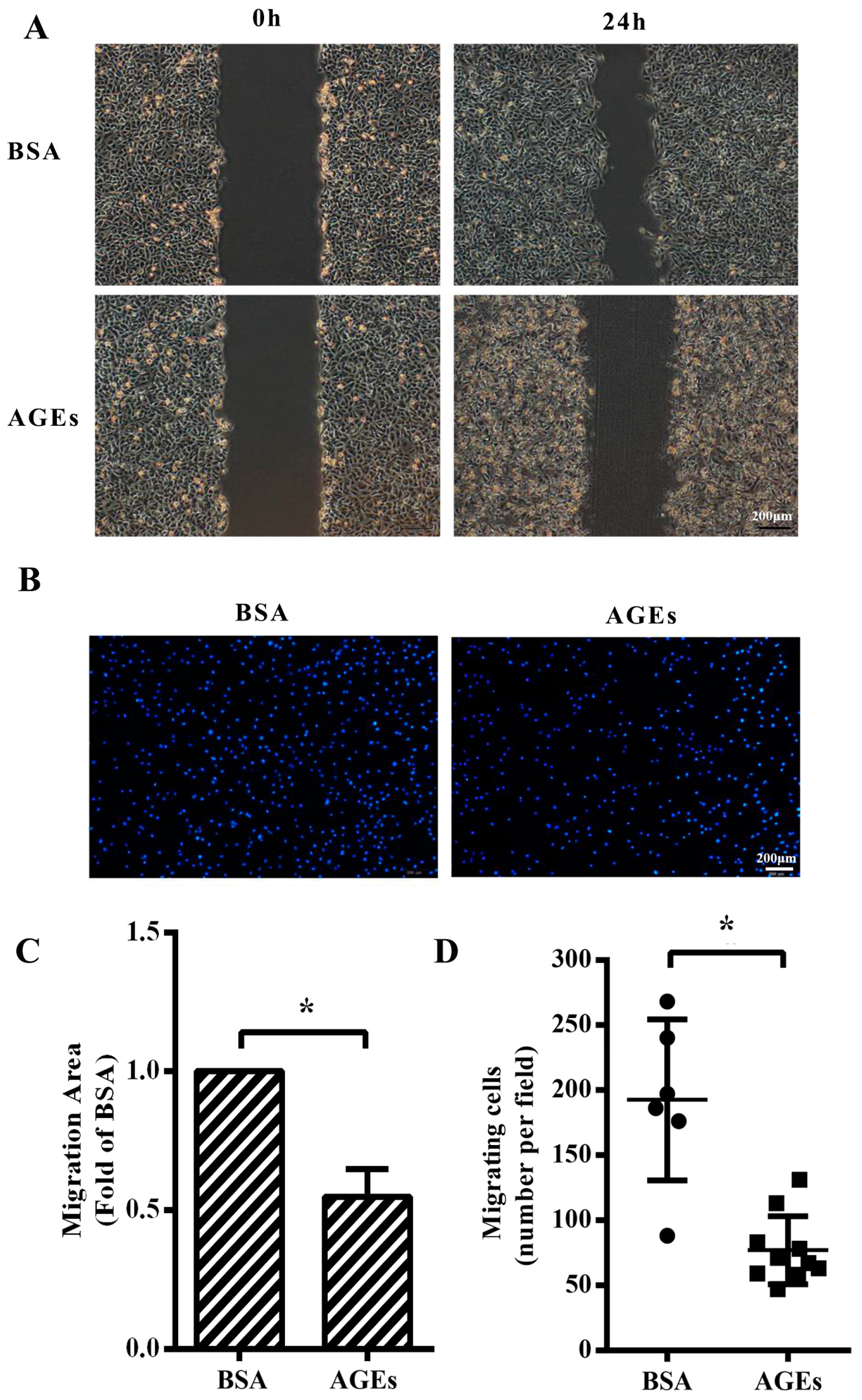
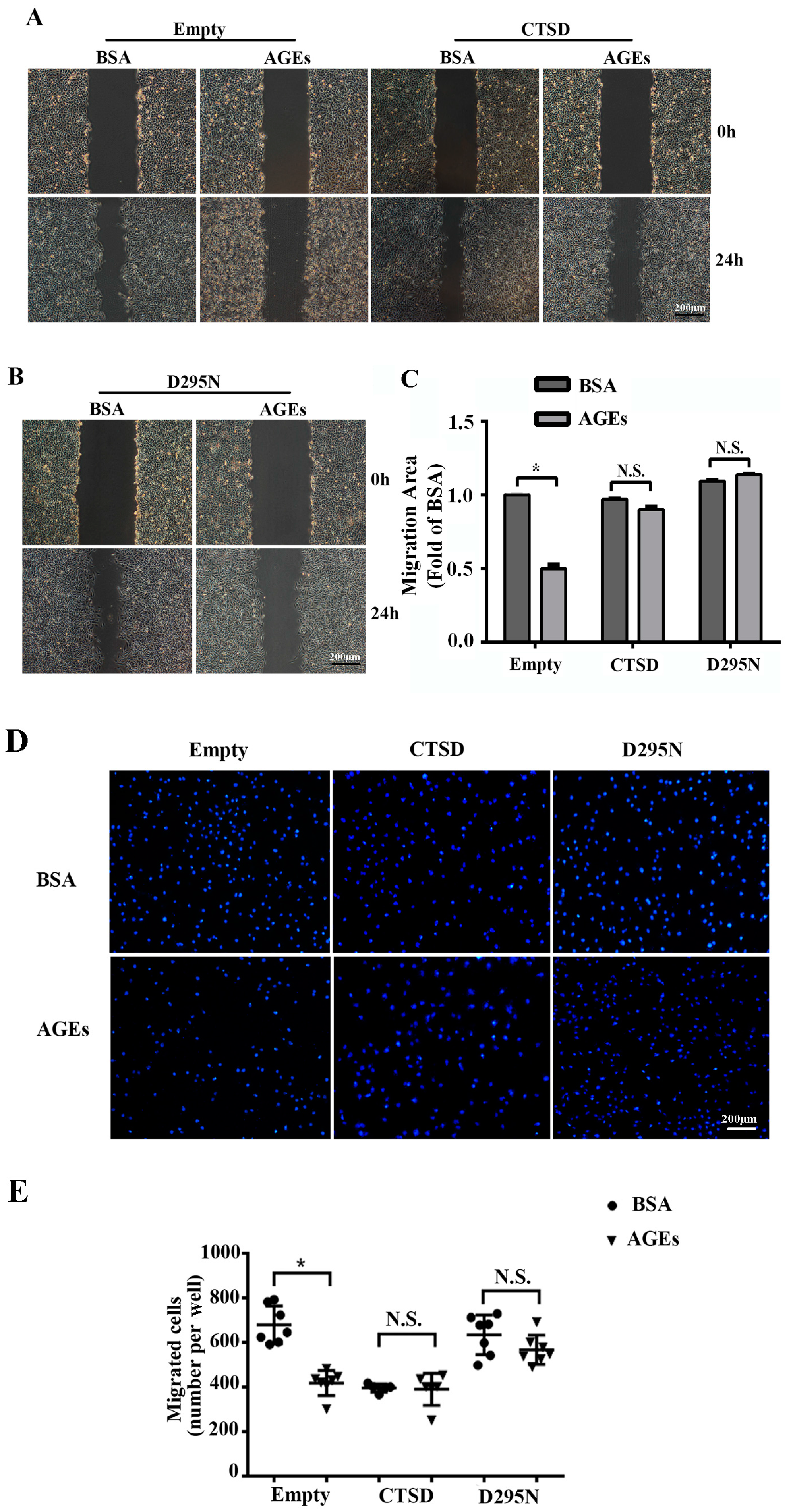
2.2. AGEs Inhibited the Migration of HUVECs
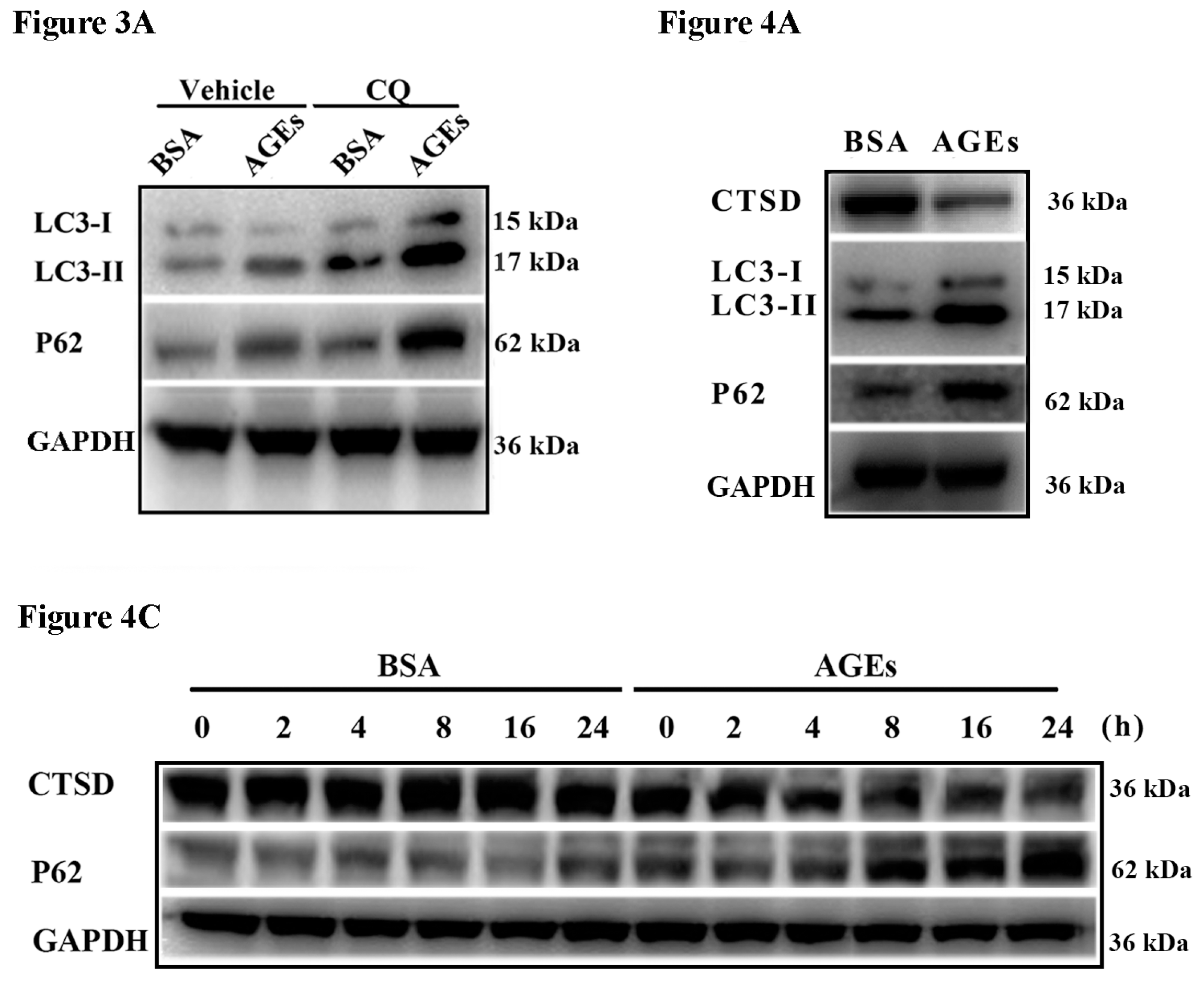
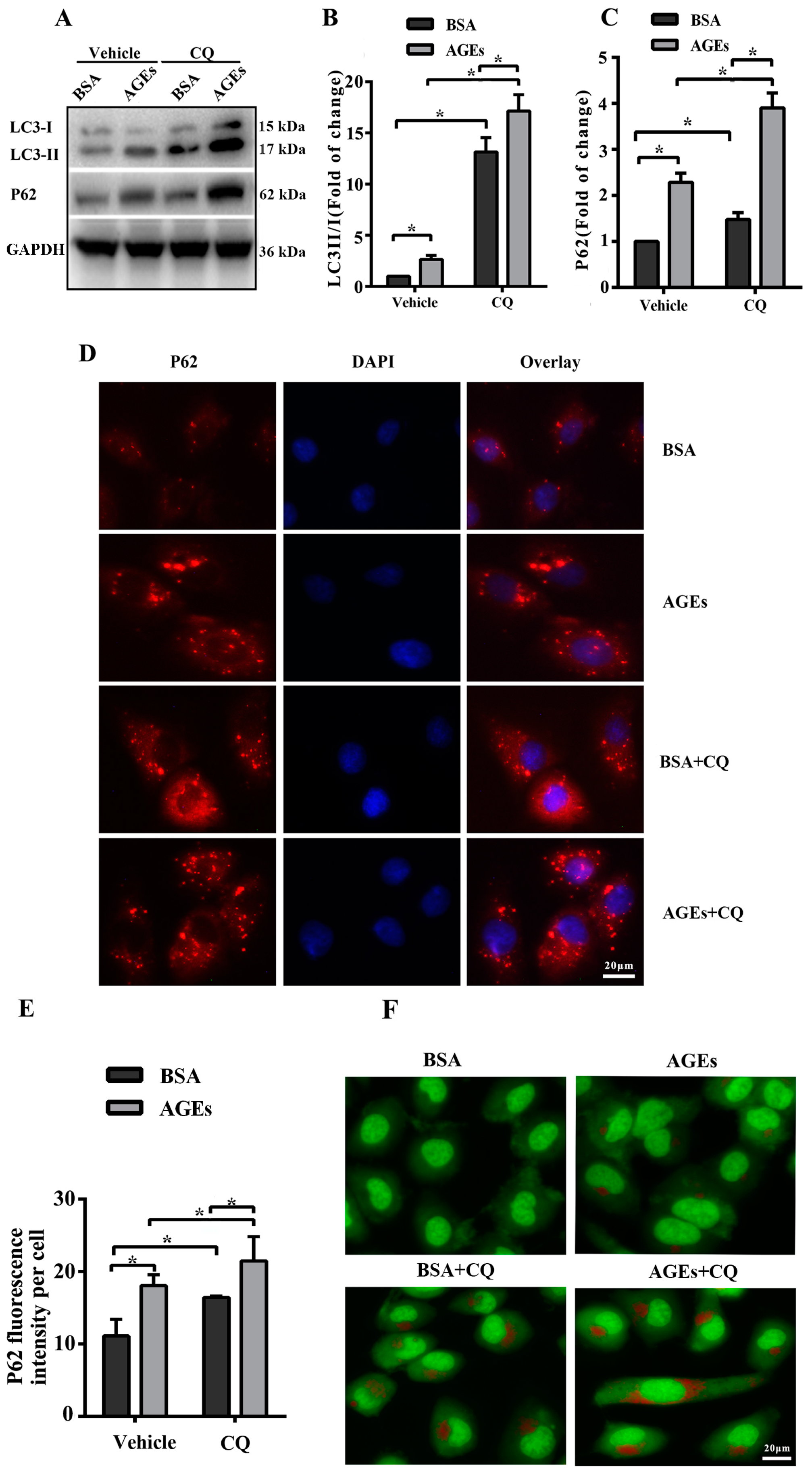
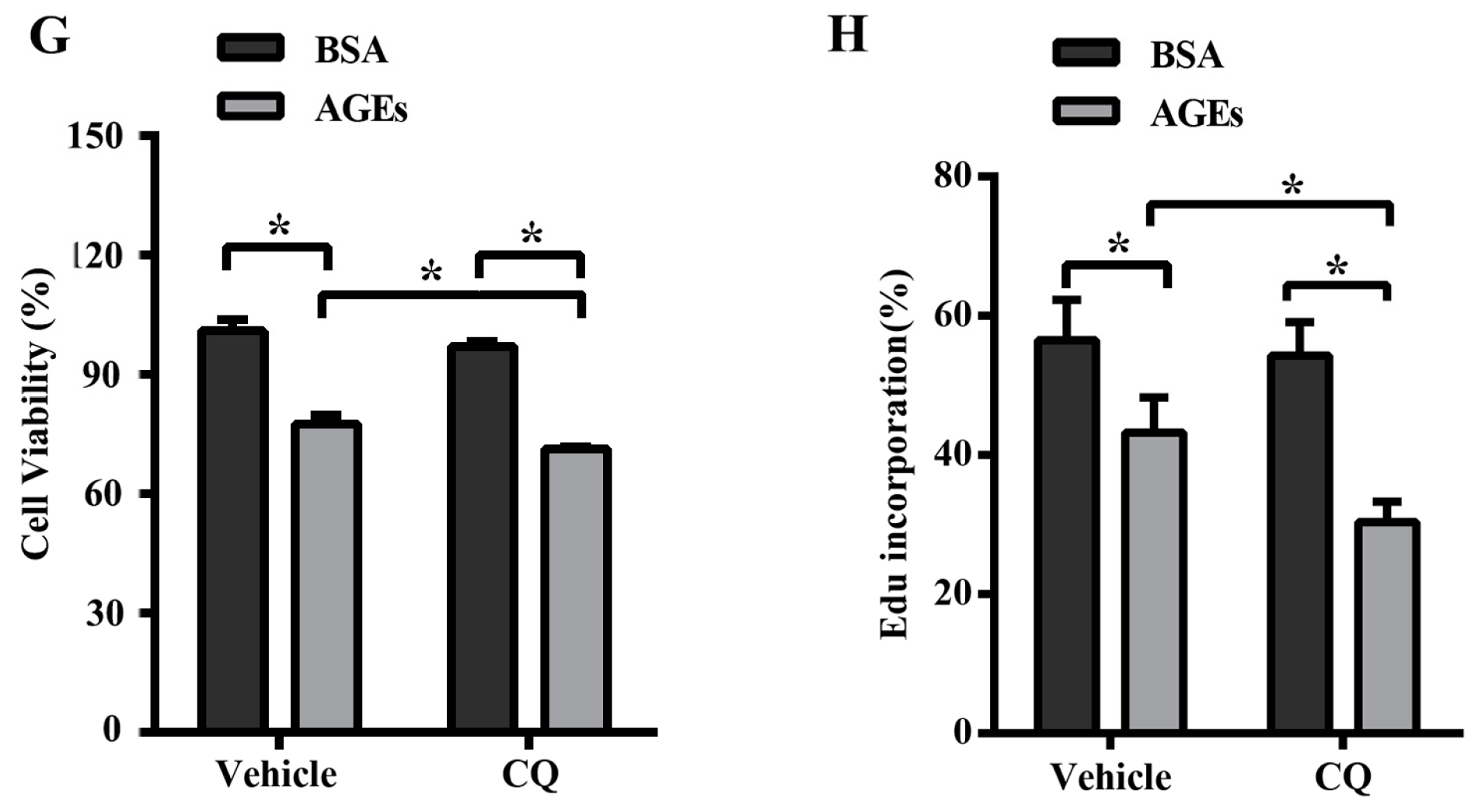
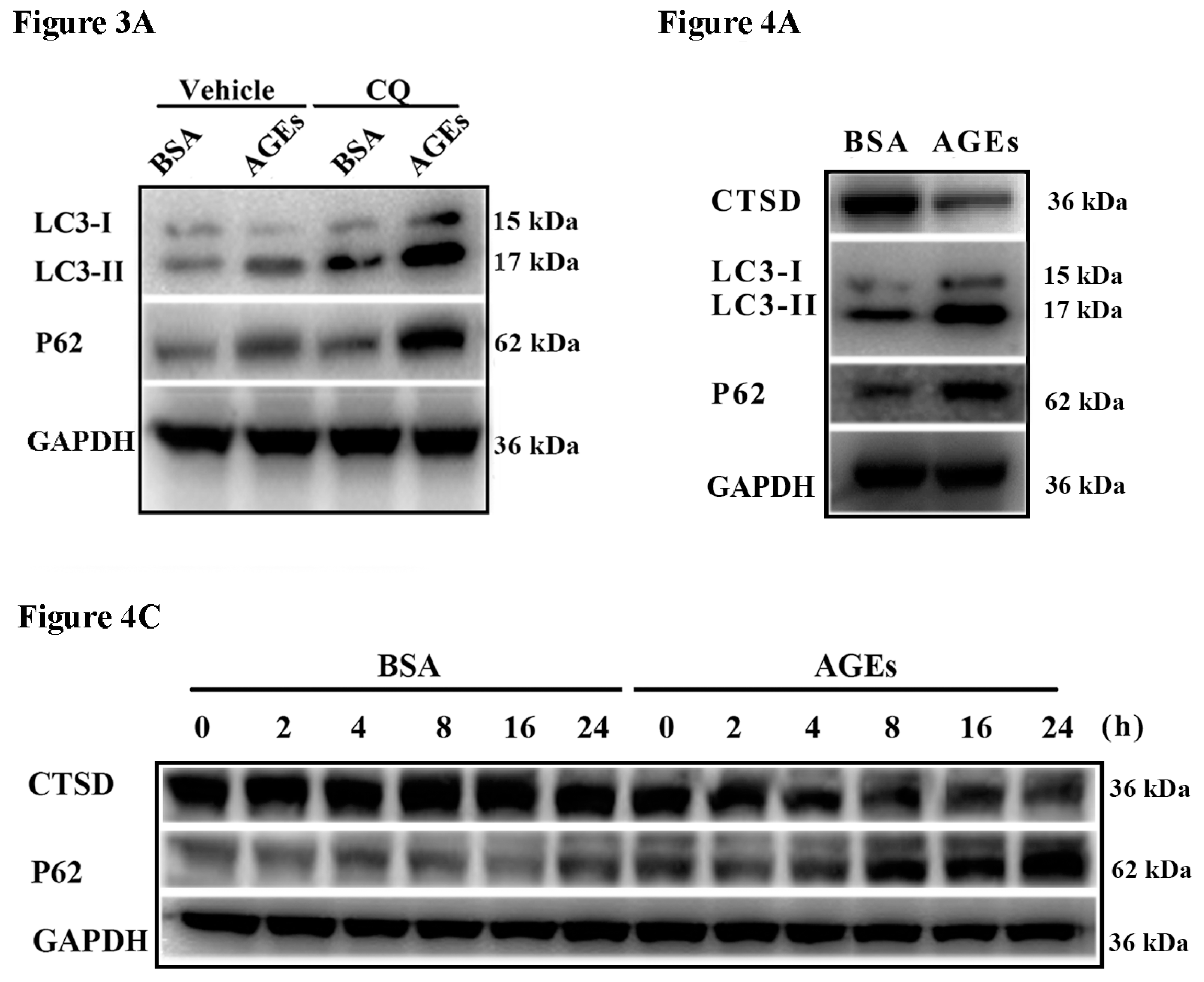
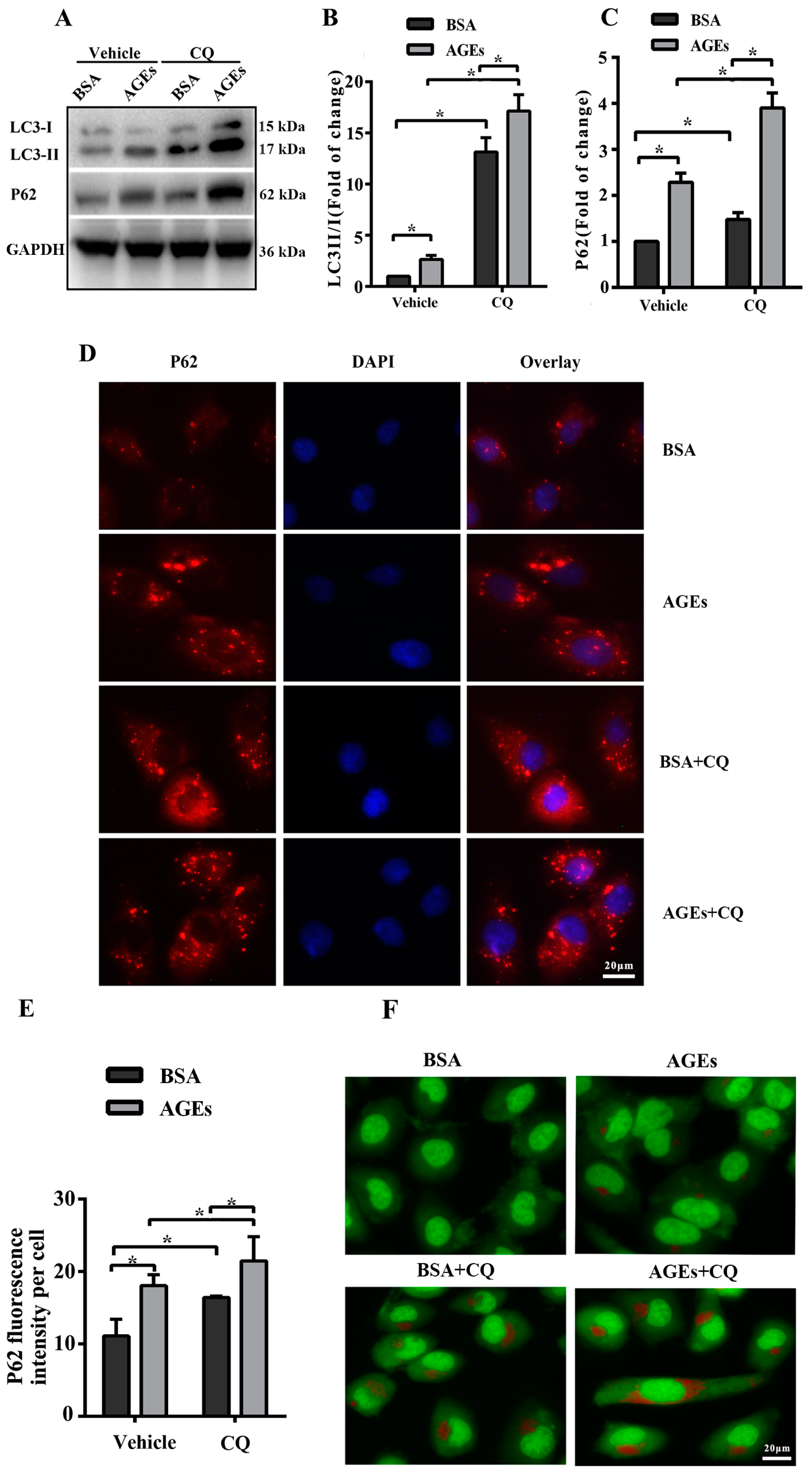
2.3. AGEs Promoted Autophagic Flux in HUVECs
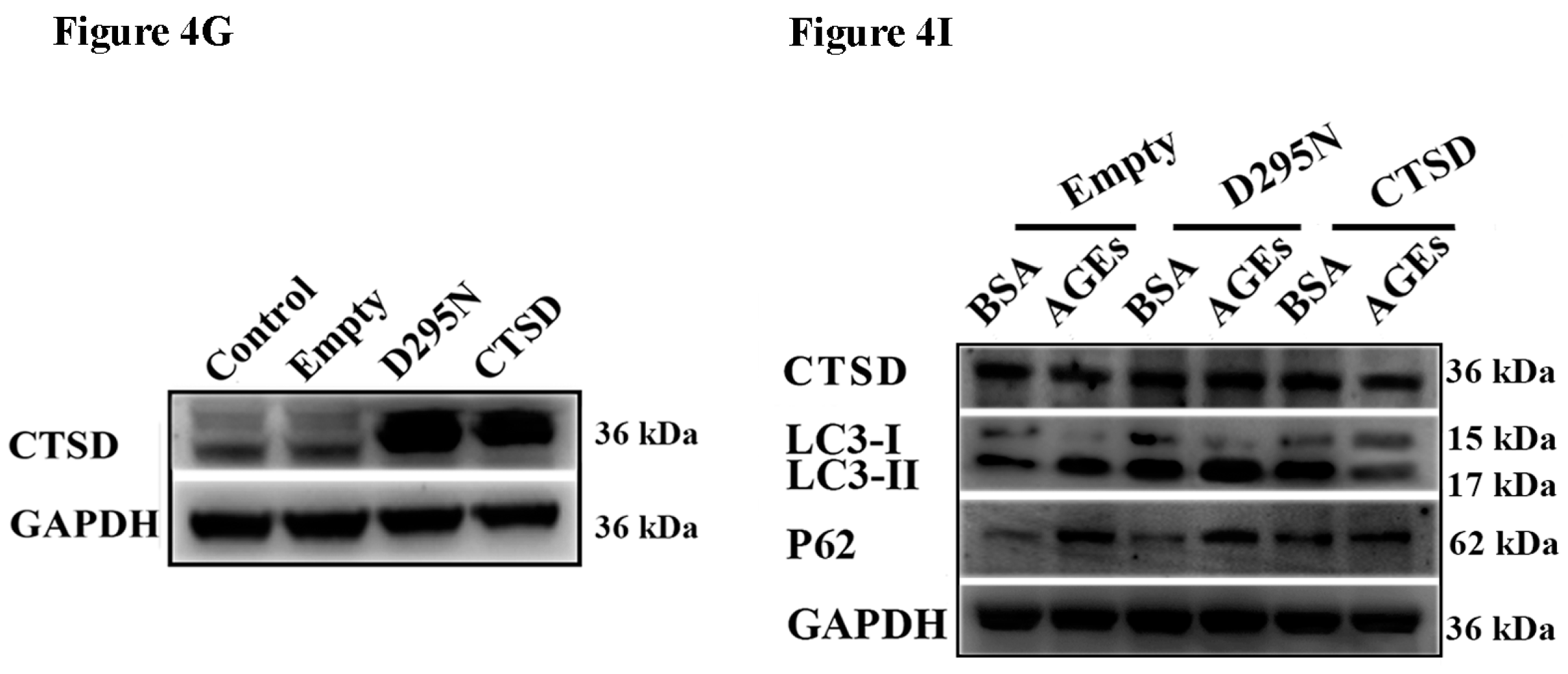
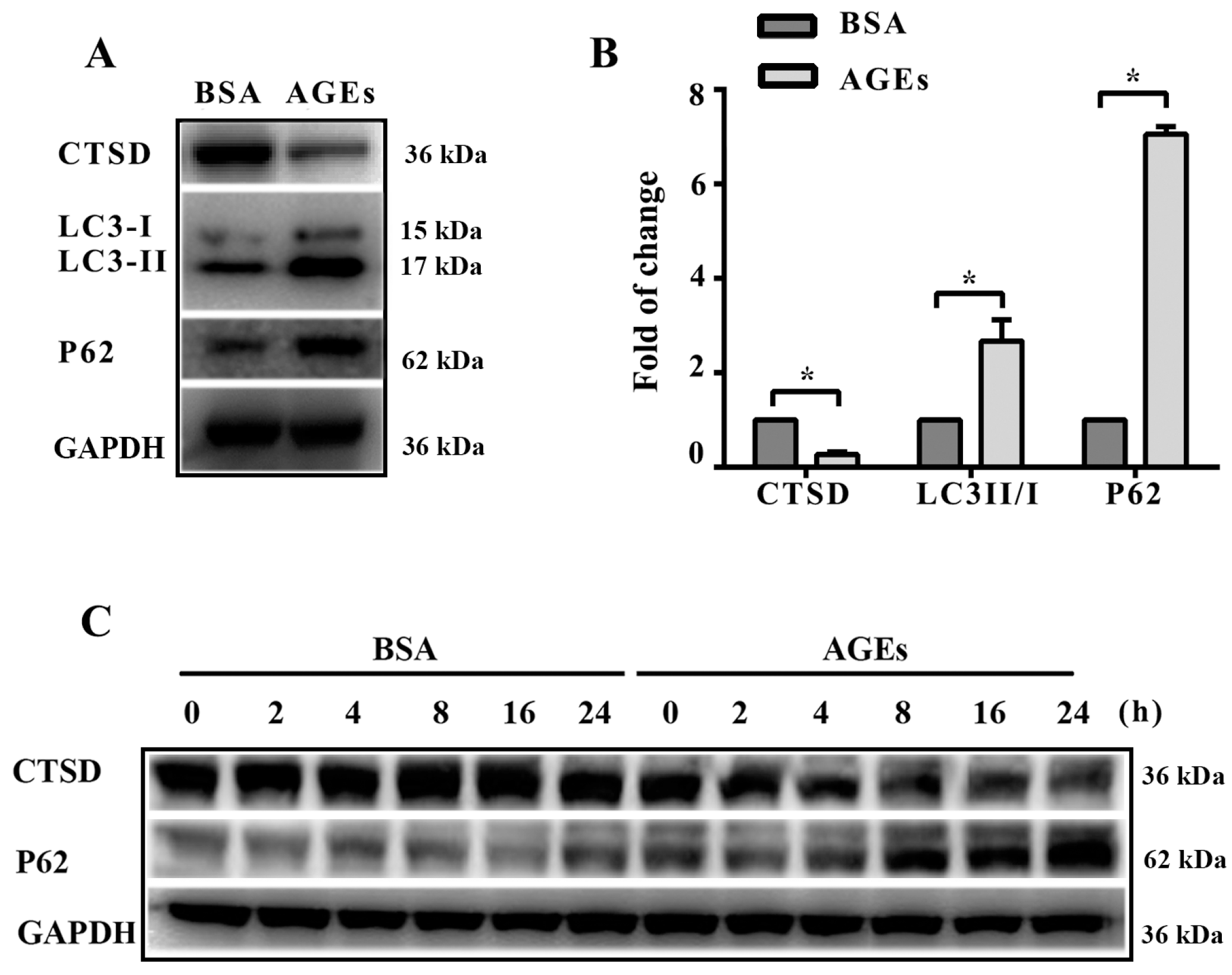
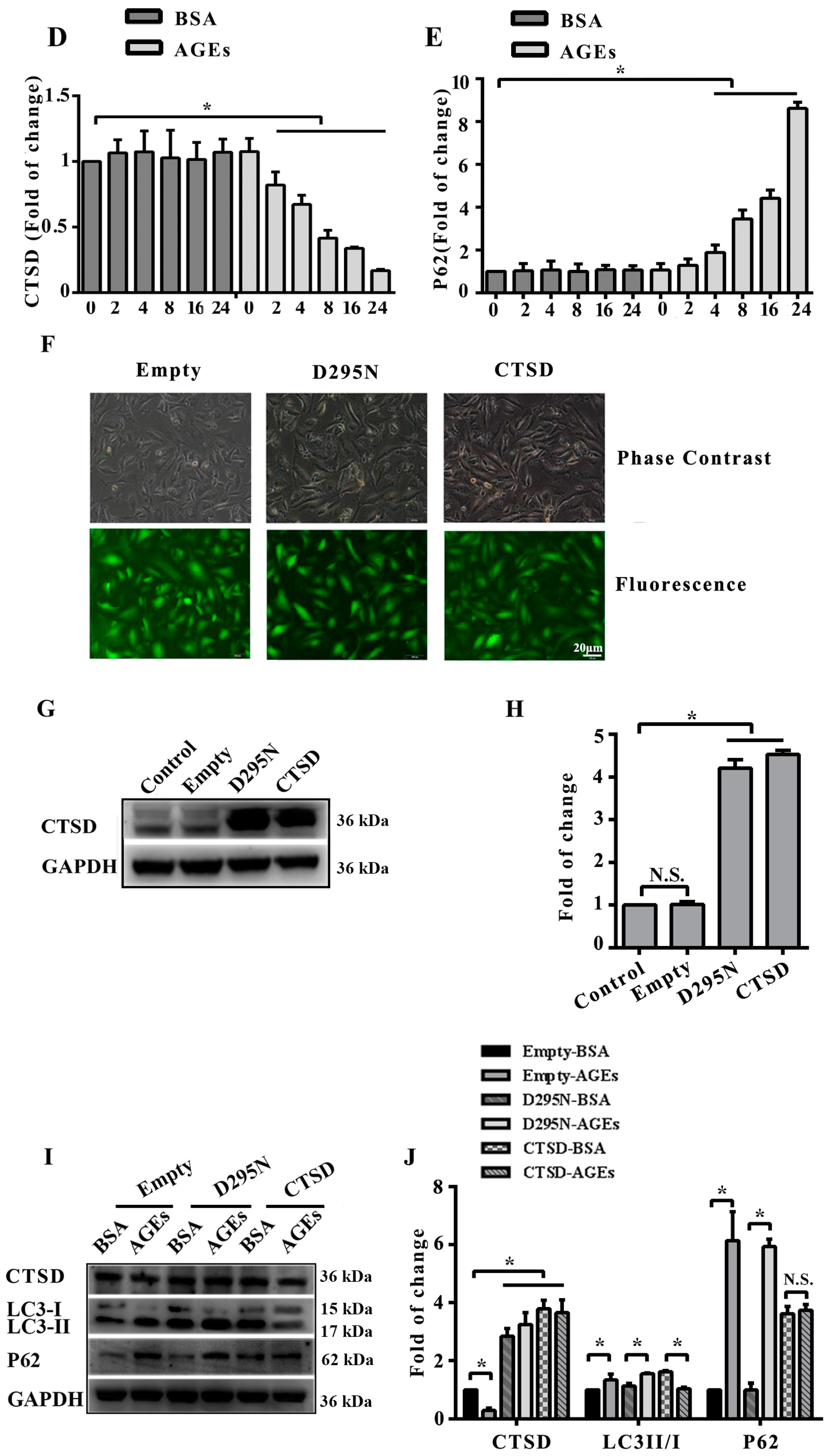
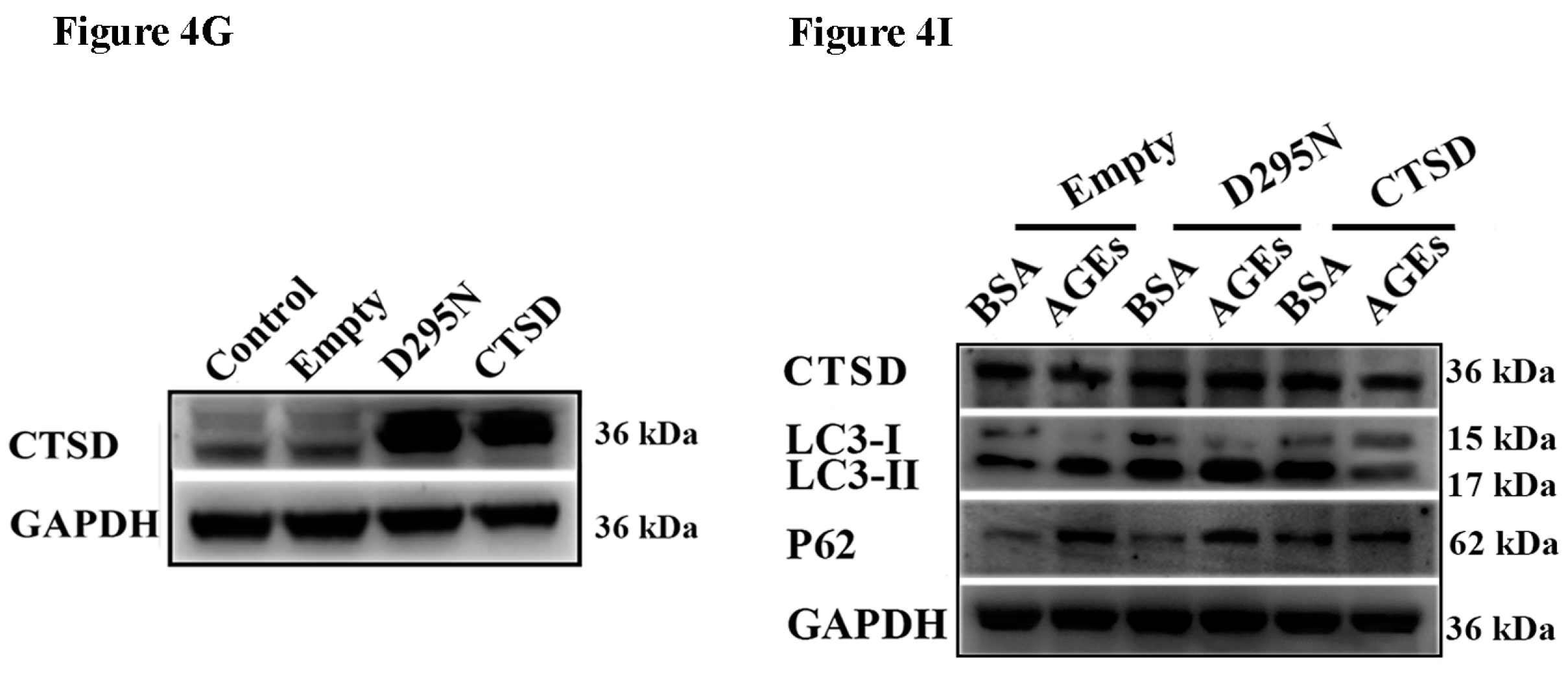
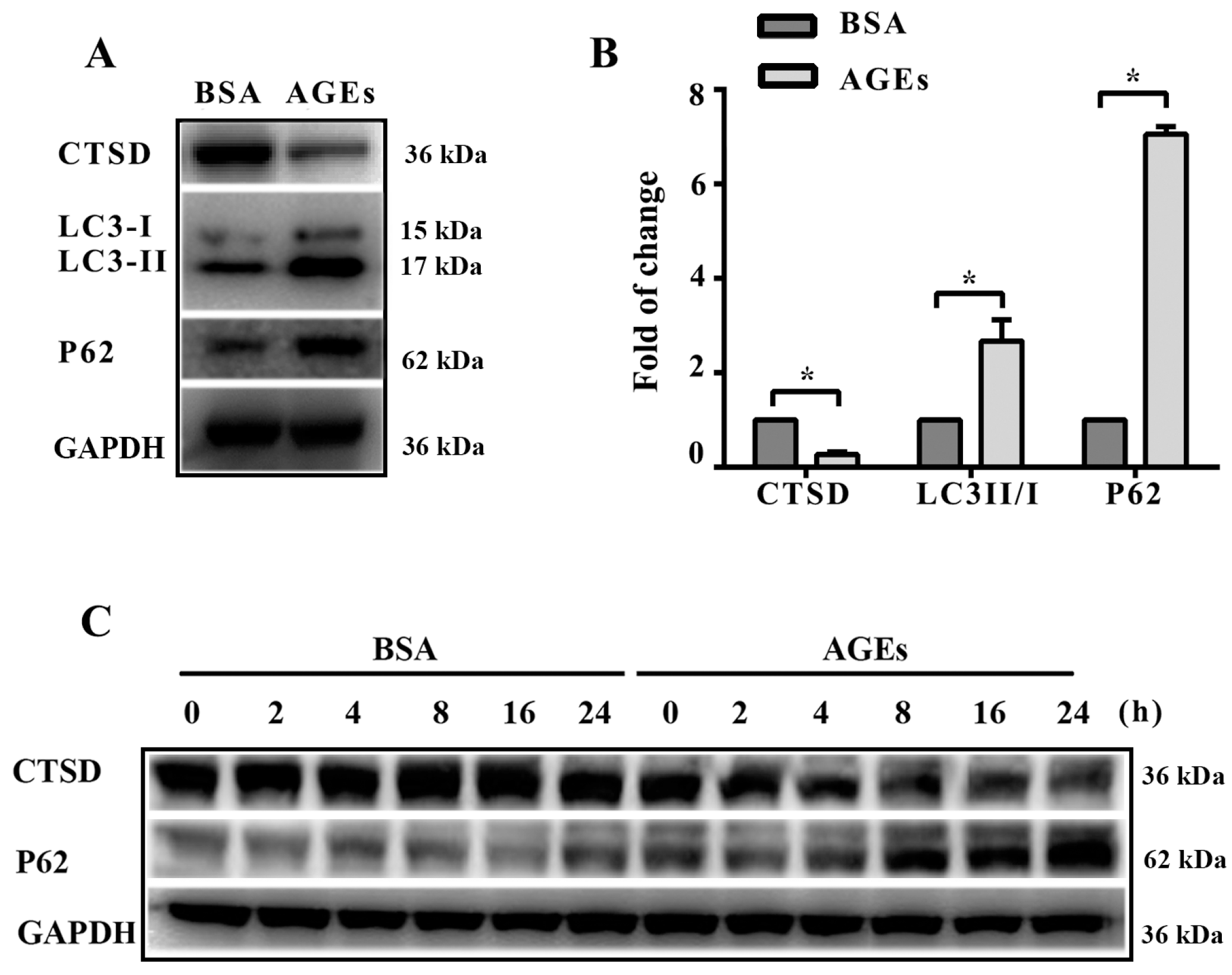
2.4. Cathepsin D (CTSD) Was Implicated in Autophagic Flux Mediated by AGEs in HUVECs
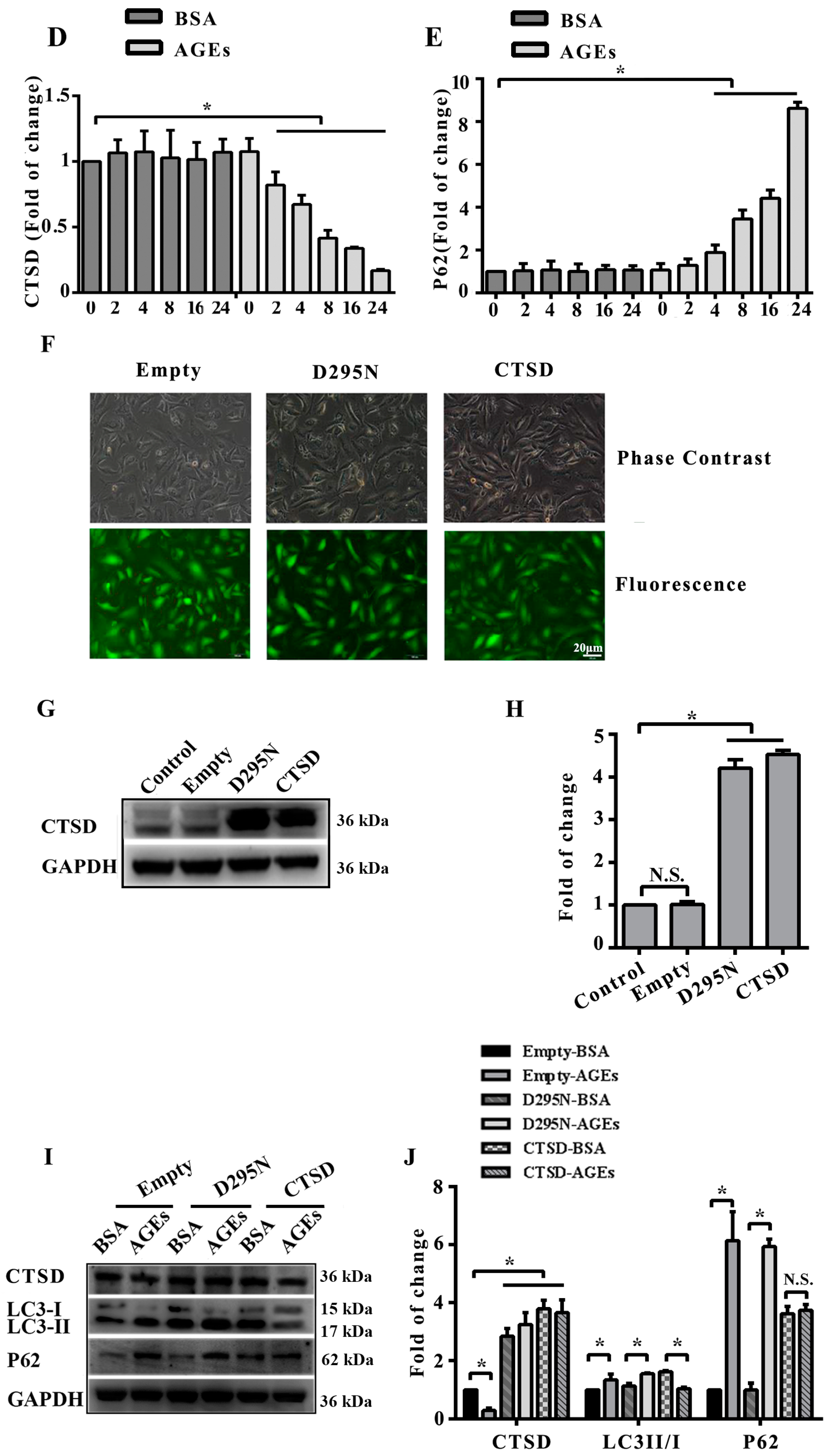
2.5. Overexpression of CTSD Recovered AGEs-Inhibited Proliferation of HUVECs
2.6. Overexpression of CTSD Recovered AGEs-Inhibited Migration of HUVECs
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Overexpression of CTSD in HUVECs Using Lentiviral Vectors
4.4. Cell Viability Assays
4.5. Growth Curve Assays Using Real-Time Cell Analyzer (RTCA)
4.6. EdU Incorporation Analysis
4.7. Scratch Wound-Healing Assay
4.8. Transwell Migration Assay
4.9. Western Blotting Analysis
4.10. Acridine Orange (AO) Dyeing Assay for Autophagy Vesicles
4.11. Immunofluorescence (IF) Staining and Fluorescence Microscopy
4.12. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AGEs | Advanced glycation end products |
| AO | Acridine orange |
| cDNA | Complementary deoxyribonucleic acid |
| cGMP | Cyclic guanosine monophosphate |
| CQ | Chloroquine |
| CTSD | Cathepsin D |
| CXCR4 | C-X-C motif chemokine receptor 4 |
