
Gypenoside XVII Prevents Atherosclerosis by Attenuating Endothelial Apoptosis and Oxidative Stress: Insight into the ERα-Mediated PI3K/Akt Pathway

Abstract
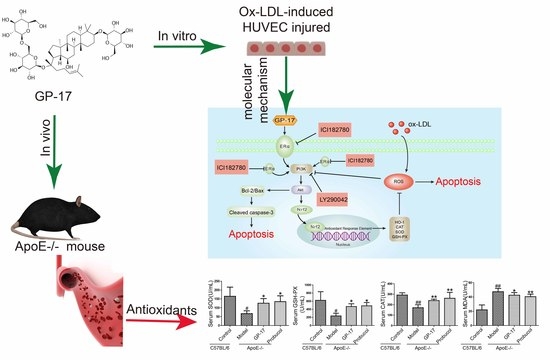
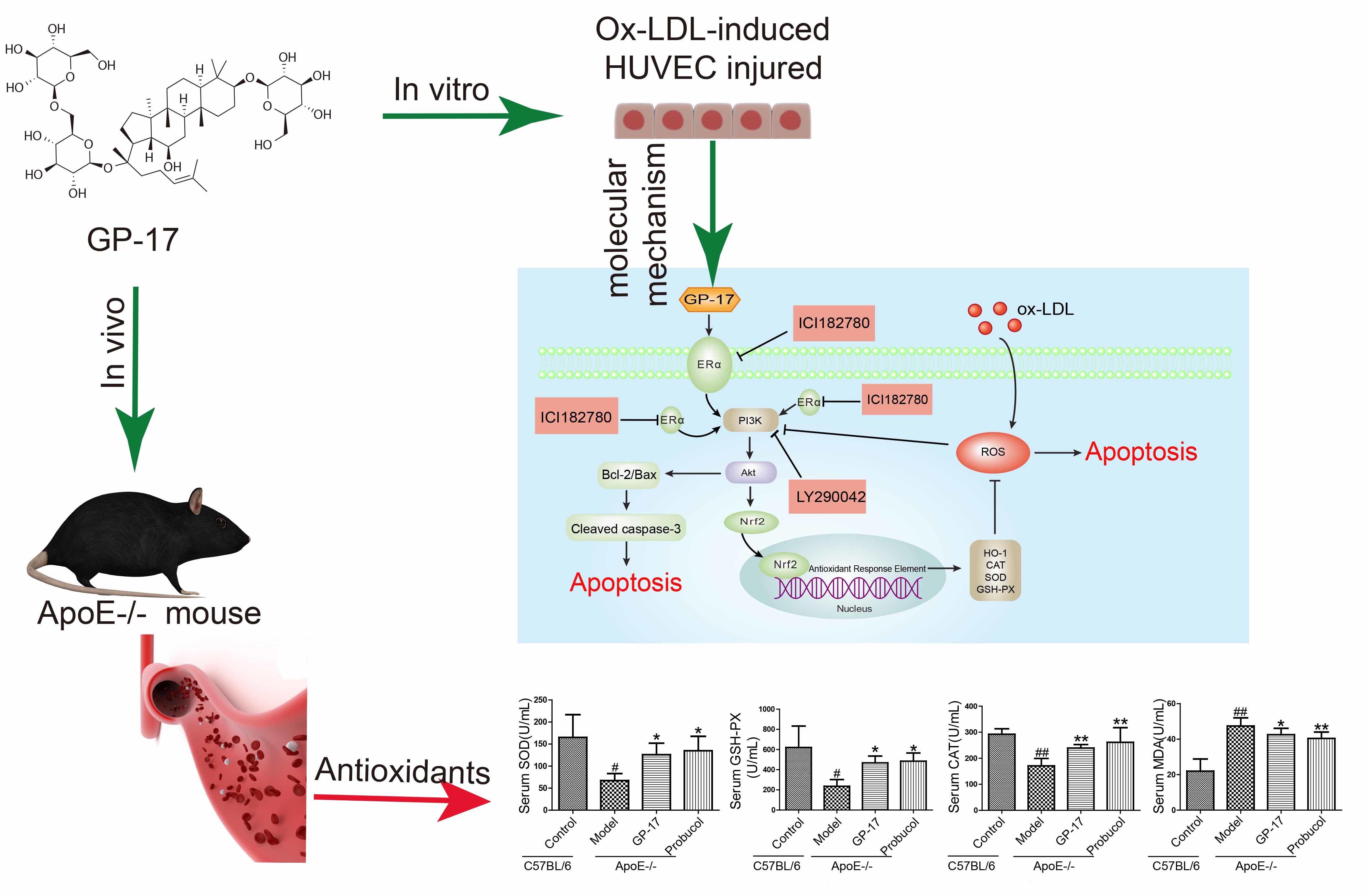
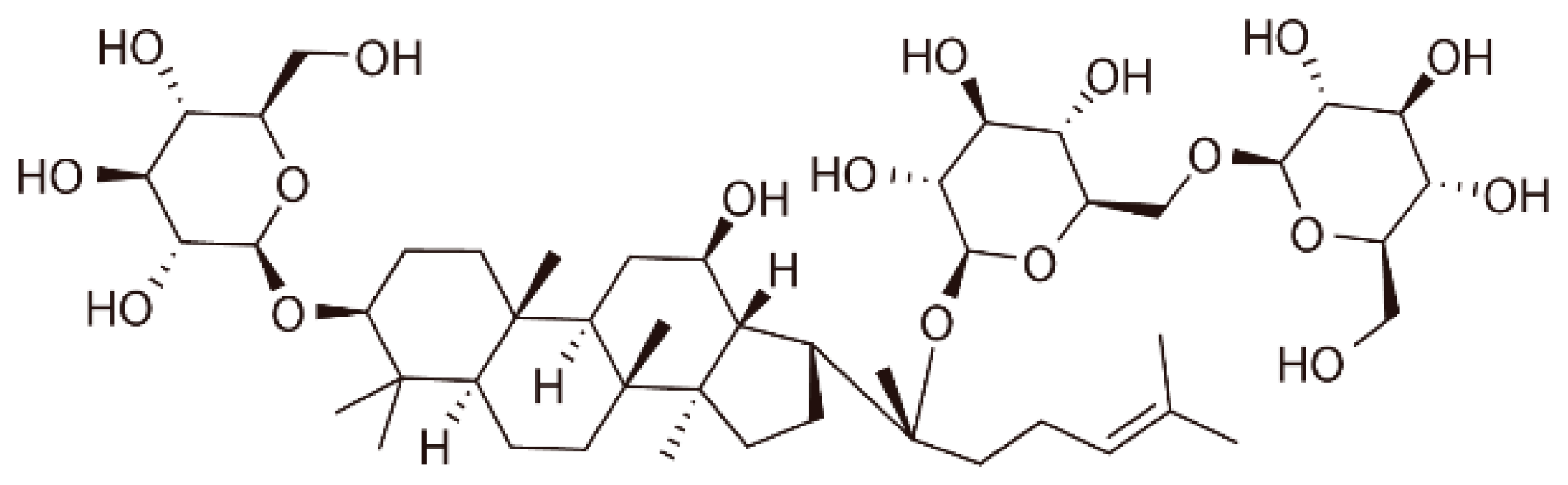
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
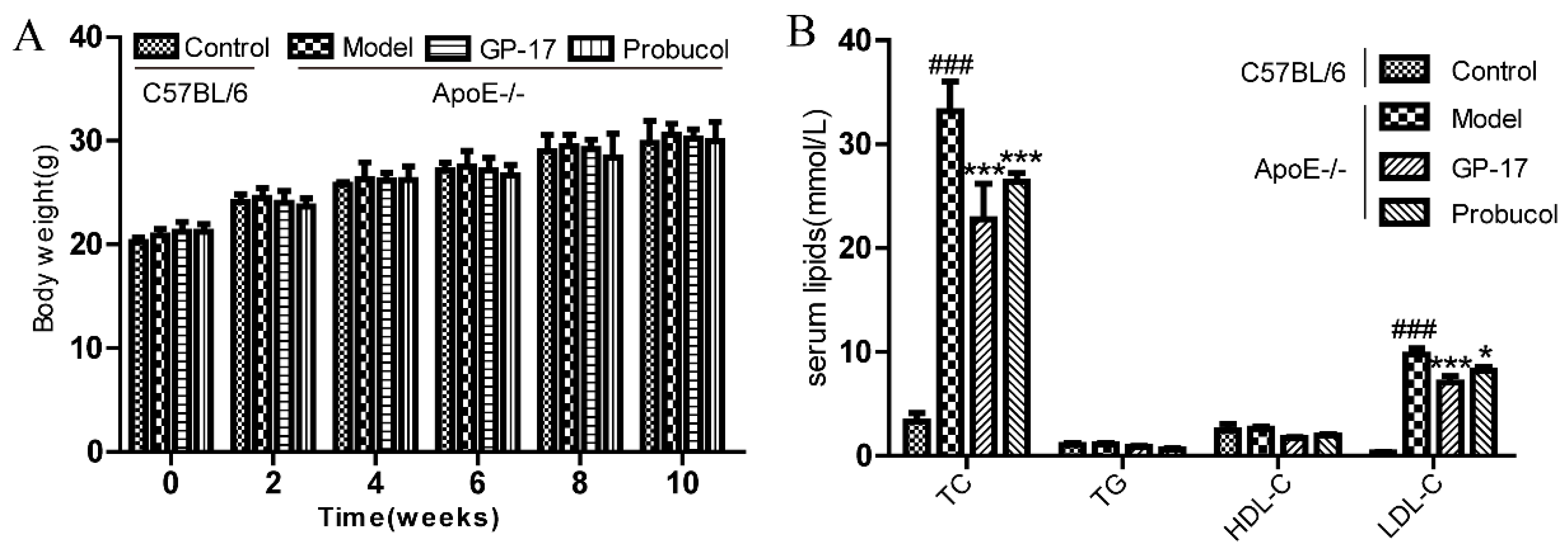
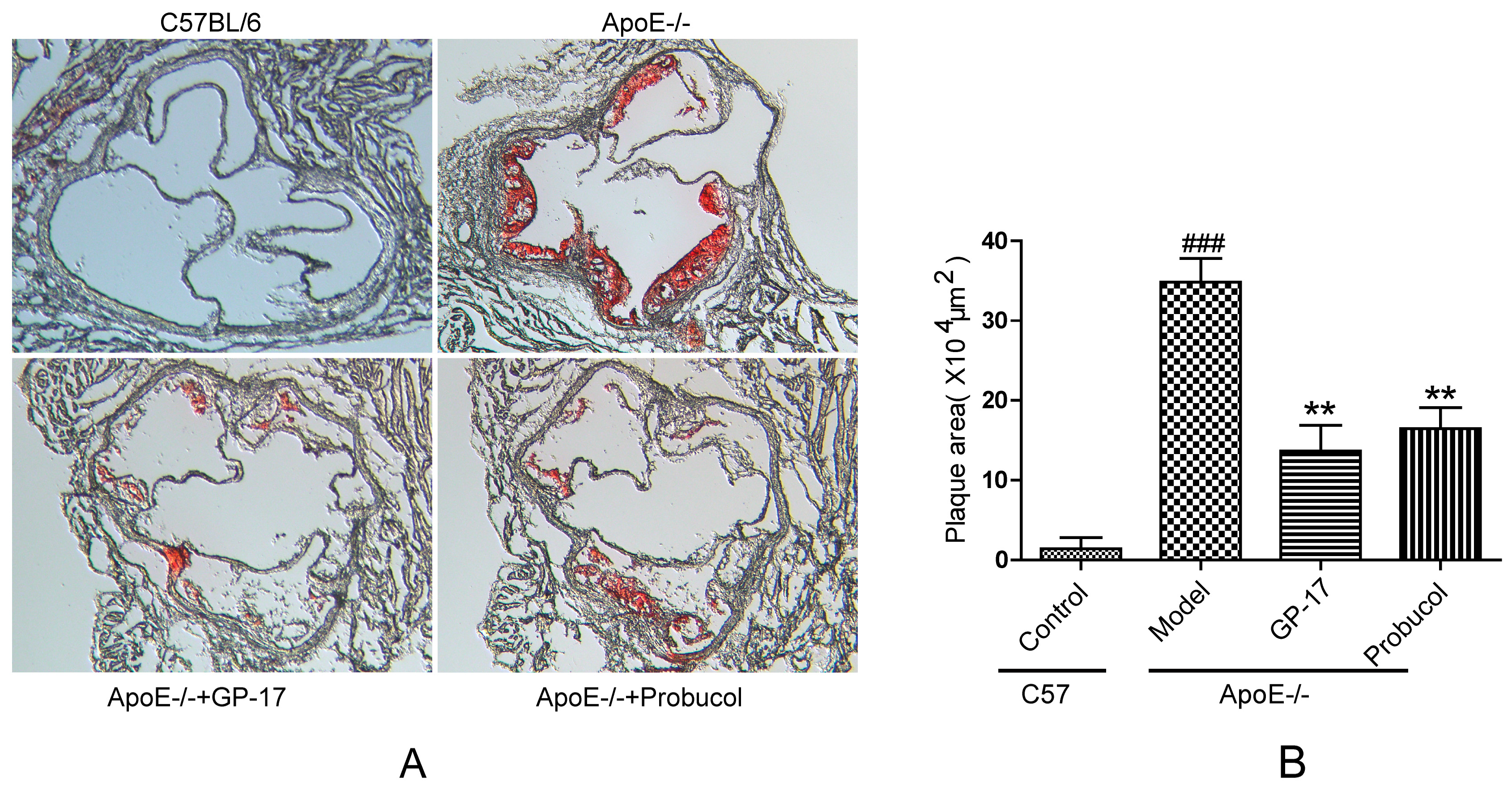
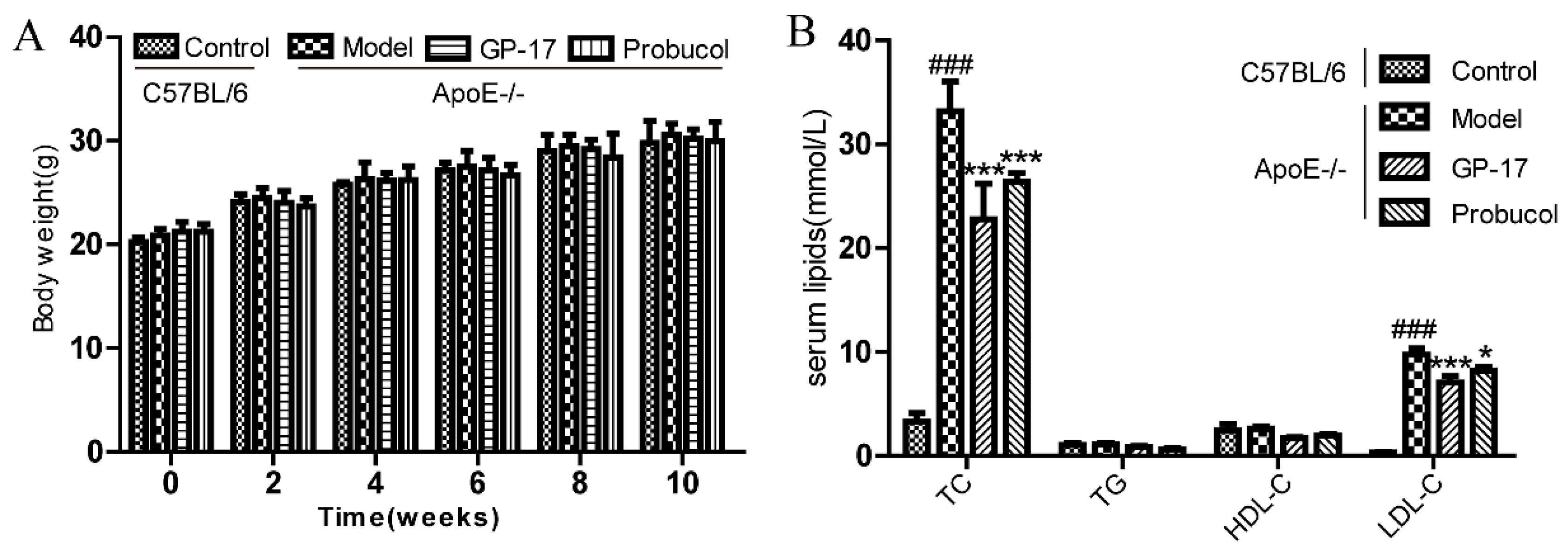
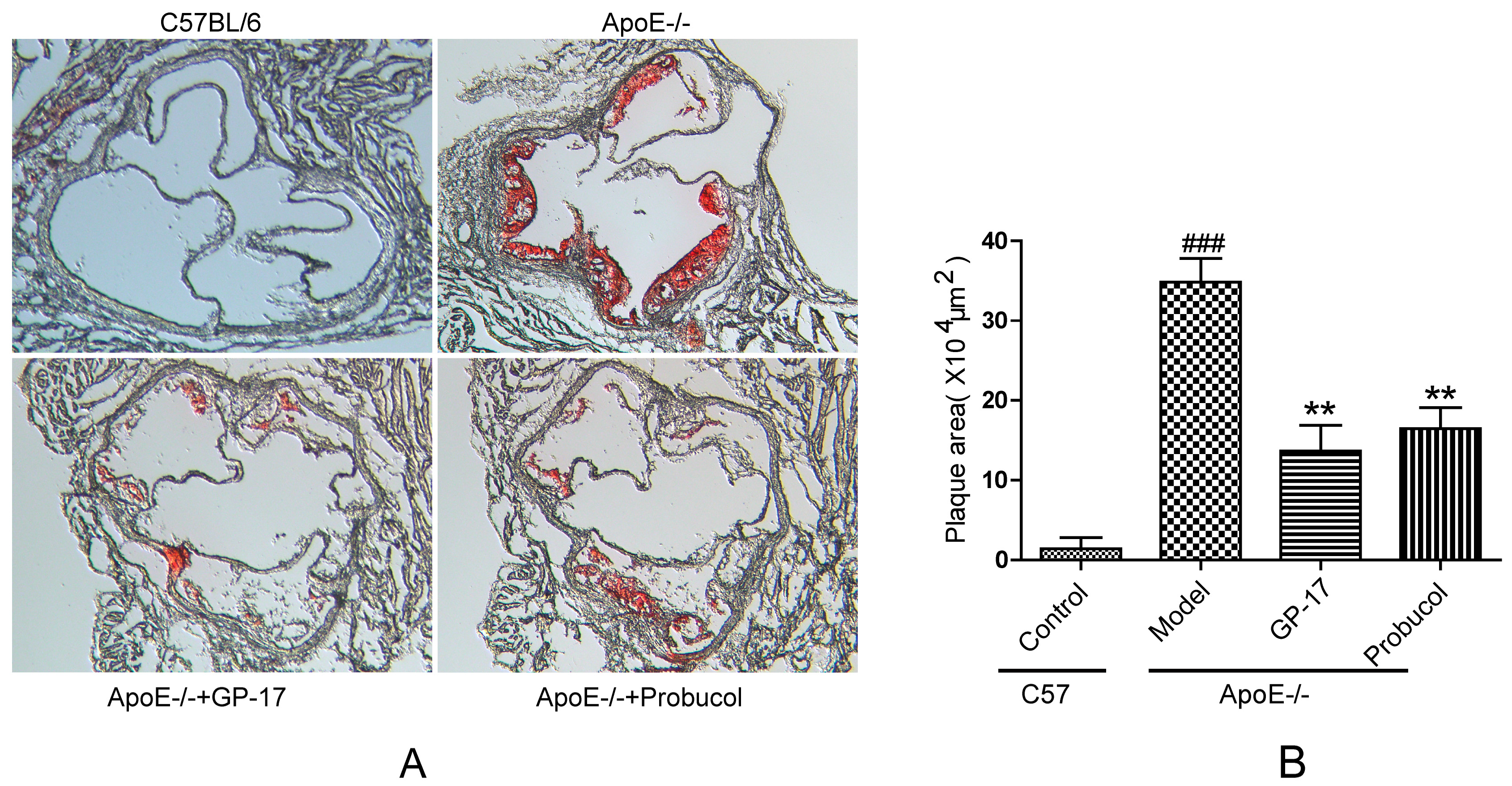
2.1. GP-17 Suppresses Atherosclerotic Development in ApoE−/− Mice
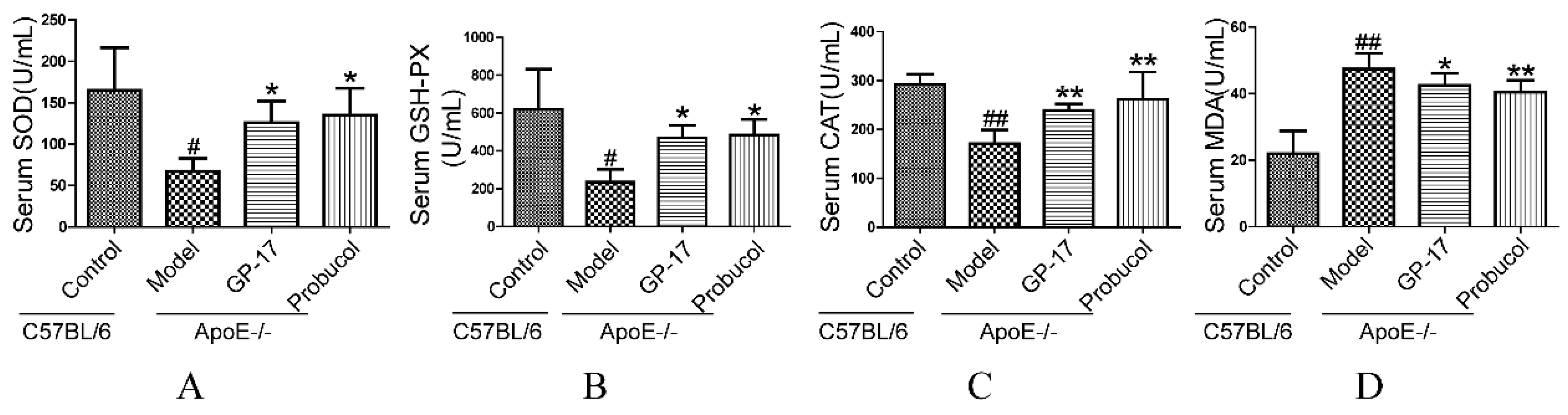
2.2. GP-17 Mitigates Oxidative Stress in Serum
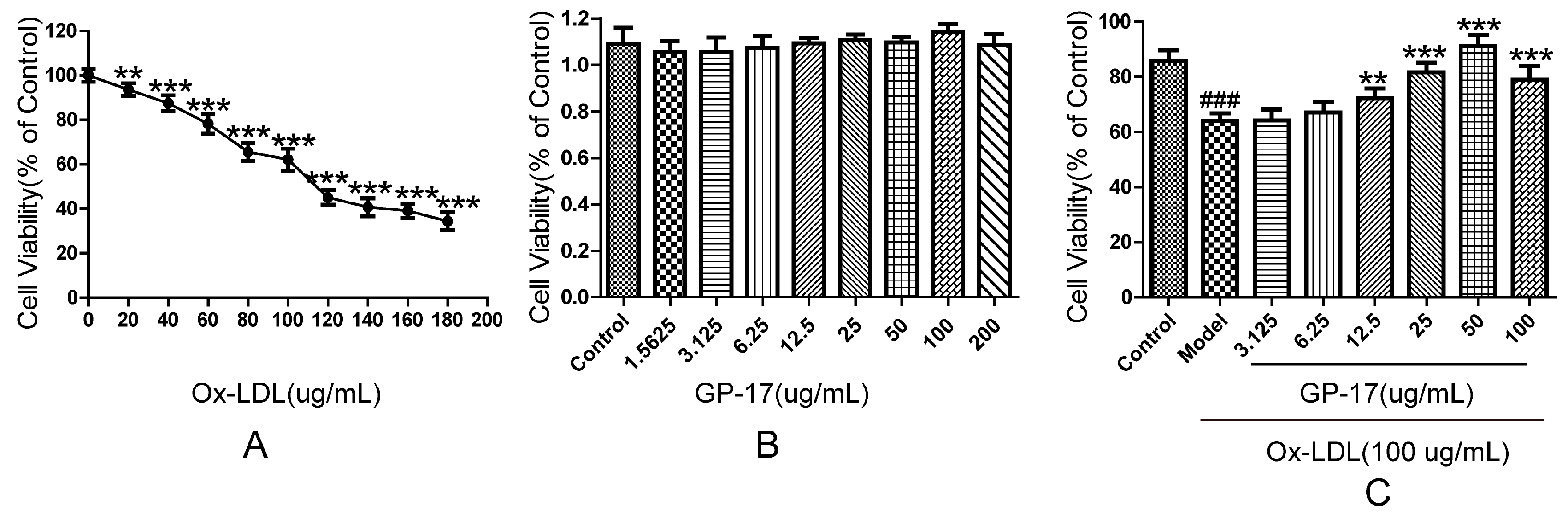
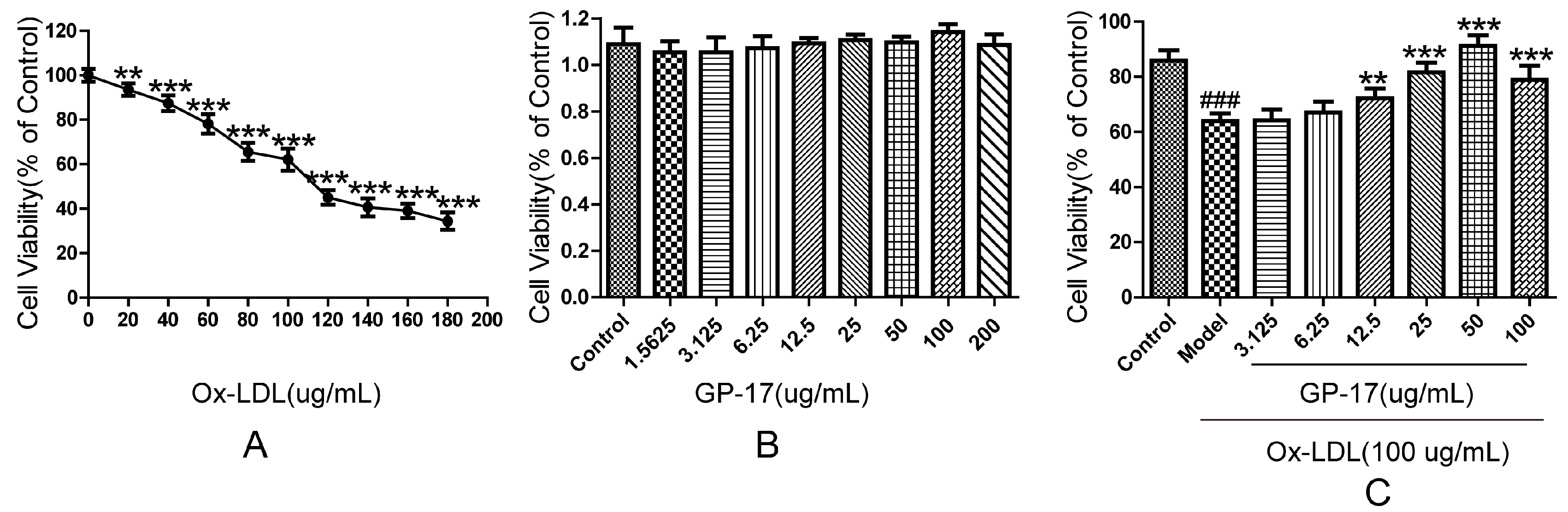
2.3. Protective Effect of GP-17 on Ox-LDL-Induced Cytotoxicity in HUVECs
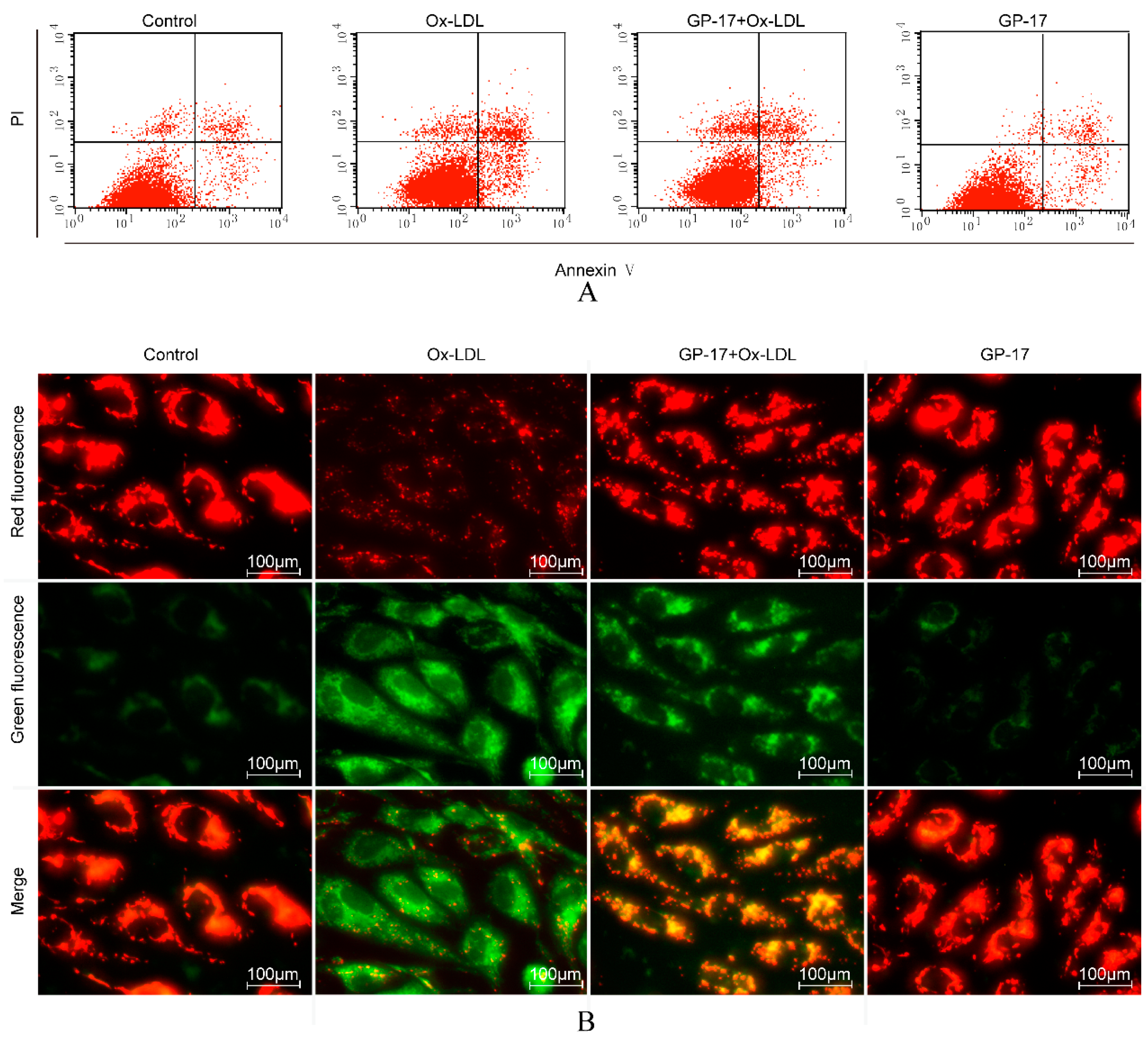
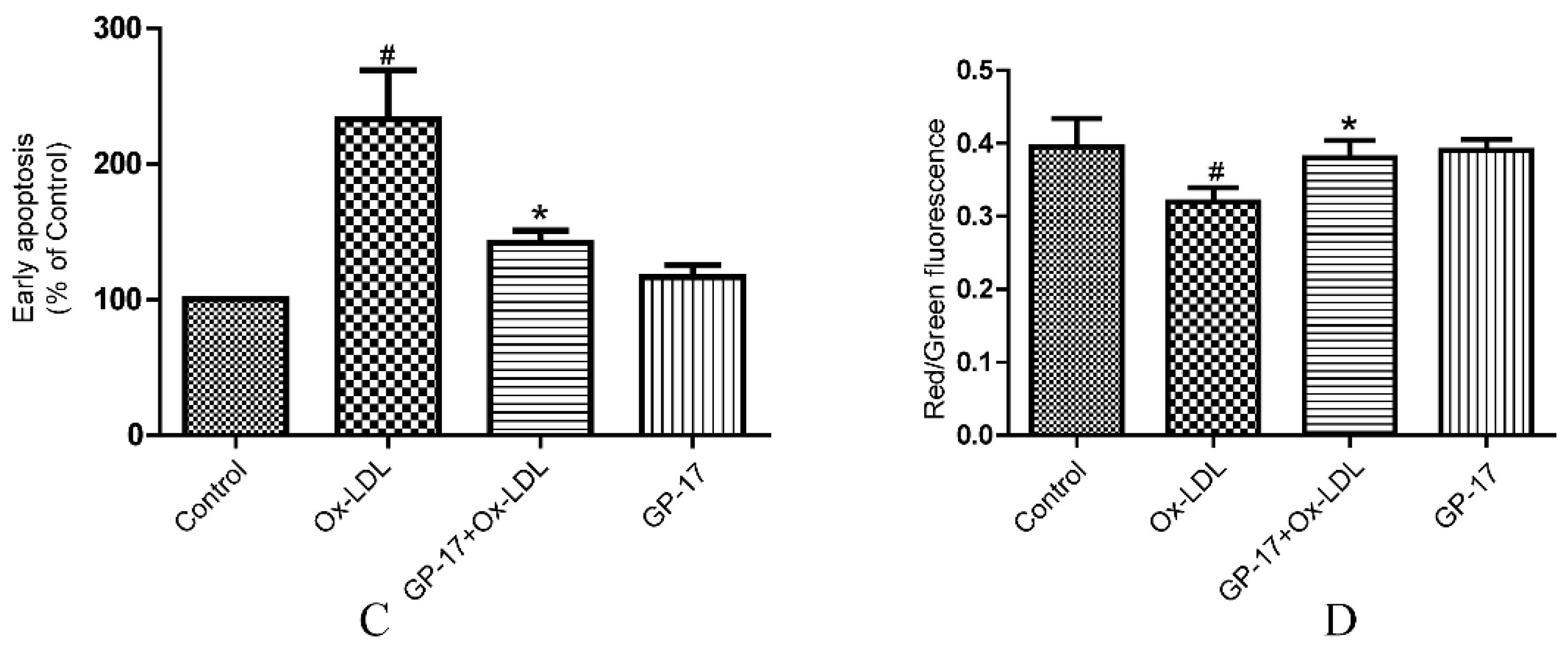
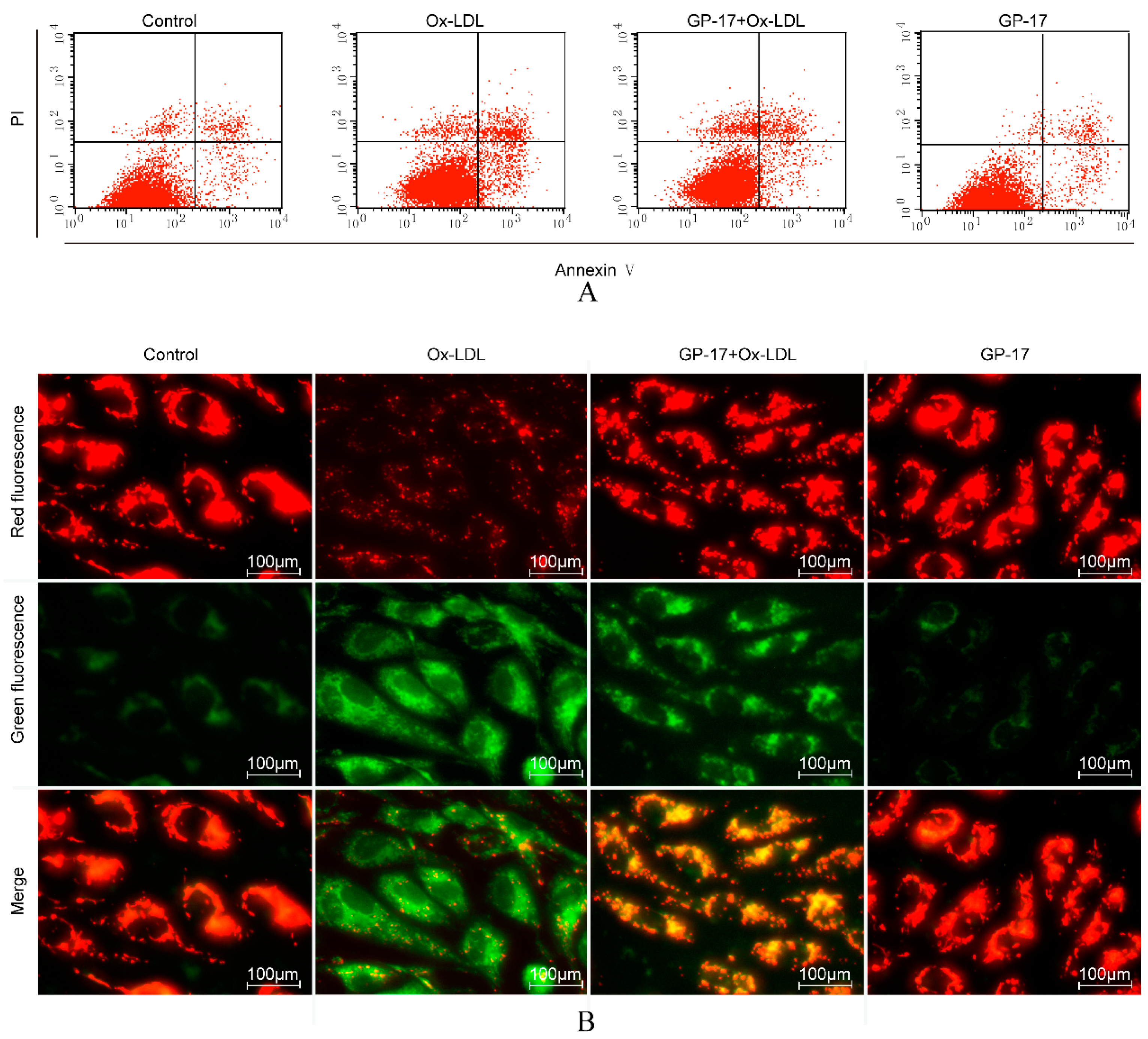
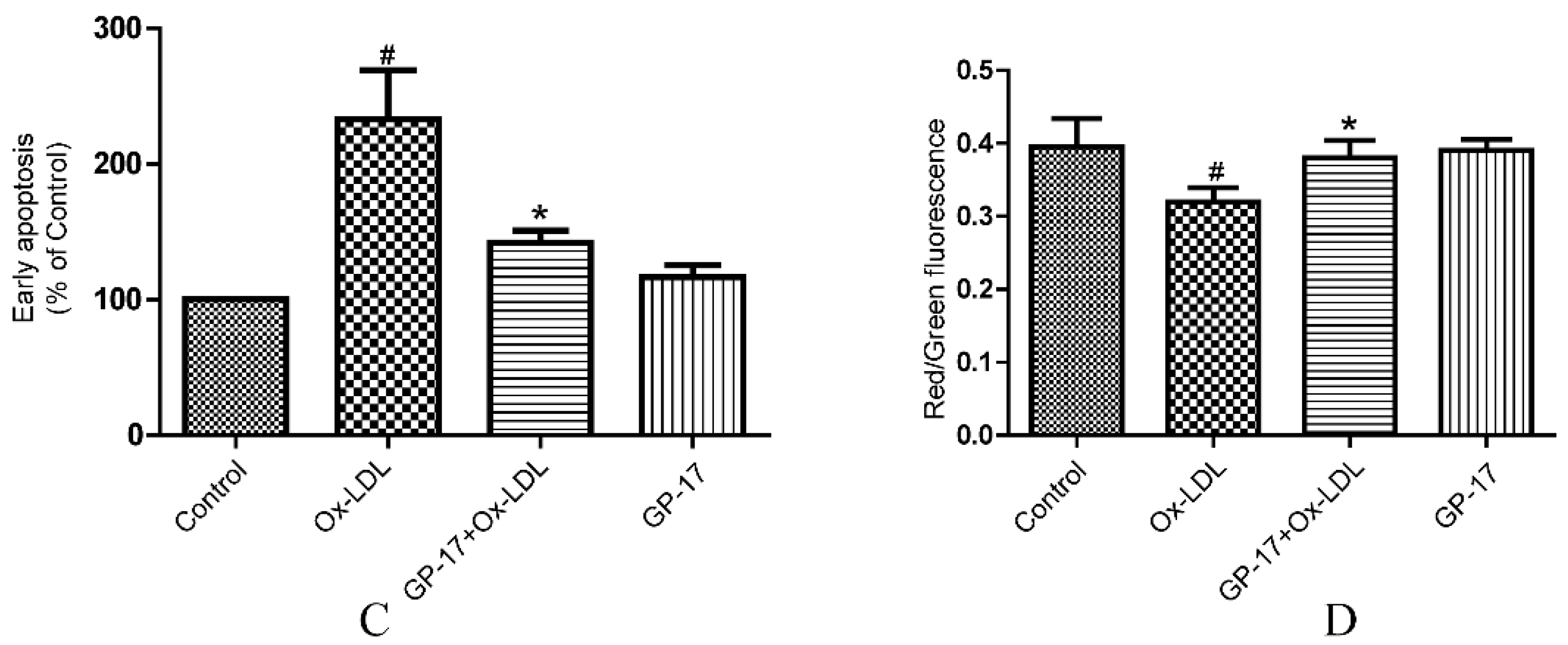
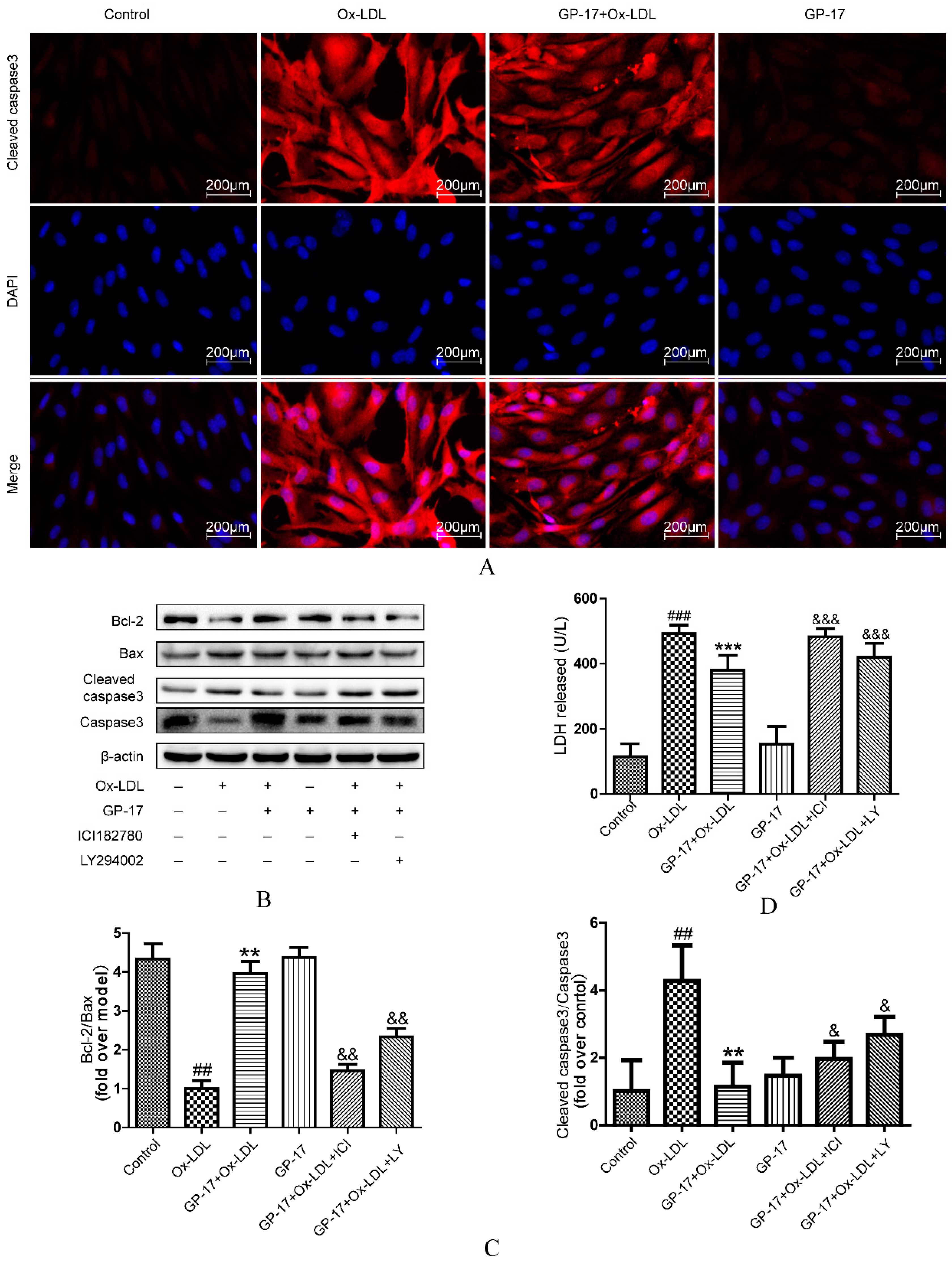
2.4. GP-17 Inhibits Ox-LDL-Induced Apoptosis in HUVECs
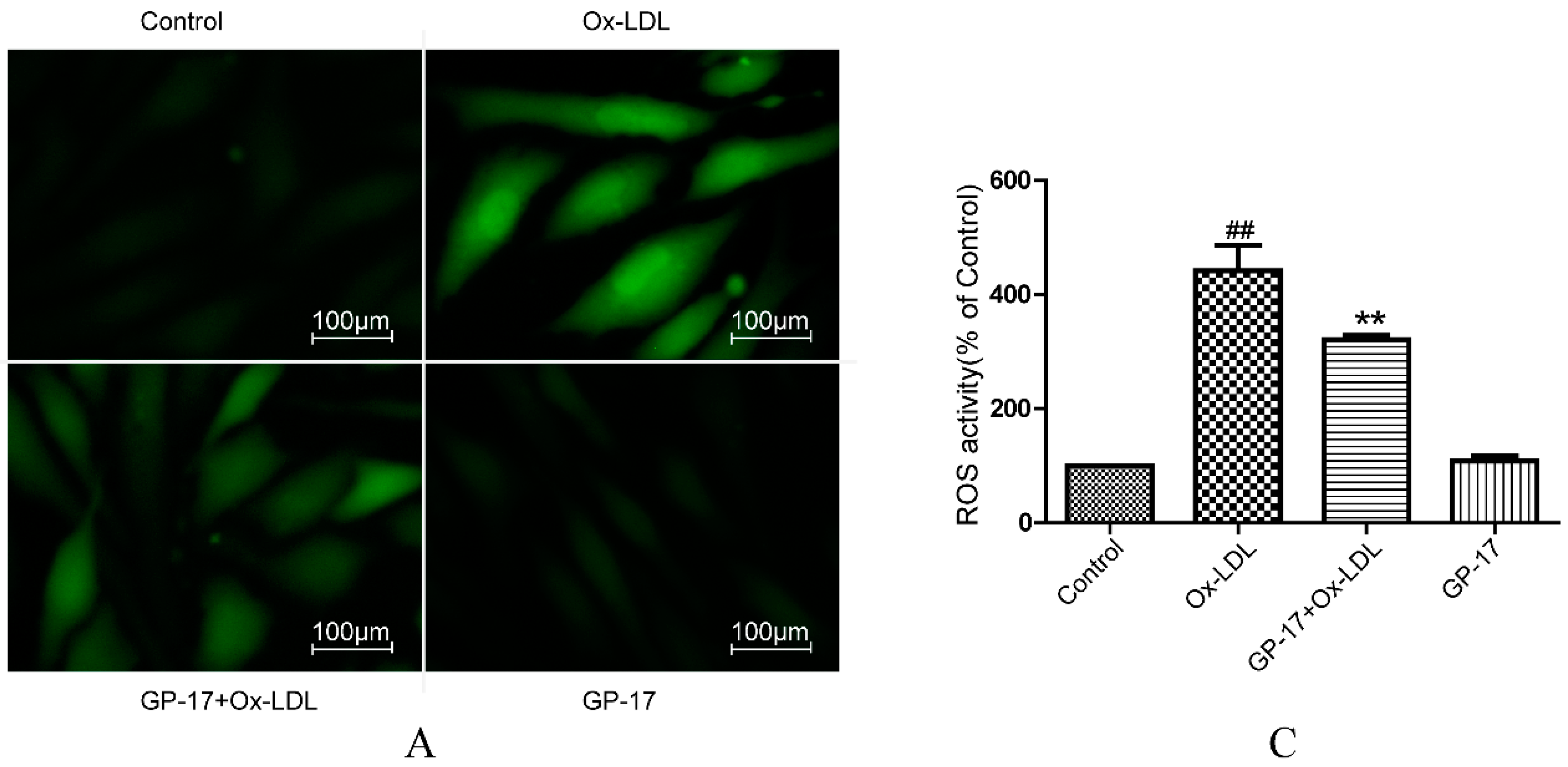
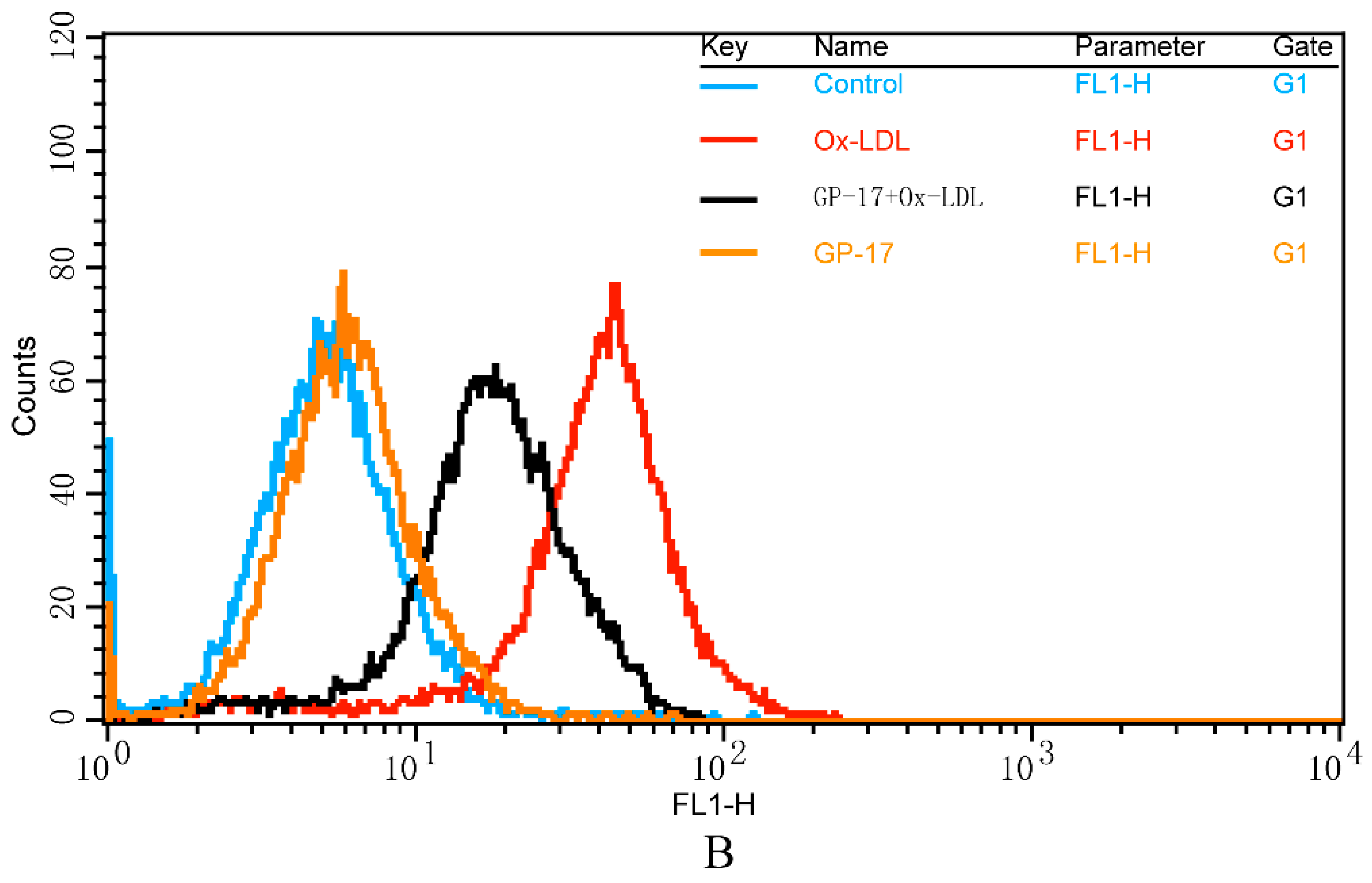
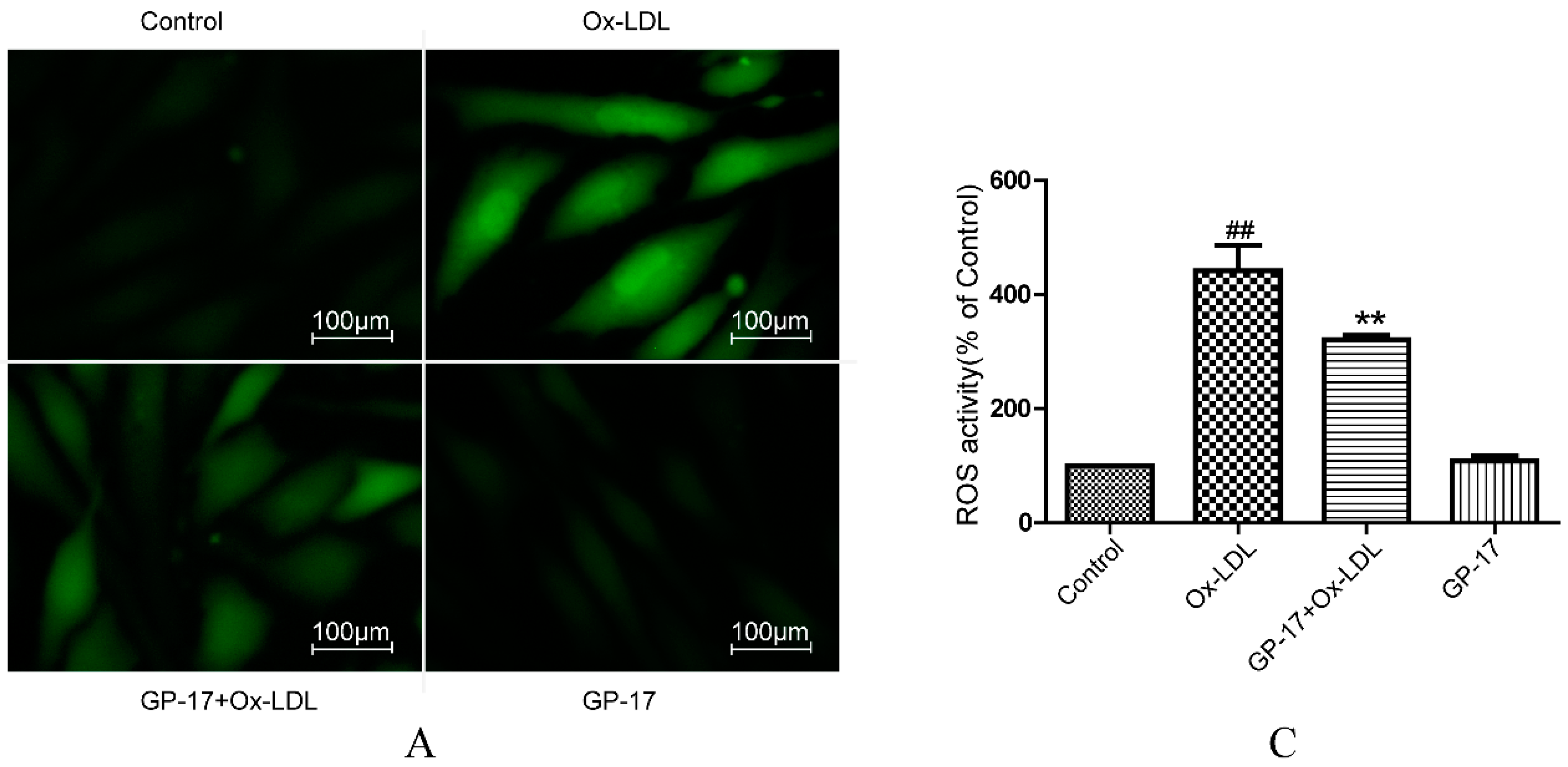
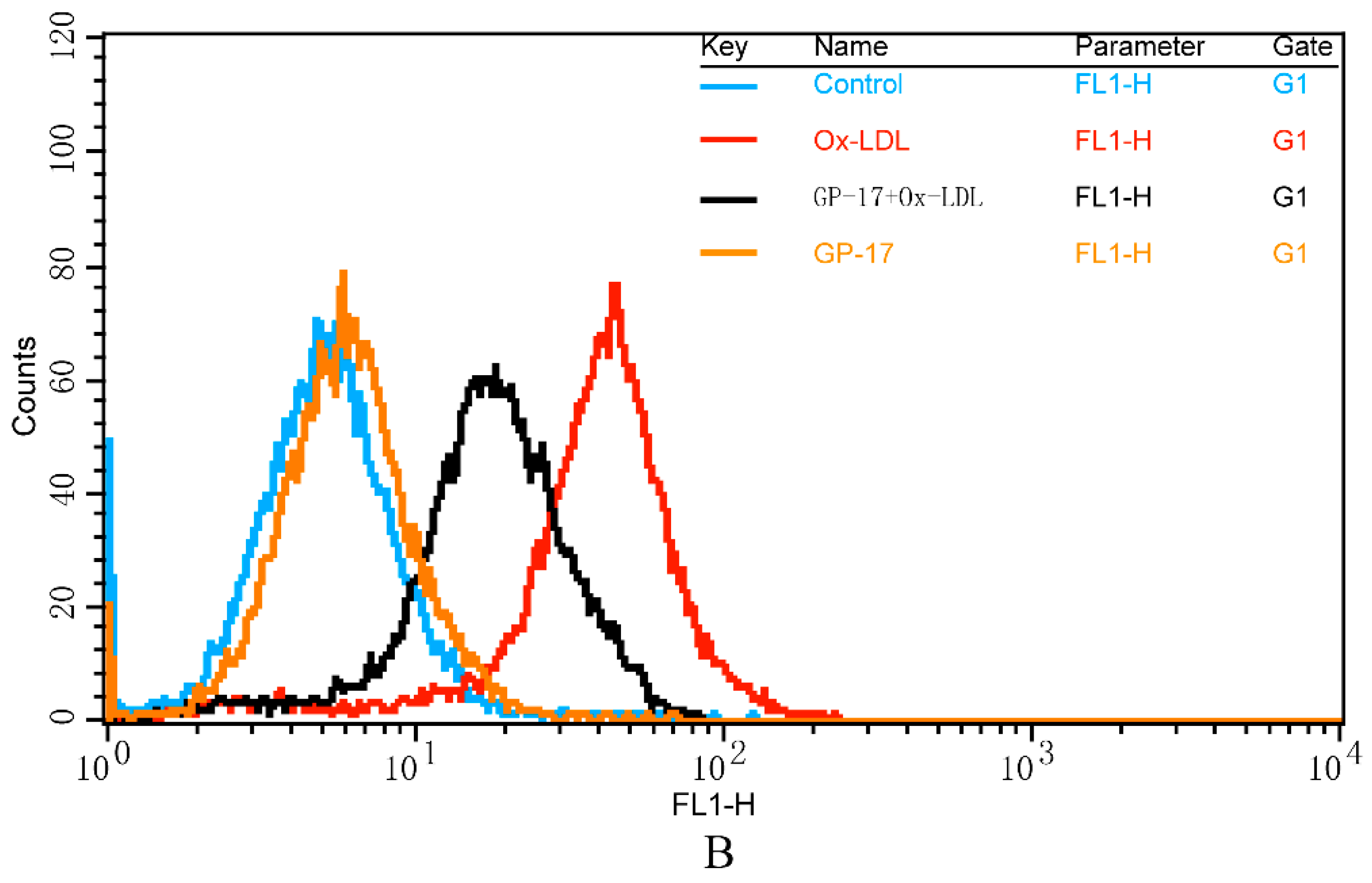
2.5. GP-17 Reduced Ox-LDL-Induced ROS Generation in HUVECs
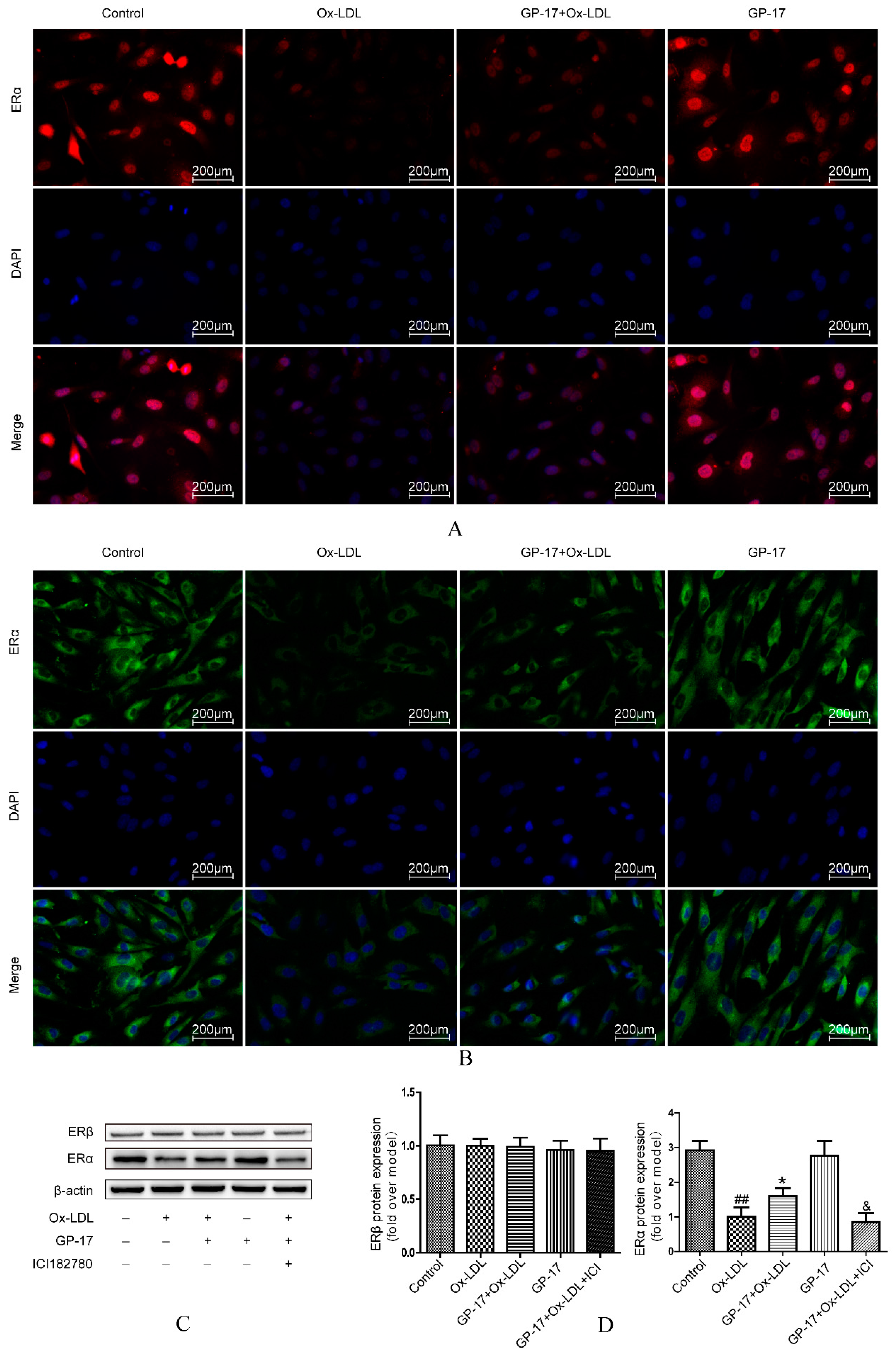
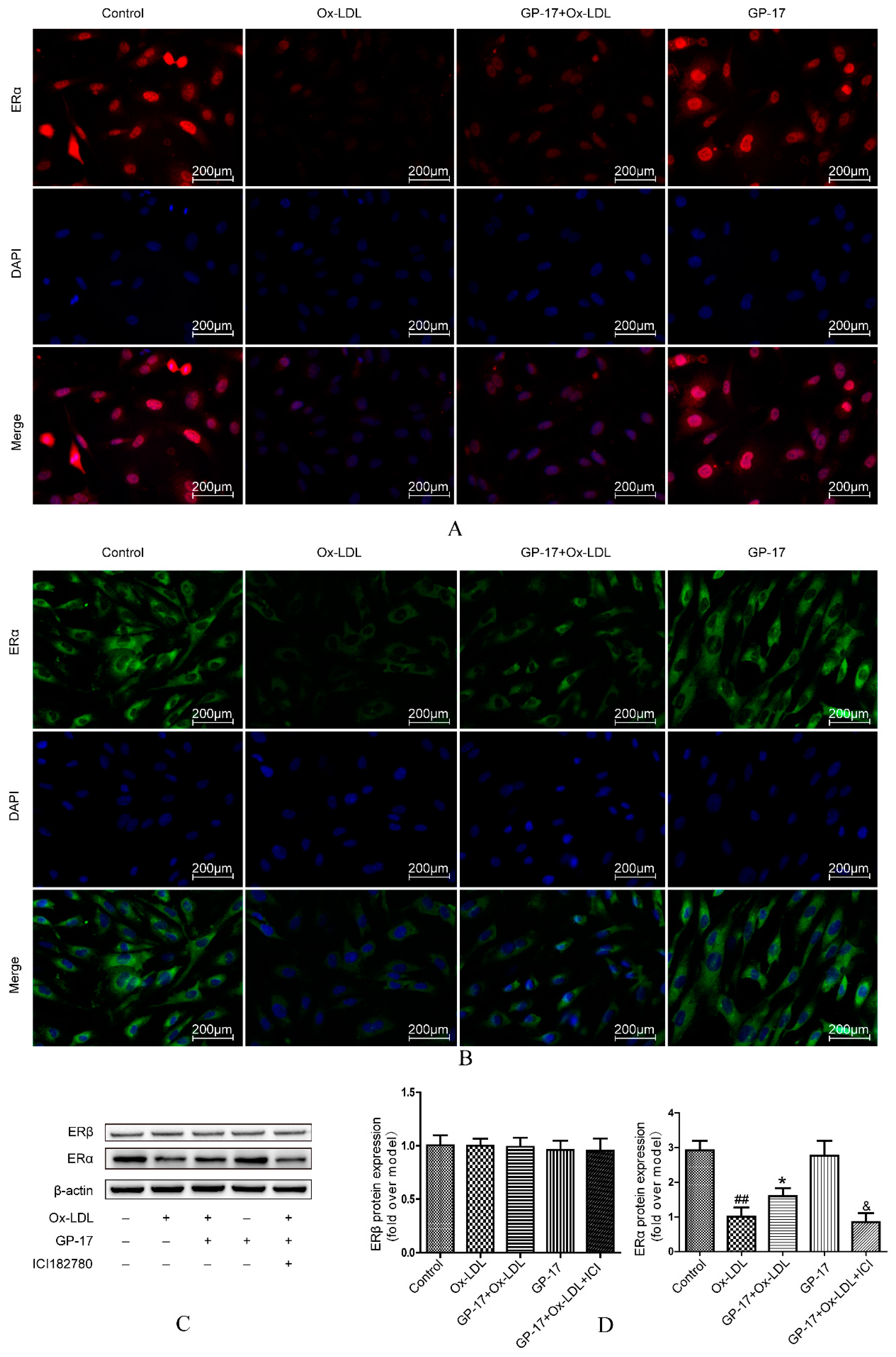
2.6. Effect of GP-17 Treatment on Expression of ERα
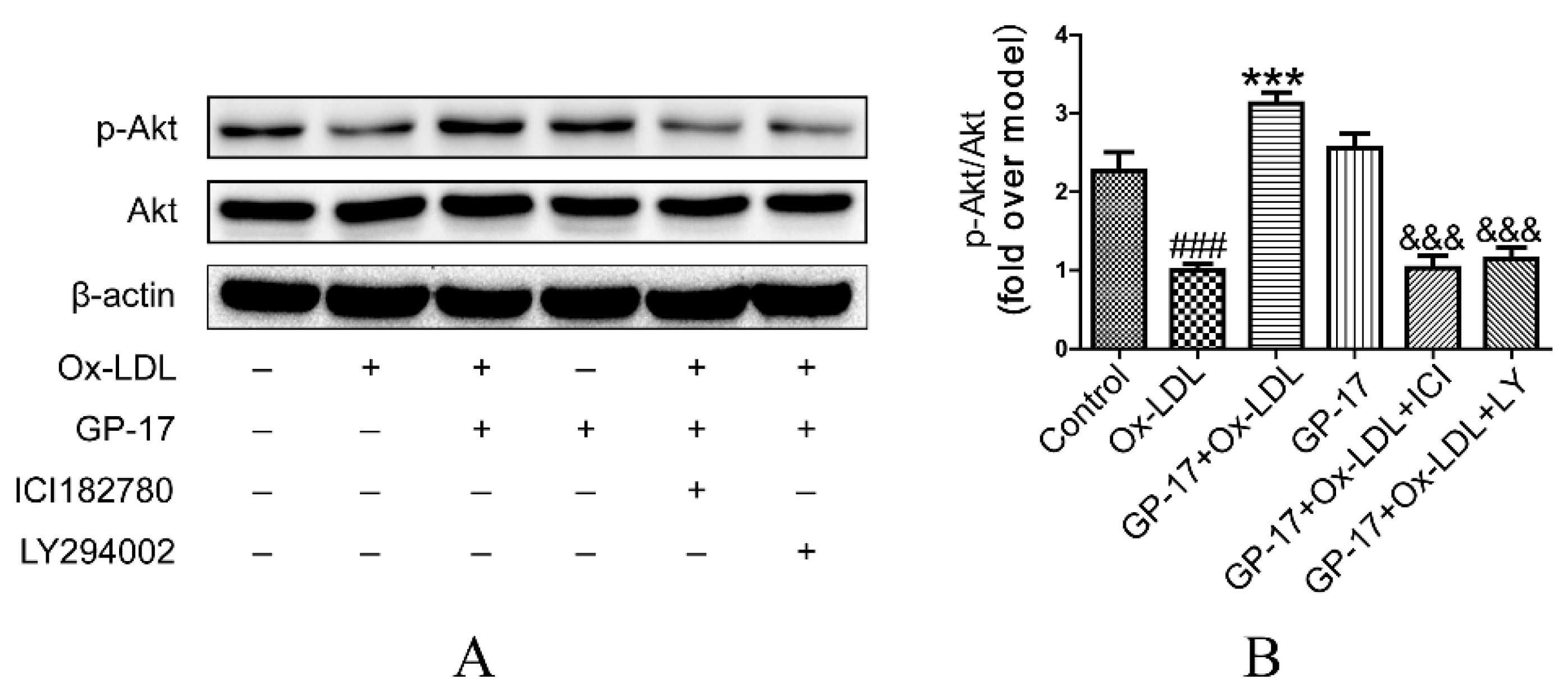
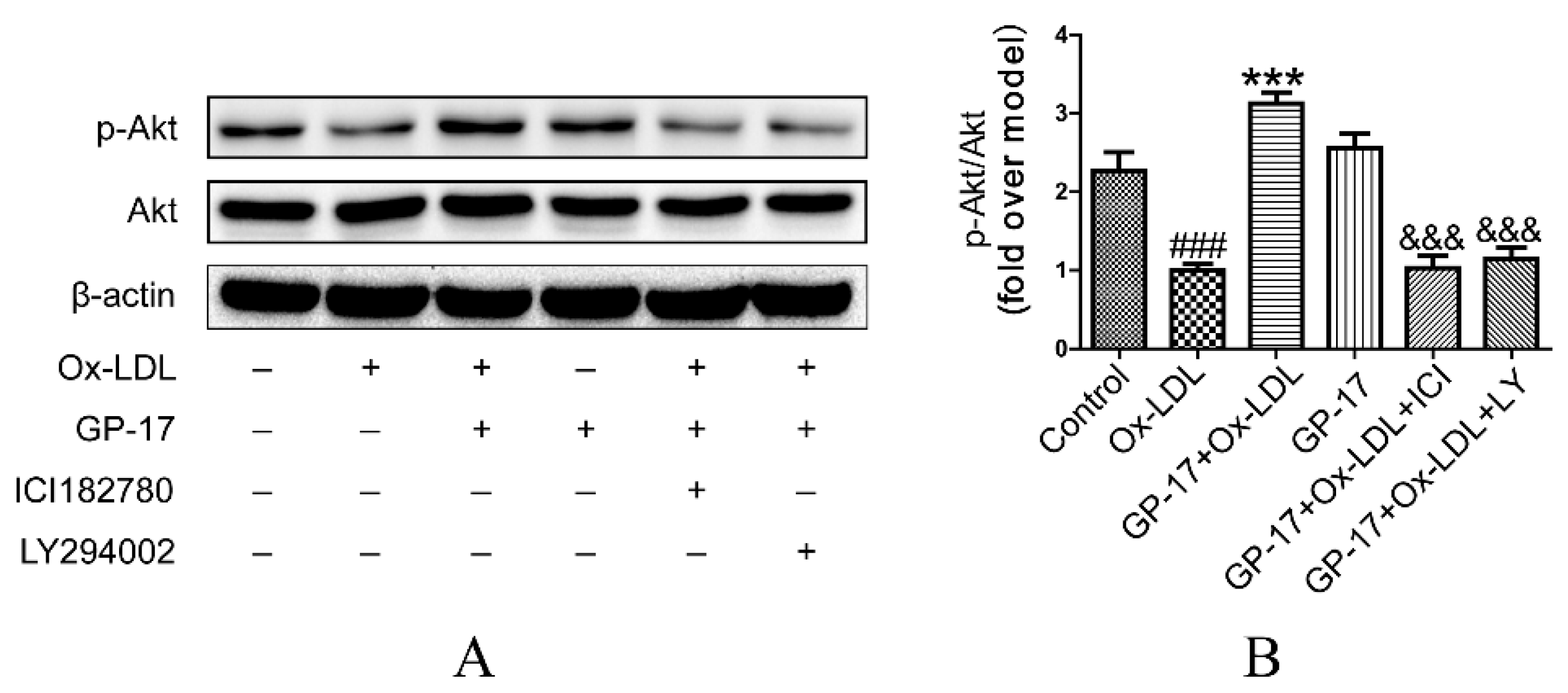
2.7. PI3K/Akt Activation by GP-17 via the ERα-Dependent Pathway
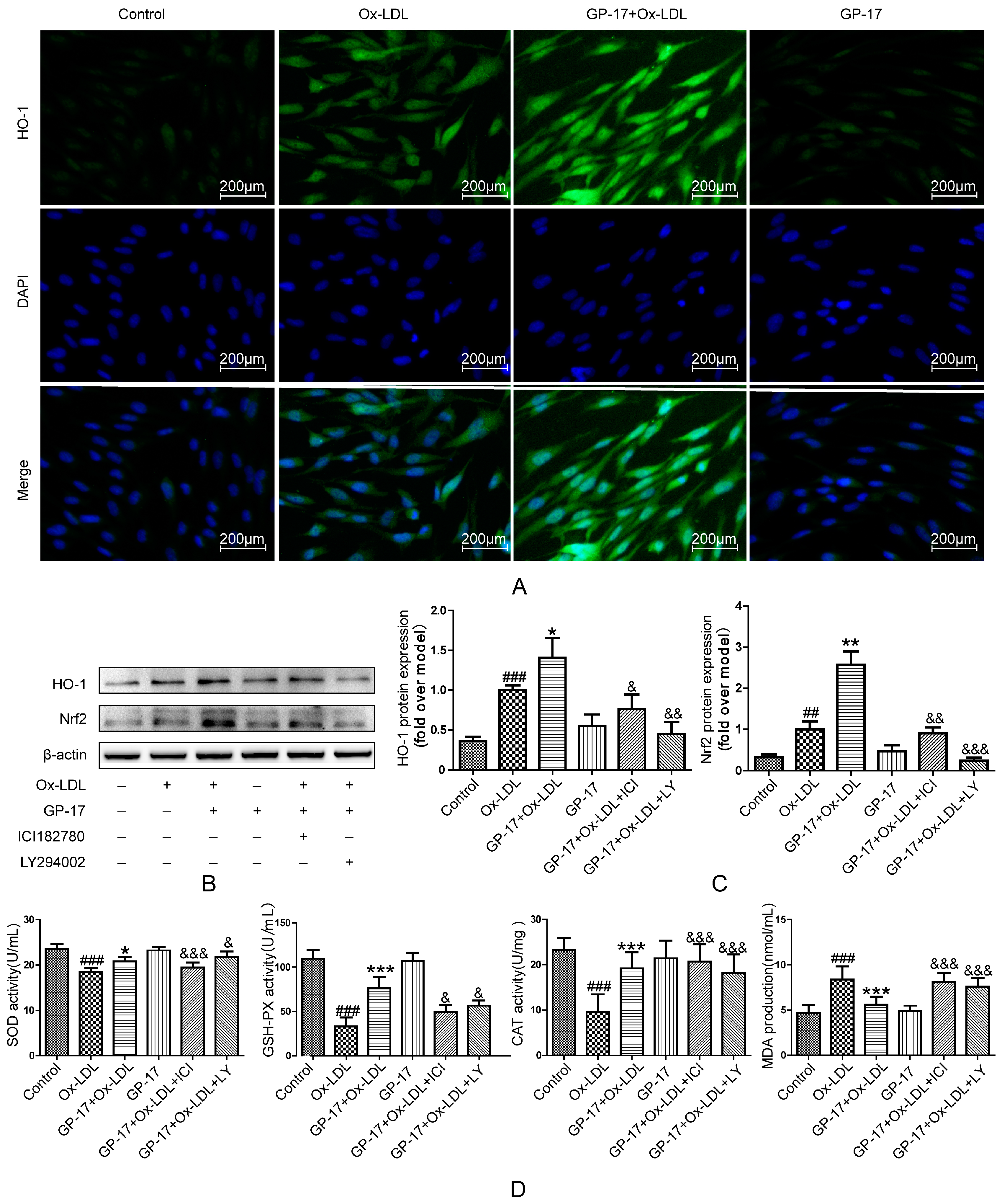
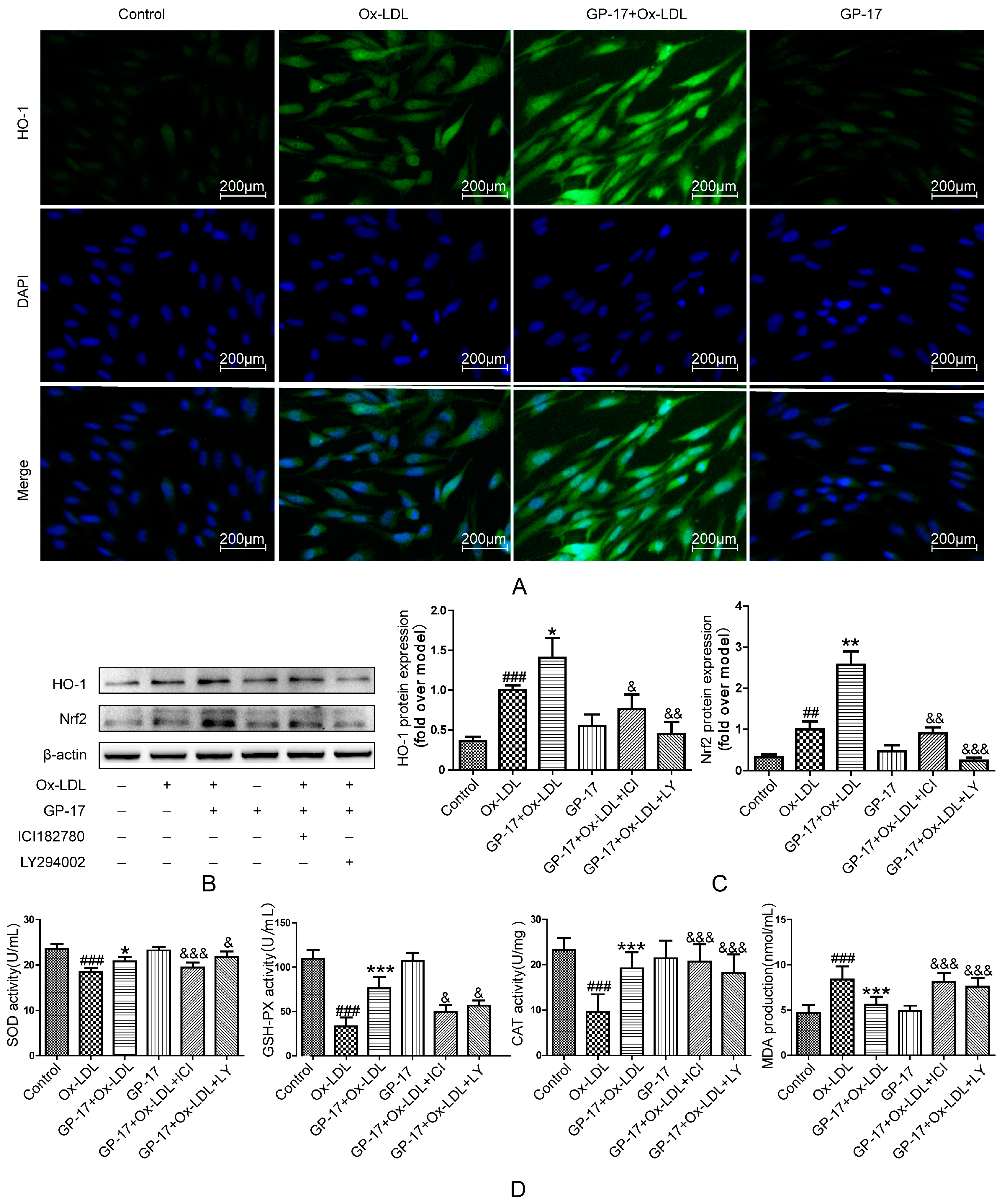
2.8. GP-17 Increases Antioxidant Defense via the ERα/PI3K/Akt Pathway in HUVECs
