
AKT2 Blocks Nucleus Translocation of Apoptosis-Inducing Factor (AIF) and Endonuclease G (EndoG) While Promoting Caspase Activation during Cardiac Ischemia

Abstract
:1. Introduction
2. Results
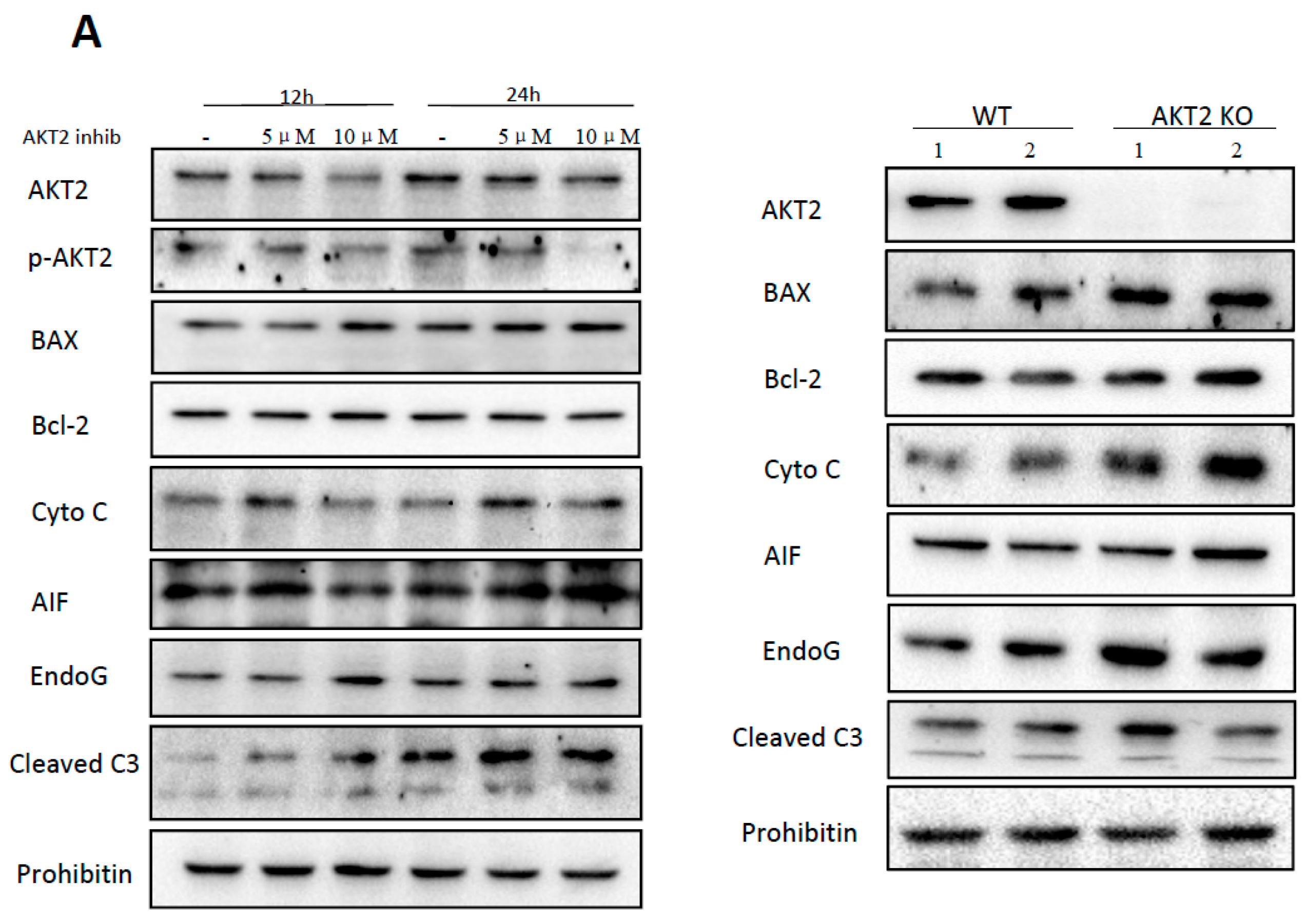
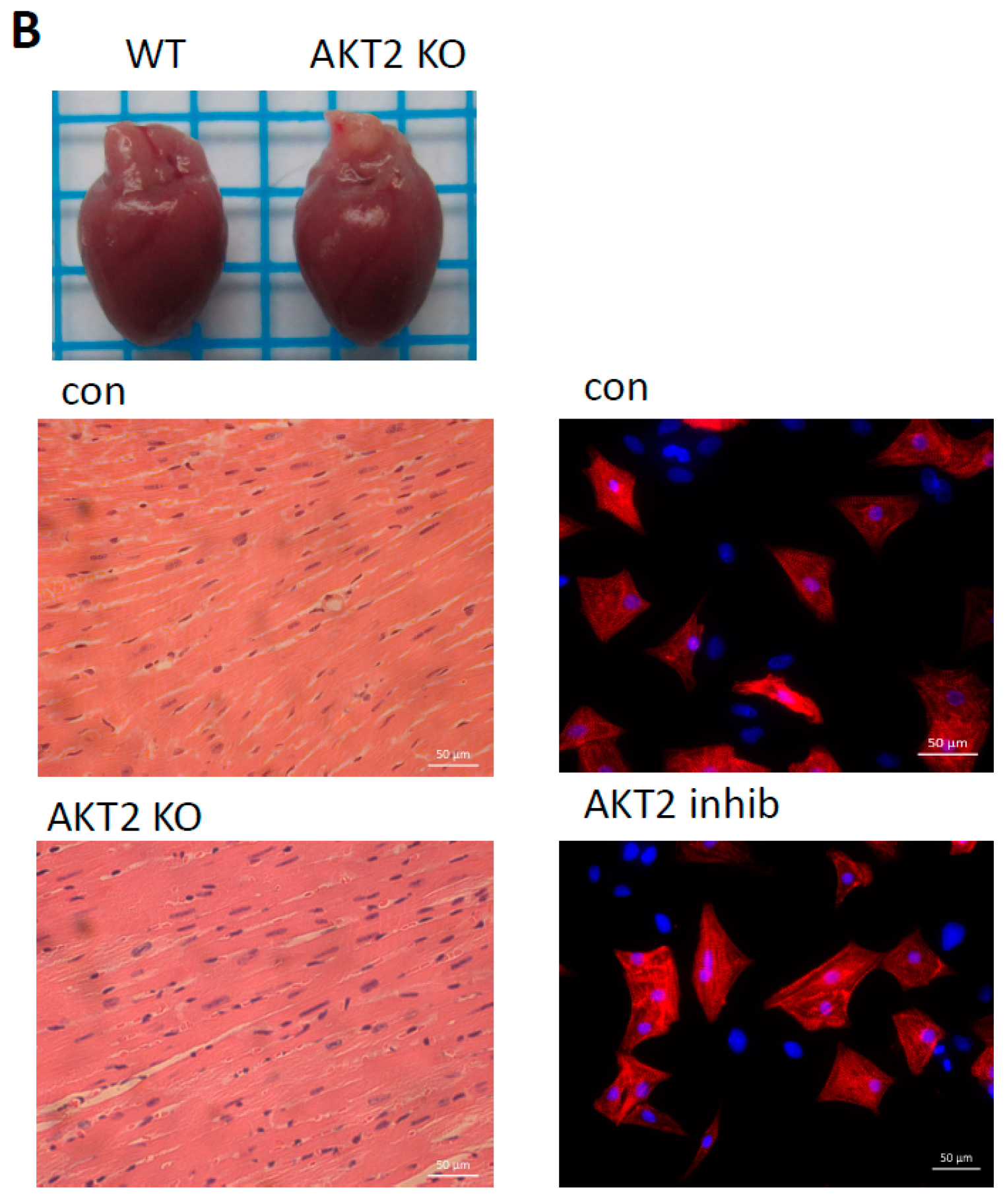
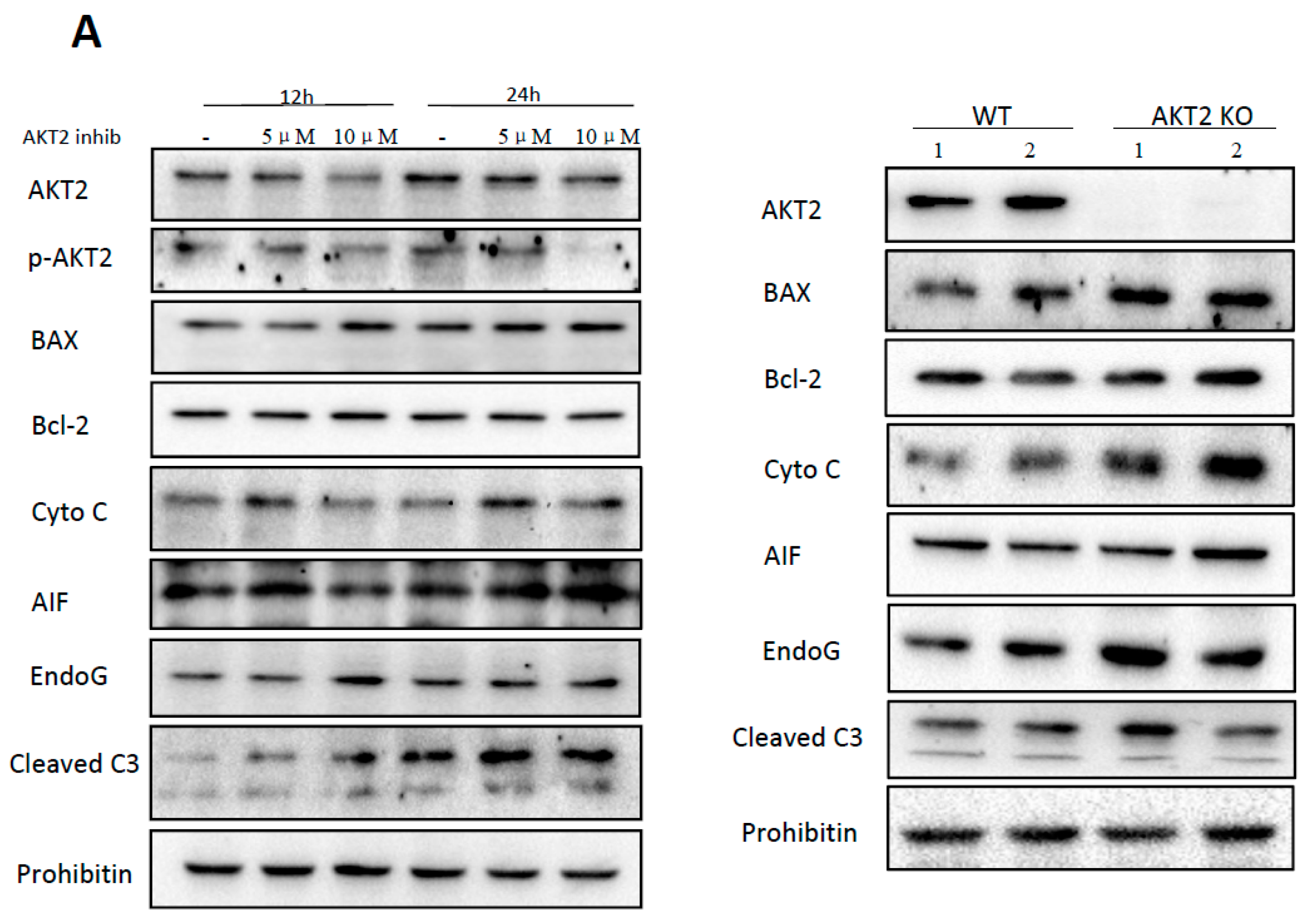