| DAPI | 4′,6-Diamidino-2-phenylindole |
| DMEM | Dulbecco’s modified eagle medium |
| DMSO | Dimethyl sulfoxide |
| ECL | Enhanced chemiluminescence |
| EdU | 5-Ethynyl-2′-deoxyuridine |
| EPCs | Endothelial progenitor cells |
| FBS | Fetal bovine serum |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GFP | Green fluorescent protein |
| HUVECs | Human umbilical vein endothelial cells |
| LC3 | Microtubule-associated protein 1 light chain 3 |
| MOI | Multiplicity of infection |
| mRNA | Messenger ribonucleic acid |
| NF-κB | Nuclear factor-κB |
| NO | Nitric oxide |
| N.S. | No statistical significance |
| ox-LDL | Oxidized low density lipoprotein |
| PBS | Phosphate buffered saline |
| PVDF | Polyvinylidene fluoride |
| RAGE | Receptor of advanced glycation end products |
| RhoA | Ras homolog gene family, member A |
| ROCK | RhoA-associated protein kinase |
| RTCA | Real-time cell analyzer |
| ROI | Region of interest |
| ROS | Reactive oxygen species |
| SDF-1 | Stromal cell derived factor-1 |
| SD | Standard deviation |
| TBS | Tris-buffered solution |
| TBS-T | Tris-buffered saline with Tween-20 |
| VSMCs | Vascular smooth muscle cells |
Appendix A
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Schmidt, A.M. Rage: A novel target for drug intervention in diabetic vascular disease. Pharm. Res. 2004, 21, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; You, S.J.; Zhang, Y.L.; Han, Q.; Cao, Y.J.; Xu, X.S.; Yang, Y.P.; Li, J.; Liu, C.F. Protective role of autophagy in age-induced early injury of human vascular endothelial cells. Mol. Med. Rep. 2011, 4, 459–464. [Google Scholar] [PubMed]