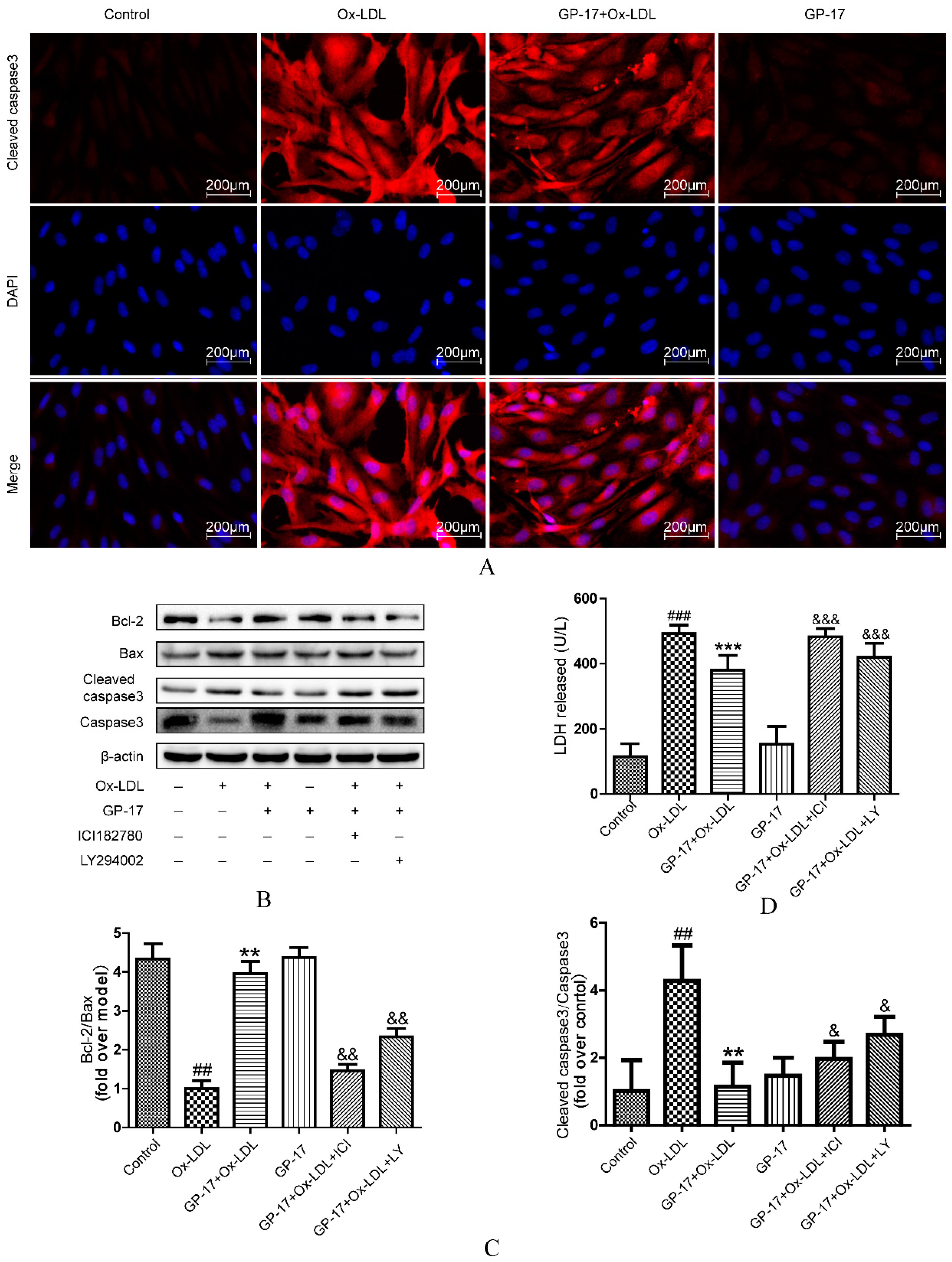
2.9. GP-17 Inhibits Ox-LDL-Induced HUVEC Apoptosis by Increasing Bcl-2 Family Proteins and Decreasing Caspase-3 Activation
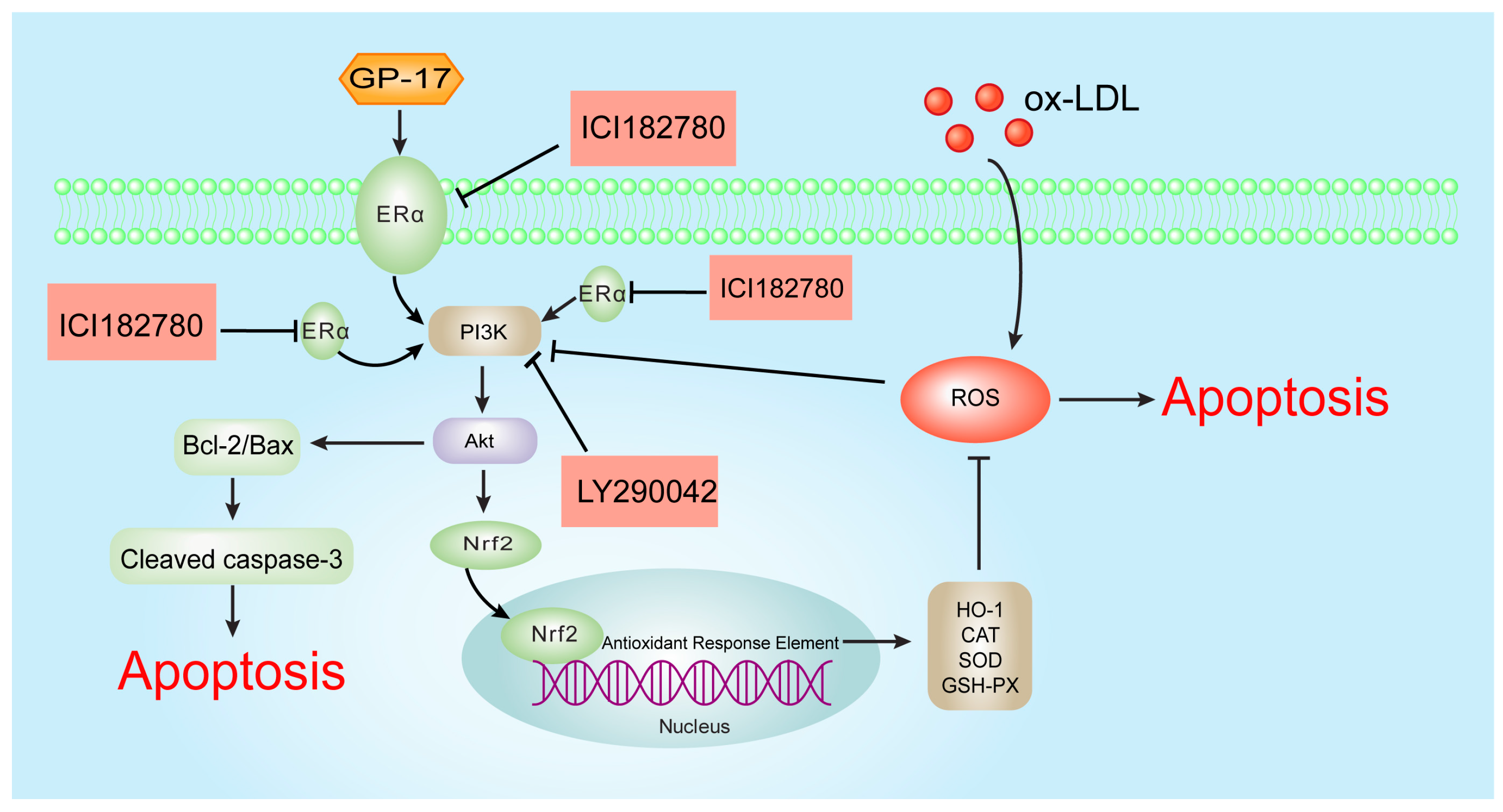
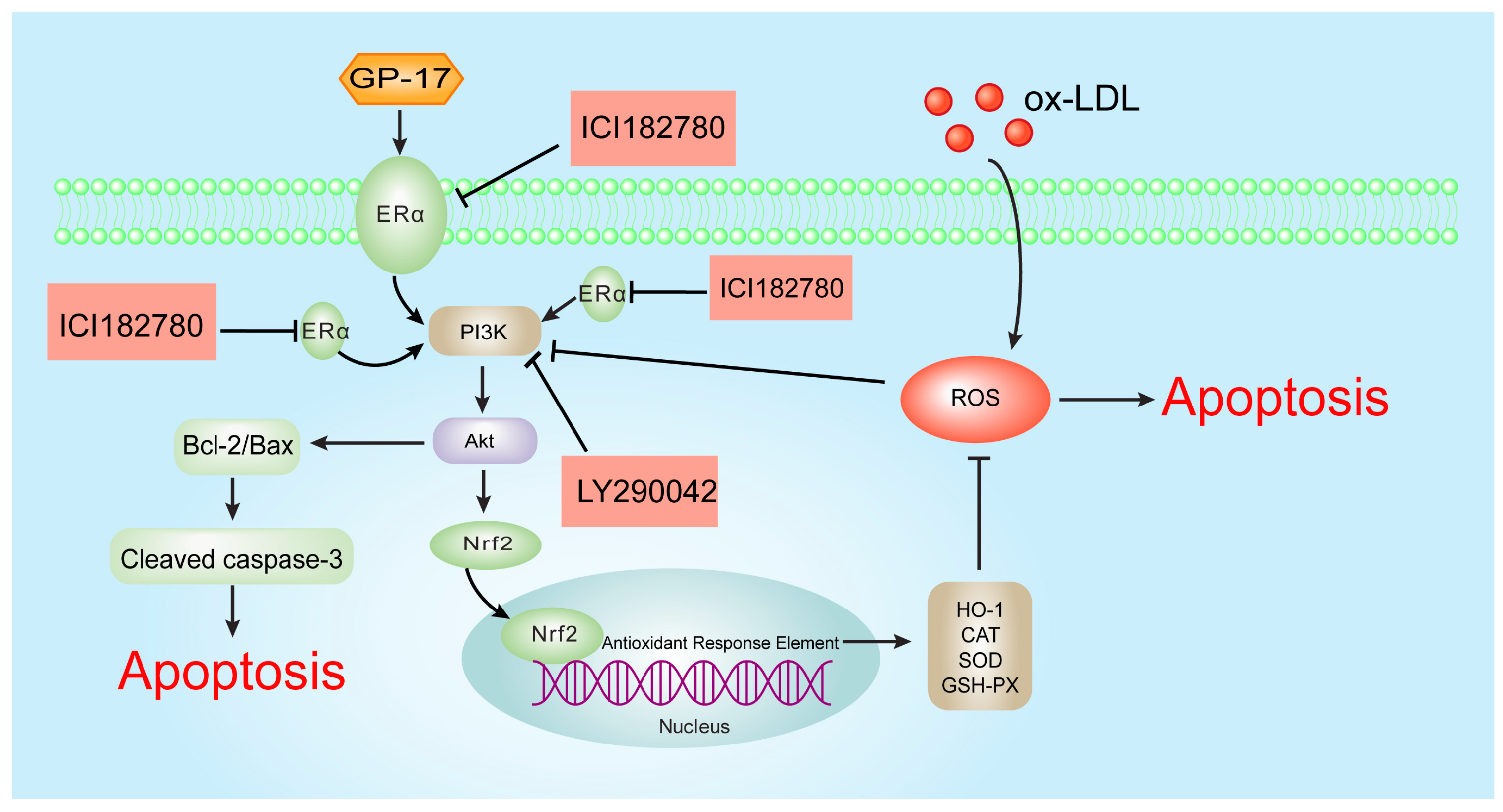
3. Discussion
4. Materials and Methods
4.1. Reagent and Materials
4.2. Experimental Animals and Drug Administration
4.3. Plasma Lipids and Oxidative Stress Analysis
4.4. Oil Red Staining
4.5. Cell Preparation and Culture
4.6. Cell Viability Assay
4.7. Cell Apoptosis Assay by Annexin V-FITC/ Propidium Iodide (PI)
4.8. Measurement of Mitochondrial Membrane Potential (ΔΨm) Disruption by Plate Reader and Fluorescence Microscopy
4.9. Measurement of ROS Activity by Plate Reader and Fluorescence Microscopy
4.10. Measurement of Caspase-3 Activity, MDA, CAT, GSH-Px, SOD Activity, LDH Release in HUVCEs
4.11. Immunocytochemical Analysis of Estrogen Receptor, HO-1 and Caspase-3
4.12. Western Blot Analysis
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| LY | LY294002 |
| ICI | ICI182780 |
| TG | Triglyceride |
| AS | Atherosclerosis |
| TC | Total cholesterol |
| CAT | Catalase |
| GP-17 | Gypenoside XVII |
| ERα | Estrogen receptor alpha |
| ERβ | Estrogen receptor beta |
| GPER | G protein-coupled estrogen receptor 1 |
| ECDs | Estrogen-dendrimer conjugates |
| ERs | Estrogen receptors |
| MDA | Malondialdehyde |
| HO-1 | Heme oxygenase 1 |
| ROS | Reactive oxygen species |
| SOD | Superoxide dismutase |
| PBS | Phosphate-buffered saline |
| LDH | Lactate dehydrogenase |
| GSH-Px | Glutathione peroxidase |
| PI3K | Phosphatidylinositide 3-kinase |
| Akt | Protein kinase B |
| DAPI | 4,6-Diamidino-2-phenylindole |
| ApoE−/− | Apolipoprotein E-deficient |