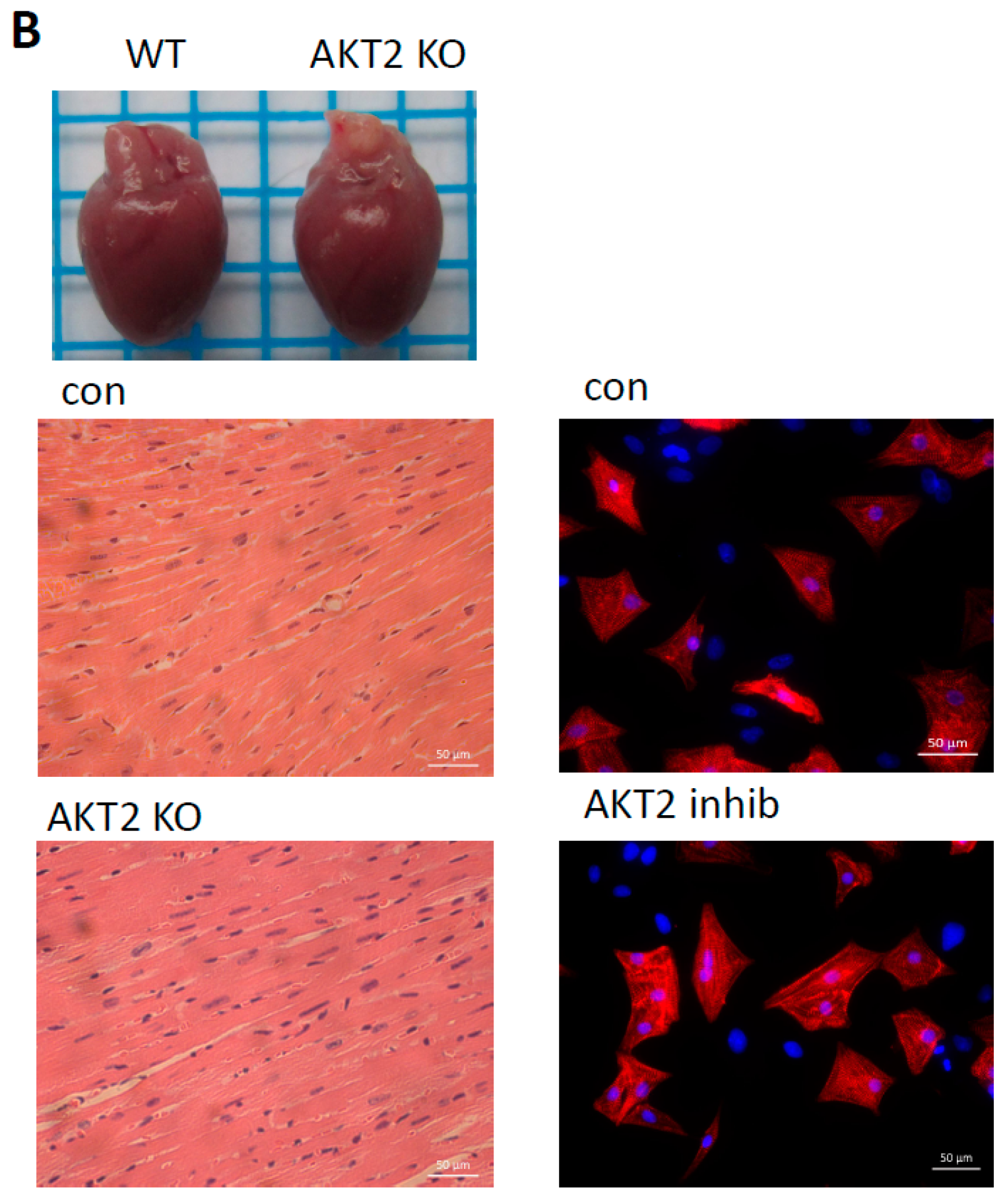
2.1. AKT2 Inhibition Induces Elevation of Apoptotic Gene