- Chen, Y.; Du, X.; Zhou, Y.; Zhang, Y.; Yang, Y.; Liu, Z.; Liu, C.; Xie, Y. Paeoniflorin protects HUVECS from age-BSA-induced injury via an autophagic pathway by acting on the rage. Int. J. Clin. Exp. Pathol. 2015, 8, 53–62. [Google Scholar] [PubMed]
- Morigi, M.; Angioletti, S.; Imberti, B.; Donadelli, R.; Micheletti, G.; Figliuzzi, M.; Remuzzi, A.; Zoja, C.; Remuzzi, G. Leukocyte-endothelial interaction is augmented by high glucose concentrations and hyperglycemia in a NF-κB-dependent fashion. J. Clin. Investig. 1998, 101, 1905–1915. [Google Scholar] [CrossRef] [PubMed]
- Sengoelge, G.; Fodinger, M.; Skoupy, S.; Ferrara, I.; Zangerle, C.; Rogy, M.; Horl, W.H.; Sunder-Plassmann, G.; Menzel, J. Endothelial cell adhesion molecule and PMNL response to inflammatory stimuli and age-modified fibronectin. Kidney Int. 1998, 54, 1637–1651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Song, Y.; Han, X.; Feng, L.; Wang, R.; Zhang, M.; Zhu, M.; Jia, X.; Hu, S. Liquiritin attenuates advanced glycation end products-induced endothelial dysfunction via rage/NF-κB pathway in human umbilical vein endothelial cells. Mol. Cell. Biochem. 2013, 374, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Sang, H.Q.; Gu, J.F.; Yuan, J.R.; Zhang, M.H.; Jia, X.B.; Feng, L. The protective effect of smilax glabra extract on advanced glycation end products-induced endothelial dysfunction in HUVECS via rage-ERK1/2-NF-κB pathway. J. Ethnopharmacol. 2014, 155, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Liu, Y.; Xiang, Y.; Lin, M.; Gao, J. Ghrelin protects human umbilical vein endothelial cells against advanced glycation end products-induced apoptosis via NO/CGMP signaling. Int. J. Clin. Exp. Med. 2015, 8, 15269–15275. [Google Scholar] [PubMed]