| HUVECs | Human umbilical vein endothelial cells |
| Ox-LDL | Oxidized low-density lipoprotein |
| LDL-C | Low-density lipoprotein cholesterol |
| HDL-C | High-density lipoprotein cholesterol |
| Nrf2 | Nuclear factor erythroid 2-related factor 2 |
| MTT | 3-(4,5-Dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide |
| JC-1 | 5,5’,6,6’-Tetrachloro-1,1’,3,3’-tetraethylbenzimidazolylcarbocyanine iodide |
References
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Napoli, C. Oxidation of LDL, atherogenesis, and apoptosis. Ann. N. Y. Acad. Sci. 2003, 1010, 698–709. [Google Scholar] [CrossRef] [PubMed]
- Botham, K.M.; Wheeler-Jones, C.P. Postprandial lipoproteins and the molecular regulation of vascular homeostasis. Prog. Lipid Res. 2013, 52, 446–464. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Wang, X.M.; Qian, D.H.; Qin, Z.X.; Jin, J.; Xu, Q.; Yuan, Q.Y.; Li, X.J.; Si, L.Y. Induction of oxidative stress by oxidized LDL via meprinalpha-activated epidermal growth factor receptor in macrophages. Cardiovasc. Res. 2013, 97, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Dandapat, A.; Hu, C.; Sun, L.; Mehta, J.L. Small concentrations of oxLDL induce capillary tube formation from endothelial cells via LOX-1-dependent redox-sensitive pathway. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2435–2442. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Mehta, J.L.; Haider, N.; Zhang, X.; Narula, J.; Li, D. Role of caspases in ox-LDL-induced apoptotic cascade in human coronary artery endothelial cells. Circ. Res. 2004, 94, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yang, J.H.; Burns, A.R.; Chen, H.H.; Tang, D.; Walterscheid, J.P.; Suzuki, S.; Yang, C.Y.; Sawamura, T.; Chen, C.H. Mediation