Expression in Myocardium without Affecting Myocyte Development
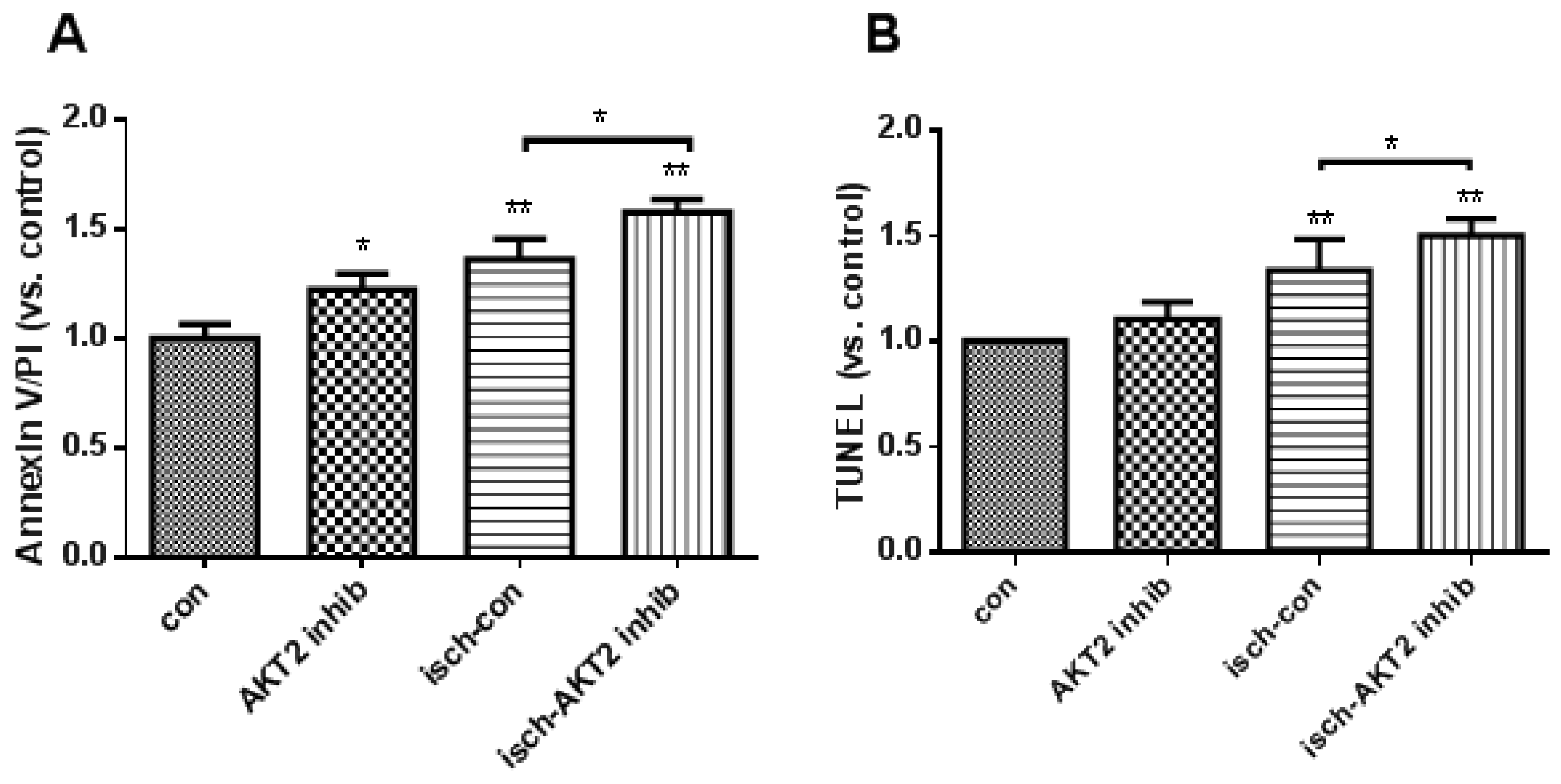