- Pan, Y.; Liang, H.; Liu, H.; Li, D.; Chen, X.; Li, L.; Zhang, C.Y.; Zen, K. Platelet-secreted microRNA-223 promotes endothelial cell apoptosis induced by advanced glycation end products via targeting the insulin-like growth factor 1 receptor. J. Immunol. 2014, 192, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Sun, H.L.; Chen, H.; Zhang, H.; Sun, J.; Zhang, Z.; Cai, D.H. Glucagon-like peptide-1 (GLP-1) protects vascular endothelial cells against advanced glycation end products (ages)-induced apoptosis. Med. Sci. Monit. 2012, 18, BR286–BR291. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, X.; Guan, X.; Cui, X.; Wang, Y.; Chu, H.; Cheng, M. Advanced glycation end products impair the migration, adhesion and secretion potentials of late endothelial progenitor cells. Cardiovasc. Diabetol. 2012, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Devi, M.S.; Sudhakaran, P.R. Differential modulation of angiogenesis by advanced glycation end products. Exp. Biol. Med. 2011, 236, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Fan, A.; Yuan, Y.; Chen, L.; Guo, X.; Huang, X.; Huang, Q. Role of moesin in advanced glycation end products-induced angiogenesis of human umbilical vein endothelial cells. Sci. Rep. 2016, 6, 22749. [Google Scholar] [CrossRef] [PubMed]