of electronegative low-density lipoprotein signaling by LOX-1: A possible mechanism of endothelial apoptosis. Circ. Res. 2009, 104, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Arnal, J.F.; Gourdy, P.; Elhage, R.; Garmy-Susini, B.; Delmas, E.; Brouchet, L.; Castano, C.; Barreira, Y.; Couloumiers, J.C.; Prats, H.; et al. Estrogens and atherosclerosis. Eur. J. Endocrinol. 2004, 150, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Arnal, J.F.; Douin-Echinard, V.; Brouchet, L.; Tremollières, F.; Laurell, H.; Lenfant, F.; Gadeau, A.P.; Guery, J.C.; Gourdy, P. Understanding the oestrogen action in experimental and clinical atherosclerosis. Fundam. Clin. Pharmacol. 2006, 20, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.H.; Hsieh, C.C.; Chou, S.Y.; Lau, Y.T. 17β-estradiol inhibits oxidized low density lipoprotein-induced generation of reactive oxygen species in endothelial cells. Life Sci. 2001, 70, 403–413. [Google Scholar] [CrossRef]
- Xu, H.S.; Duan, J.; Dai, S.; Wu, Y.; Sun, R.; Ren, J. Phytoestrogen alpha-zearalanol antagonizes oxidized LDL-induced inhibition of nitric oxide production and stimulation of endothelin-1 release in human umbilical vein endothelial cells. Endocrine 2004, 25, 235–245. [Google Scholar] [CrossRef]
- Barton, M. Cholesterol and atherosclerosis: Modulation by oestrogen. Curr. Opin. Lipidol. 2013, 24, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Menazza, S.; Murphy, E. The expanding complexity of estrogen receptor signaling in the Cardiovascular System. Circ. Res. 2016, 118, 994–1007. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Morton, J.S.; Davidge, S.T. Mechanisms of estrogen effects on the endothelium: An overview. Can. J. Cardiol. 2014, 30, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Simoncini, T.; Hafezi-Moghadam, A.; Brazil, D.P.; Ley, K.; Chin, W.W.; Liao, J.K. Interaction of oestrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature 2000, 407, 538–541. [Google Scholar] [PubMed]