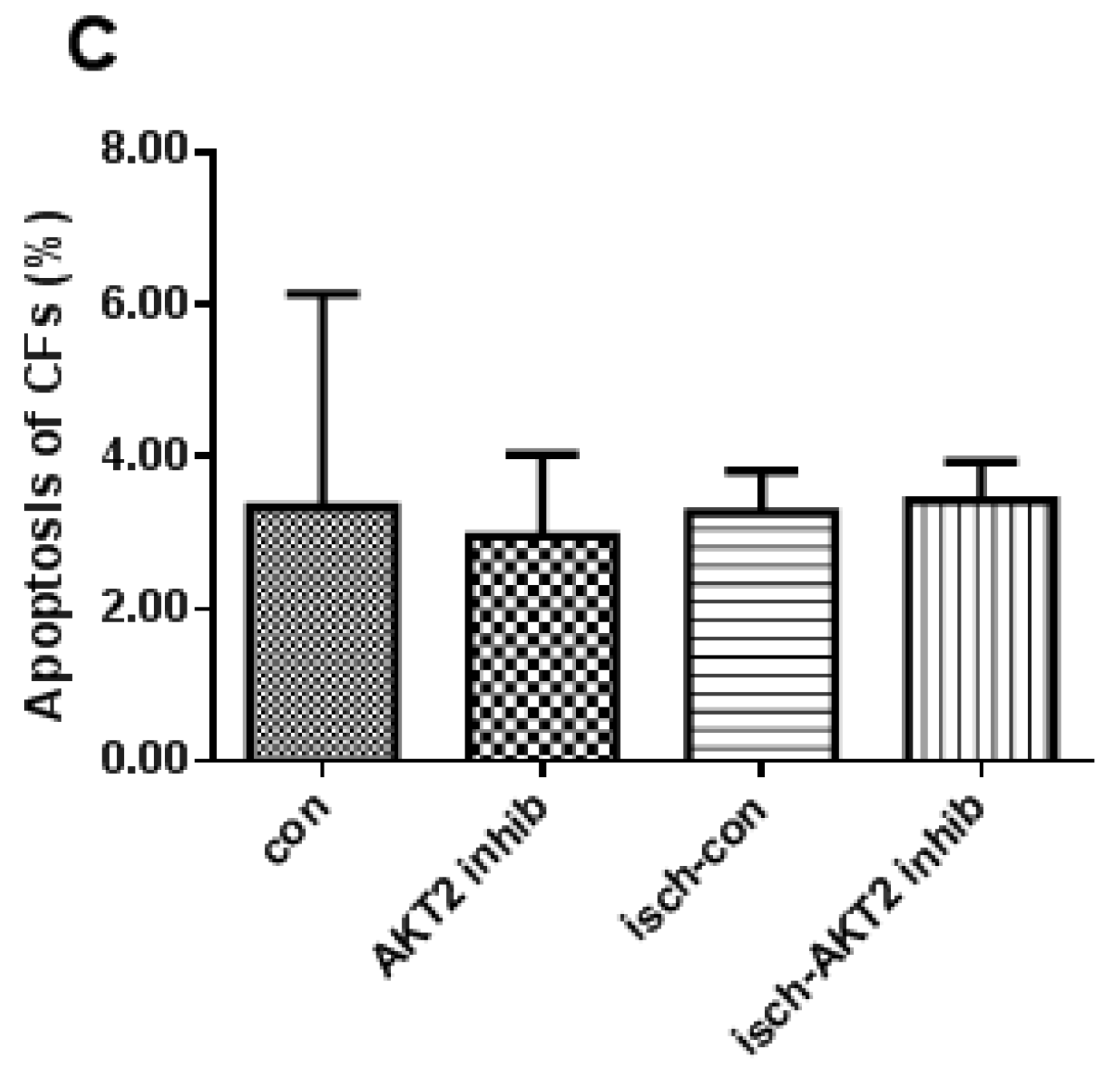
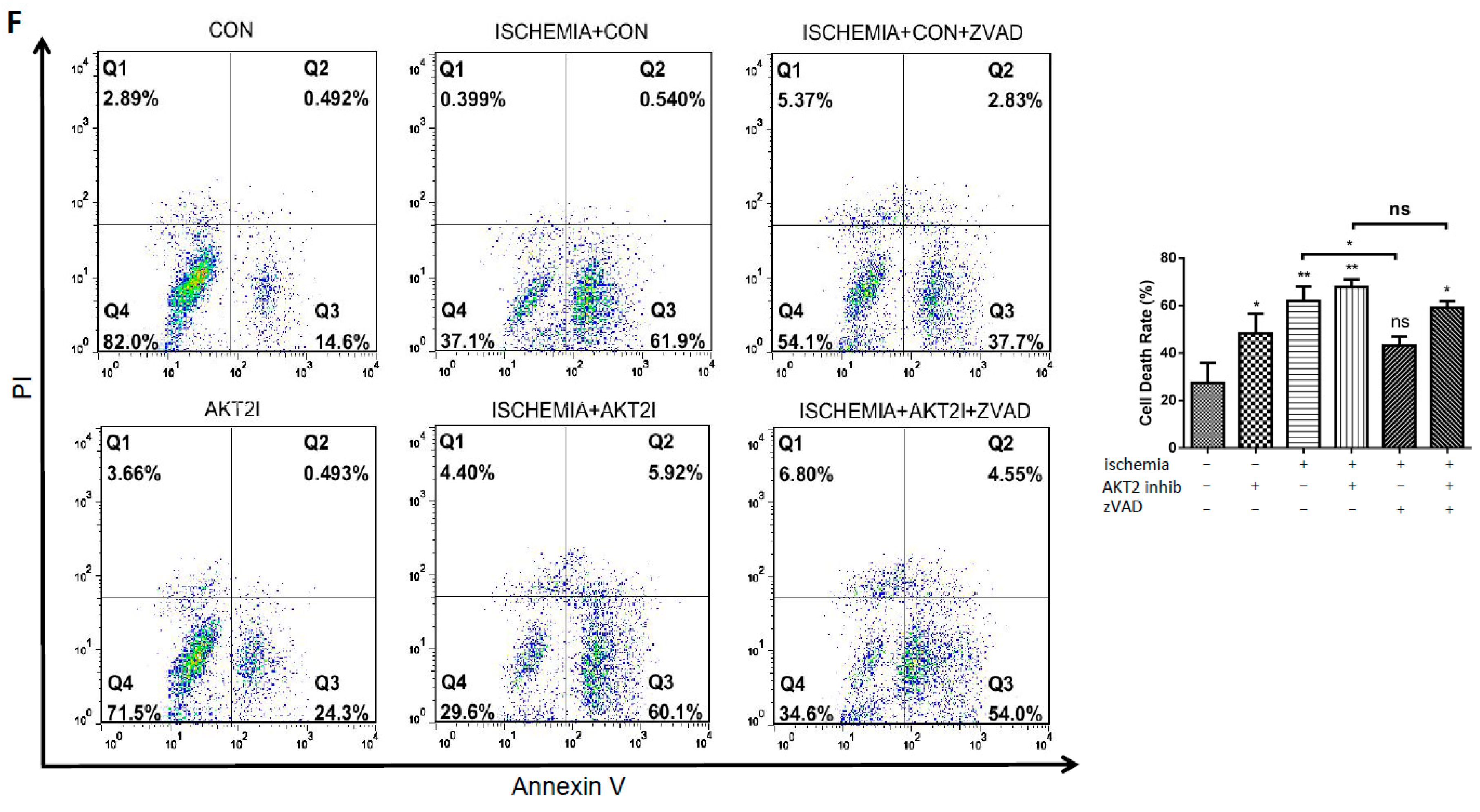