- Tezuka, M.; Koyama, N.; Morisaki, N.; Saito, Y.; Yoshida, S.; Araki, N.; Horiuchi, S. Angiogenic effects of advanced glycation end products of the maillard reaction on cultured human umbilical cord vein endothelial cells. Biochem. Biophys. Res. Commun. 1993, 193, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Sarkar, S.; Davies, J.E.; Futter, M.; Garcia-Arencibia, M.; Green-Thompson, Z.W.; Jimenez-Sanchez, M.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol. Rev. 2010, 90, 1383–1435. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Cao, Y.J.; Zhang, X.; Liu, H.H.; Tong, T.; Xiao, G.D.; Yang, Y.P.; Liu, C.F. The autophagy-lysosome pathway: A novel mechanism involved in the processing of oxidized LDL in human vascular endothelial cells. Biochem. Biophys. Res. Commun. 2010, 394, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Patschan, S.; Chen, J.; Polotskaia, A.; Mendelev, N.; Cheng, J.; Patschan, D.; Goligorsky, M.S. Lipid mediators of autophagy in stress-induced premature senescence of endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1119–H1129. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, Y. Autophagic cell death and its execution by lysosomal cathepsins. Arch. Histol. Cytol. 2001, 64, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Guo, X.; Chang, Y.; Li, C.; Meng, X.; Li, S.; Du, Z.X.; Wang, H.Q.; Sun, Y. Advanced glycation end products promote proliferation and suppress autophagy via reduction of cathepsin D in rat vascular smooth muscle cells. Mol. Cell. Biochem. 2015, 403, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.M.; Shin, D.M.; Yuk, J.M.; Shi, G.; Choi, D.K.; Lee, S.H.; Huang, S.M.; Kim, J.M.; Kim, C.D.; Lee, J.H.; et al. Autophagy negatively regulates keratinocyte inflammatory responses via scaffolding protein p62/SQSTM1. J. Immunol. 