- Meng, X.; Wang, M.; Sun, G.; Ye, J.; Zhou, Y.; Dong, X.; Wang, T.; Lu, S.; Sun, X. Attenuation of Aβ25-35-induced parallel autophagic and apoptotic cell death by gypenoside XVII through the estrogen receptor-dependent activation of Nrf2/ARE pathways. Toxicol. Appl. Pharmacol. 2014, 279, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell. Mol. Med. 2005, 9, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Schenck-Gustafsson, K.; Brincat, M.; Erel, C.T.; Gambacciani, M.; Lambrinoudaki, I.; Moen, M.H.; Tremollieres, F.; Vujovic, S.; Rozenberg, S.; Rees, M.; et al. EMAS position statement: Managing the menopause in the context of coronary heart disease. Maturitas 2011, 68, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Cos, P.; De Bruyne, T.; Apers, S.; Vanden Berghe, D.; Pieters, L.; Vlietinck, A.J. Phytoestrogens: Recent developments. Planta Med. 2003, 69, 589–599. [Google Scholar] [PubMed]
- Gencel, V.B.; Benjamin, M.M.; Bahou, S.N.; Khalil, R.A. Vascular effects of phytoestrogens and alternative menopausal hormone therapy in cardiovascular disease. Mini Rev. Med. Chem. 2012, 12, 149–174. [Google Scholar] [CrossRef] [PubMed]
- Aktan, F.; Henness, S.; Roufogalis, B.D.; Ammit, A.J. Gypenosides derived from Gynostemma pentaphyllum suppress NO synthesis in murine macrophages by inhibiting iNOS enzymatic activity and attenuating NF-κB-mediated iNOS protein expression. Nitric Oxide 2003, 8, 235–242. [Google Scholar] [CrossRef]