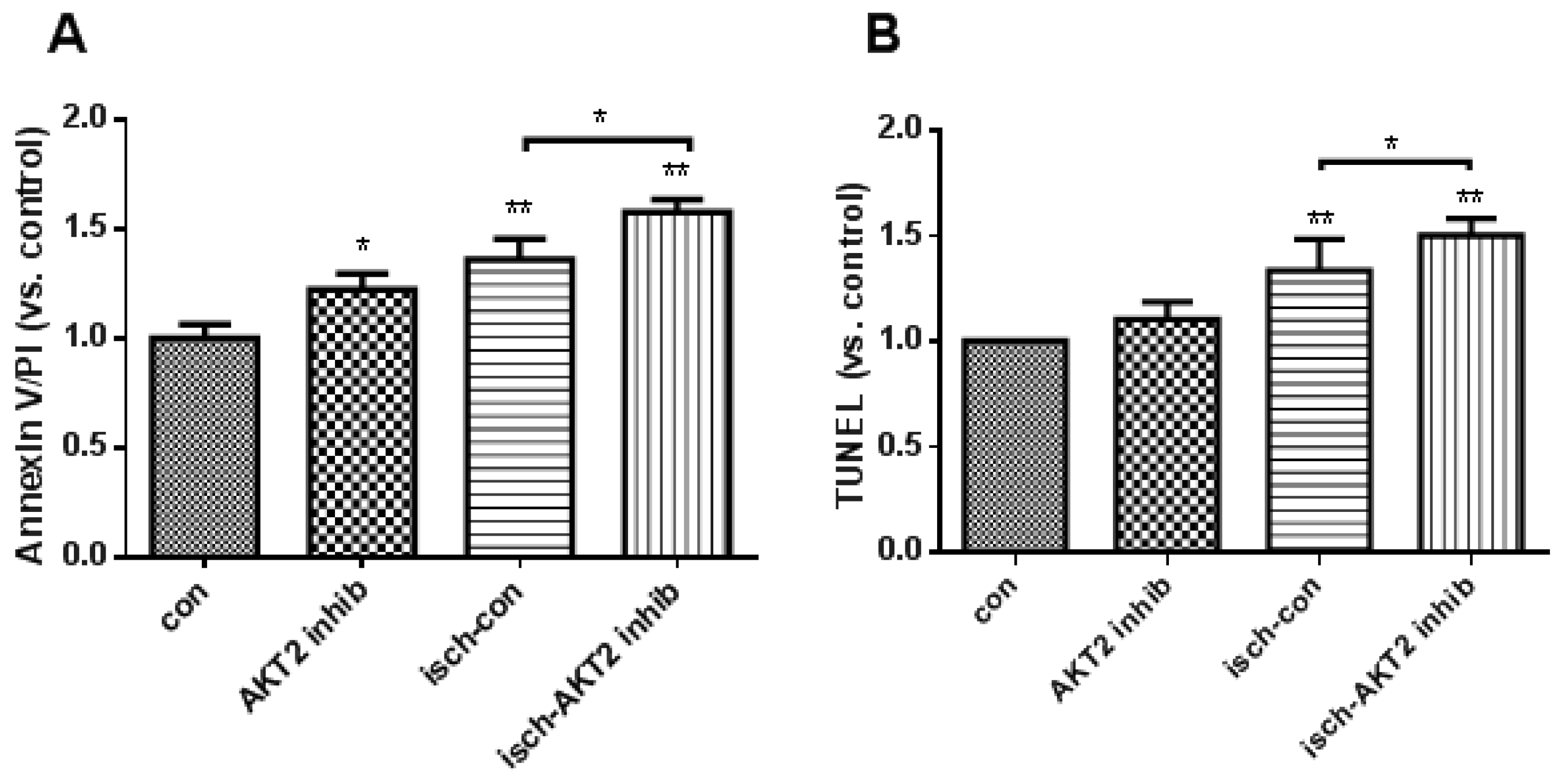
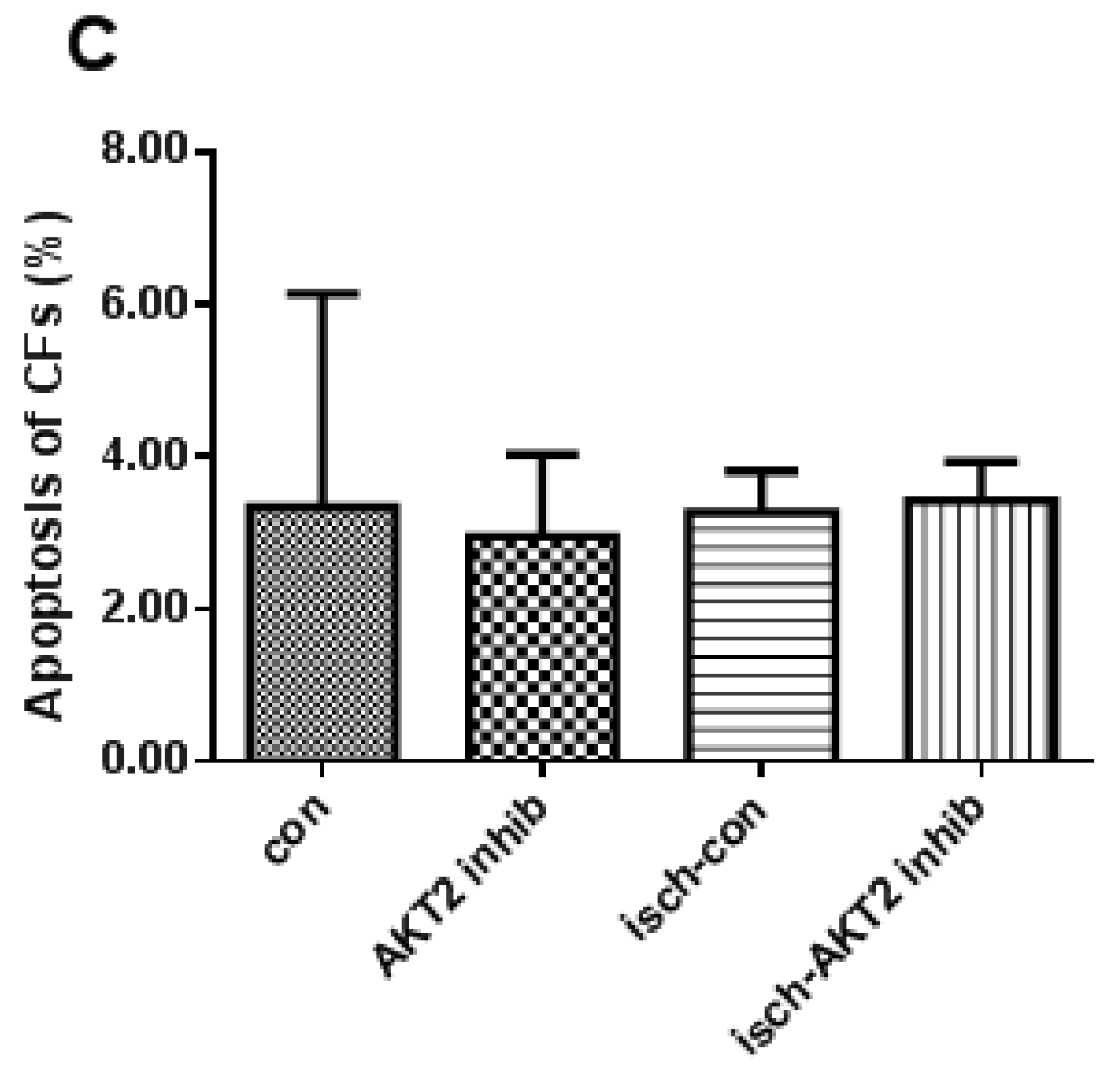
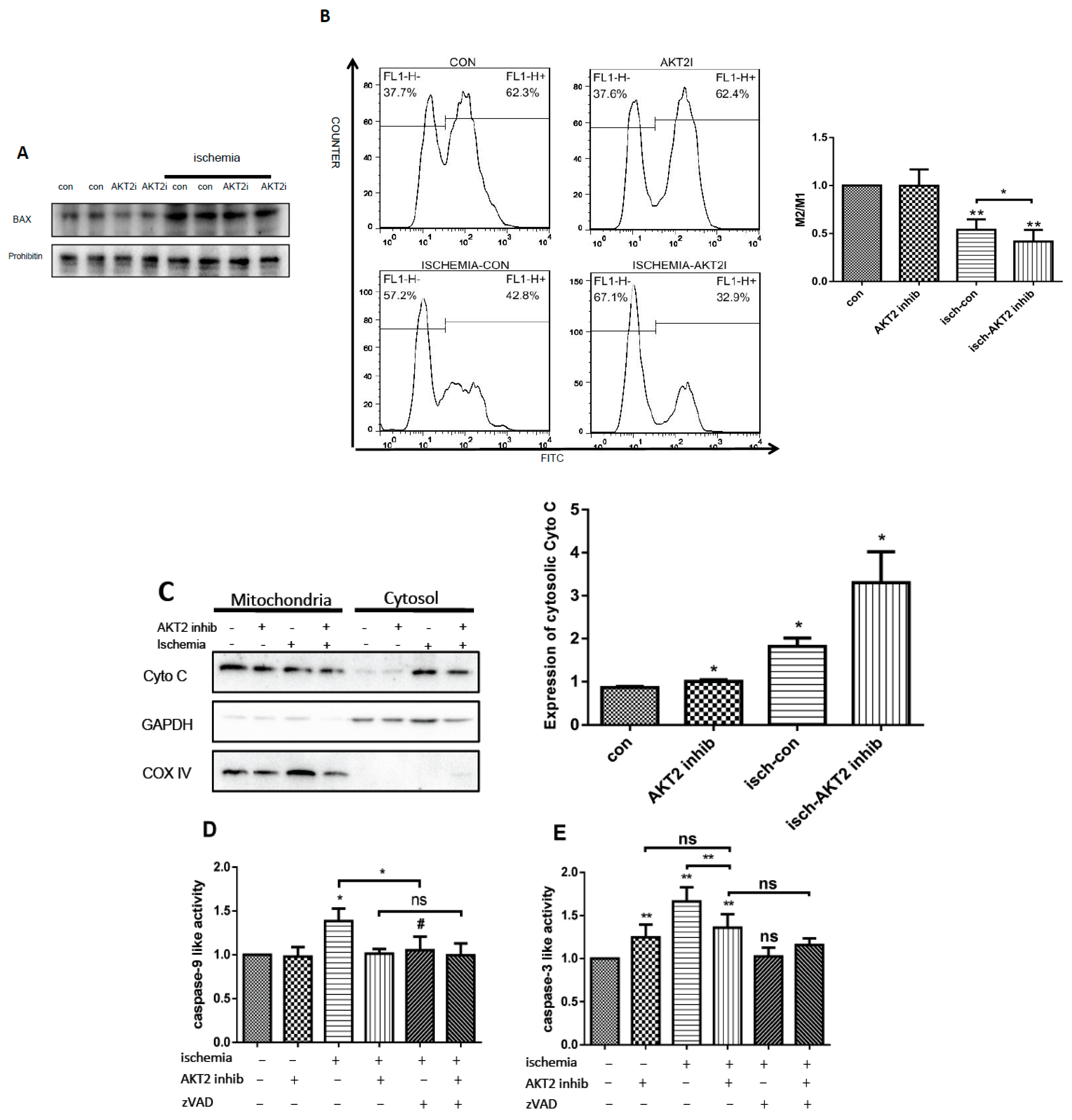