2011, 186, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- Gump, J.M.; Thorburn, A. Sorting cells for basal and induced autophagic flux by quantitative ratiometric flow cytometry. Autophagy 2014, 10, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, R.A.; Andres, A.M.; Sin, J.; Taylor, D.P. Untangling autophagy measurements: All fluxed up. Circ. Res. 2015, 116, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Hah, Y.S.; Noh, H.S.; Ha, J.H.; Ahn, J.S.; Hahm, J.R.; Cho, H.Y.; Kim, D.R. Cathepsin D inhibits oxidative stress-induced cell death via activation of autophagy in cancer cells. Cancer Lett. 2012, 323, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Song, M.; Yu, S.; Gao, P.; Yu, Y.; Wang, H.; Huang, L. Advanced glycation endproducts alter functions and promote apoptosis in endothelial progenitor cells through receptor for advanced glycation endproducts mediate overpression of cell oxidant stress. Mol. Cell. Biochem. 2010, 335, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Glenn, J.V.; Mahaffy, H.; Wu, K.; Smith, G.; Nagai, R.; Simpson, D.A.; Boulton, M.E.; Stitt, A.W. Advanced glycation end product (age) accumulation on bruch’s membrane: Links to age-related rpe dysfunction. Investig. Ophthalmol. Vis. Sci. 2009, 50, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Boulton, M.; Moriarty, P.; Jarvis-Evans, J.; Marcyniuk, B. Regional variation and age-related changes of lysosomal enzymes in the human retinal pigment epithelium. Br. J. Ophthalmol. 1994, 78, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S.; Ernst, L.; Grotzinger, N.; Hohn, A.; Breusing, N.; Reinheckel, T.; Grune, T. Cathepsin d is one of the major enzymes involved in intracellular degradation of age-modified proteins. Free Radic. Res. 2010, 44, 1013–1026. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J.; Bateman, A. Merops: The peptidase database. Nucleic Acids Res. 2010, 38, D227–D233. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Kanneganti, T.D. Regulation of lysosomal dynamics and autophagy by CTSB/CATHEPSIN B. Autophagy 2016, 12, 2504–2505. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Lee, C.C.; Lin, H.H.; Chang, J.Y. Cathepsin s attenuates endosomal EGFR signalling: A mechanical rationale for the combination of cathepsin S and EGFR tyrosine kinase inhibitors. Sci. Rep. 2016, 6, 29256. [Google Scholar] [CrossRef] [PubMed]
- Pillay, C.S.; Elliott, E.; Dennison, C. Endolysosomal proteolysis and its regulation. Biochem. J. 2002, 363, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Li, Y.; Ye, N.; Guo, X.; Li, Z.; Sun, G.; Sun, Y. Atorvastatin inhibits the apoptosis of human umbilical vein endothelial cells induced by angiotensin ii via the lysosomal-mitochondrial axis. Apoptosis 2016, 21, 977–996. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Liu, B.; Kong, D.; Li, S.; Li, C.; Wang, H.; Sun, Y. Atorvastatin calcium inhibits phenotypic modulation of PDGF-BB-induced VSMCS via down-regulation the AKT signaling pathway. PLoS ONE 2015, 10, e0122577. [Google Scholar] [CrossRef] [PubMed]
- Marlina, S.; Shu, M.H.; AbuBakar, S.; Zandi, K. Development of a real-time cell analysing (RTCA) method as a fast and accurate screen for the selection of chikungunya virus replication inhibitors. Parasit. Vectors 2015, 8, 579. [Google Scholar] [CrossRef] [PubMed]