- Huang, T.H.; Razmovski-Naumovski, V.; Salam, N.K.; Duke, R.K.; Tran, V.H.; Duke, C.C.; Roufogalis, B.D. A novel LXR-α activator identified from the natural product Gynostemma pentaphyllum. Biochem. Pharmacol. 2005, 70, 1298–1308. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.H.; Tran, V.H.; Roufogalis, B.D.; Li, Y. Gypenoside XLIX, A naturally occurring gynosaponin, PPAR-α dependently inhibits LPS-induced tissue factor expression and activity in human THP-1 monocytic cells. Toxicol. Appl. Pharmacol. 2007, 218, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Bairey Merz, C.N.; Johnson, B.D.; Braunstein, G.D.; Pepine, C.J.; Reis, S.E.; Paul-Labrador, M.; Hale, G.; Sharaf, B.L.; Bittner, V.; Sopko, G.; et al. Phytoestrogens and lipoproteins in women. J. Clin. Endocrinol. Metab. 2006, 91, 2209–2213. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.Z.; Zhong, W.; Ghavideldarestani, M.; Saurabh, R.; Lindow, S.W.; Atkin, S.L. Multiple mechanisms of soy isoflavones against oxidative stress-induced endothelium injury. Free Radic. Biol. Med. 2009, 47, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Kładna, A.; Berczyński, P.; Kruk, I.; Piechowska, T.; Aboul-Enein, H.Y. Studies on the antioxidant properties of some phytoestrogens. Luminescence 2016, 31, 1201–1206. [Google Scholar] [CrossRef] [PubMed]
- Wing, L.Y.; Chen, Y.C.; Shih, Y.Y.; Cheng, J.C.; Lin, Y.J.; Jiang, M.J. Effects of oral estrogen on aortic ROS-generating and -scavenging enzymes and atherosclerosis in apoE-deficient mice. Exp. Biol. Med. 2009, 234, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Russell, K.S.; Haynes, M.P.; Sinha, D.; Clerisme, E.; Bender, J.R. Human vascular endothelial cells contain membrane binding sites for estradiol, which mediate rapid intracellular signaling. Proc. Natl. Acad. Sci. USA 2000, 97, 5930–5935. [Google Scholar] [CrossRef] [PubMed]
- Oche, B.; Chen, L.; Ma, Y.K.; Yang, Y.; Li, C.X.; Geng, X.; Qiu, L.Z.; Gao, X.M.; Wang, H. Cryptotanshinone and wogonin up-regulate eNOS in vascular endothelial cells via ERα and down-regulate iNOS in LPS stimulated vascular smooth muscle cells via ERβ. Arch. Pharm. Res. 2016, 39, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Simoncini, T.; Rabkin, E.; Liao, J.K. Molecular basis of cell membrane estrogen receptor interaction with phosphatidylinositol 3-kinase in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.X.; Wei, L.H.; Tu, Z.; Sun, P.M.; Wang, J.L.; Zhao, D.; Li, X.P.; Tang, J.M. 17 Beta-estradiol activates PI3K/Akt signaling pathway by estrogen receptor (ER)-dependent and ER-independent mechanisms in endometrial cancer cells. J. Steroid Biochem. Mol. Biol. 2006, 99, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Salvayre, R.; Negre-Salvayre, A.; Camaré, C. Oxidative theory of atherosclerosis and antioxidants. Biochimie 2016, 125, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K. Redox-derived damage-associated molecular patterns: Ligand function of lipid peroxidation adducts. Redox Biol. 2013, 1, 94–96. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.C.; Chou, F.P.; Sheu, W.H.; Hsu, S.L.; Lee, W.J. Protective effects of magnolol against oxidized LDL-induced apoptosis in endothelial cells. Arch. Toxicol. 2007, 81, 421–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, Y.P.; Jeong, H.G. Mechanism of phytoestrogen puerarin-mediated cytoprotection following oxidative injury: Estrogen receptor-dependent up-regulation of PI3K/Akt and HO-1. Toxicol. Appl. Pharmacol. 