- Ke, N.; Wang, X.; Xu, X.; Abassi, Y.A. The xcelligence system for real-time and label-free monitoring of cell viability. Methods Mol. Biol. 2011, 740, 33–43. [Google Scholar] [PubMed]
- Li, Z.; Berk, M.; McIntyre, T.M.; Gores, G.J.; Feldstein, A.E. The lysosomal-mitochondrial axis in free fatty acid-induced hepatic lipotoxicity. Hepatology 2008, 47, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Thome, M.P.; Filippi-Chiela, E.C.; Villodre, E.S.; Migliavaca, C.B.; Onzi, G.R.; Felipe, K.B.; Lenz, G. Ratiometric analysis of acridine orange staining in the study of acidic organelles and autophagy. J. Cell Sci. 2016, 129, 4622–4632. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Chang, Y.; Ye, N.; Dai, D.; Chen, Y.; Zhang, N.; Sun, G.; Sun, Y. Advanced Glycation End Products Inhibit the Proliferation of Human Umbilical Vein Endothelial Cells by Inhibiting Cathepsin D. Int. J. Mol. Sci. 2017, 18, 436. https://doi.org/10.3390/ijms18020436
Li Y, Chang Y, Ye N, Dai D, Chen Y, Zhang N, Sun G, Sun Y. Advanced Glycation End Products Inhibit the Proliferation of Human Umbilical Vein Endothelial Cells by Inhibiting Cathepsin D. International Journal of Molecular Sciences. 2017; 18(2):436. https://doi.org/10.3390/ijms18020436
Chicago/Turabian StyleLi, Yuan, Ye Chang, Ning Ye, Dongxue Dai, Yintao Chen, Naijin Zhang, Guozhe Sun, and Yingxian Sun. 2017. "Advanced Glycation End Products Inhibit the Proliferation of Human Umbilical Vein Endothelial Cells by Inhibiting Cathepsin D" International Journal of Molecular Sciences 18, no. 2: 436. https://doi.org/10.3390/ijms18020436