2008, 233, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Luo, Y.; Liang, T.; Wang, M.; Zhao, J.; Sun, G.; Sun, X. Gypenoside XVII Enhances Lysosome Biogenesis and Autophagy Flux and Accelerates Autophagic Clearance of Amyloid-beta through TFEB Activation. J. Alzheimers Dis. 2016, 52, 1135–1150. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.S.; Yang, X.Y.; Zheng, T.; Li, W.J.; Wu, D.; Chi, J.Y.; Bian, F.; Bai, X.L.; Wu, G.J.; Zhang, Y.Z.; et al. Salidroside improves endothelial function and alleviates atherosclerosis by activating a mitochondria-related AMPK/PI3K/Akt/eNOS pathway. Vascul. Pharmacol. 2015, 72, 141–152. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, K.; Zhang, H.; Luo, Y.; Zhang, J.; Wang, M.; Liao, P.; Cao, L.; Guo, P.; Sun, G.; Sun, X. Gypenoside XVII Prevents Atherosclerosis by Attenuating Endothelial Apoptosis and Oxidative Stress: Insight into the ERα-Mediated PI3K/Akt Pathway. Int. J. Mol. Sci. 2017, 18, 77. https://doi.org/10.3390/ijms18020077
Yang K, Zhang H, Luo Y, Zhang J, Wang M, Liao P, Cao L, Guo P, Sun G, Sun X. Gypenoside XVII Prevents Atherosclerosis by Attenuating Endothelial Apoptosis and Oxidative Stress: Insight into the ERα-Mediated PI3K/Akt Pathway. International Journal of Molecular Sciences. 2017; 18(2):77. https://doi.org/10.3390/ijms18020077
Chicago/Turabian StyleYang, Ke, Haijing Zhang, Yun Luo, Jingyi Zhang, Min Wang, Ping Liao, Li Cao, Peng Guo, Guibo Sun, and Xiaobo Sun. 2017. "Gypenoside XVII Prevents Atherosclerosis by Attenuating Endothelial Apoptosis and Oxidative Stress: Insight into the ERα-Mediated PI3K/Akt Pathway" International Journal of Molecular Sciences 18, no. 2: 77. https://doi.org/10.3390/ijms18020077