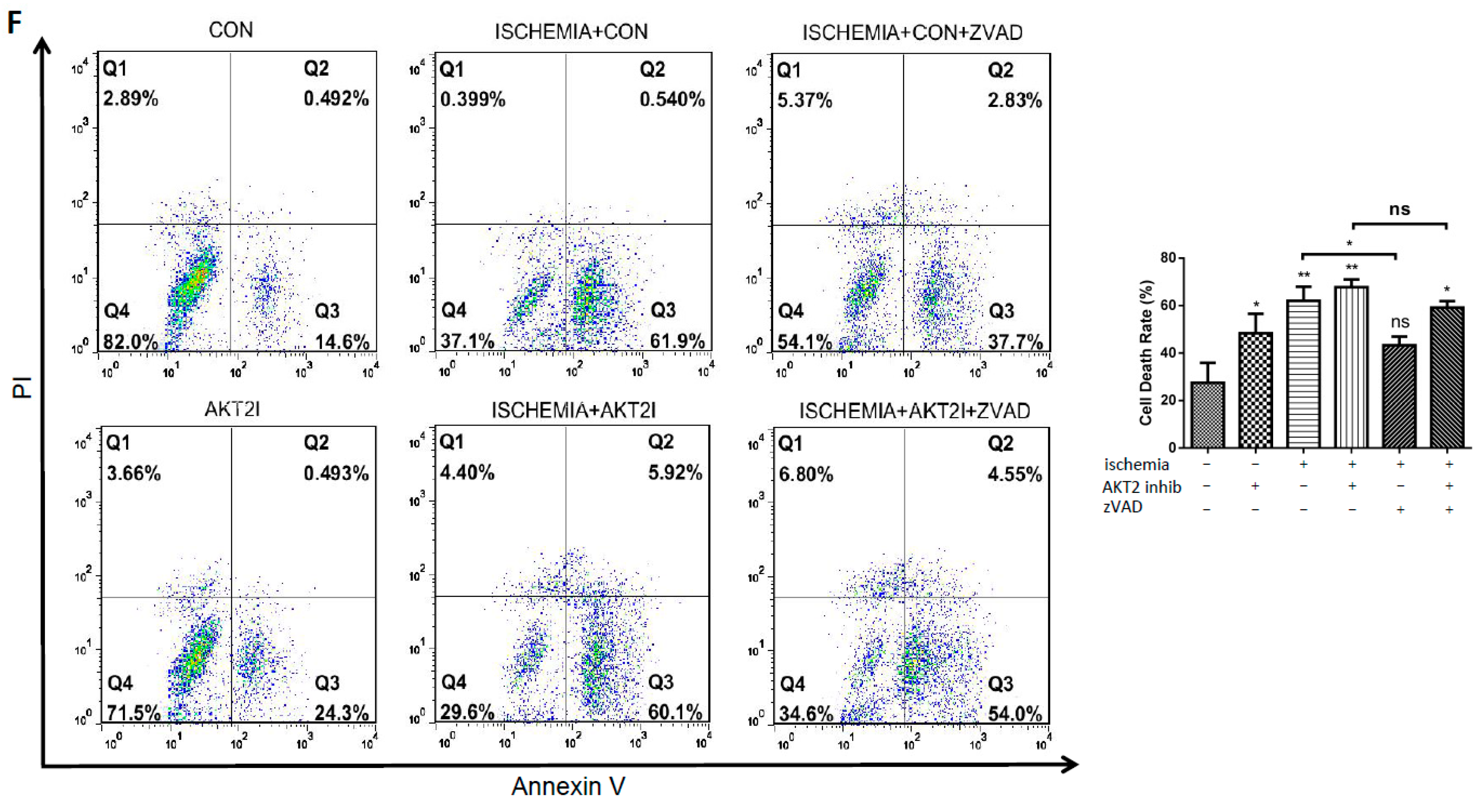
2.2. Blockage of AKT2 Facilitates Apoptosis in Cardiomyocytes
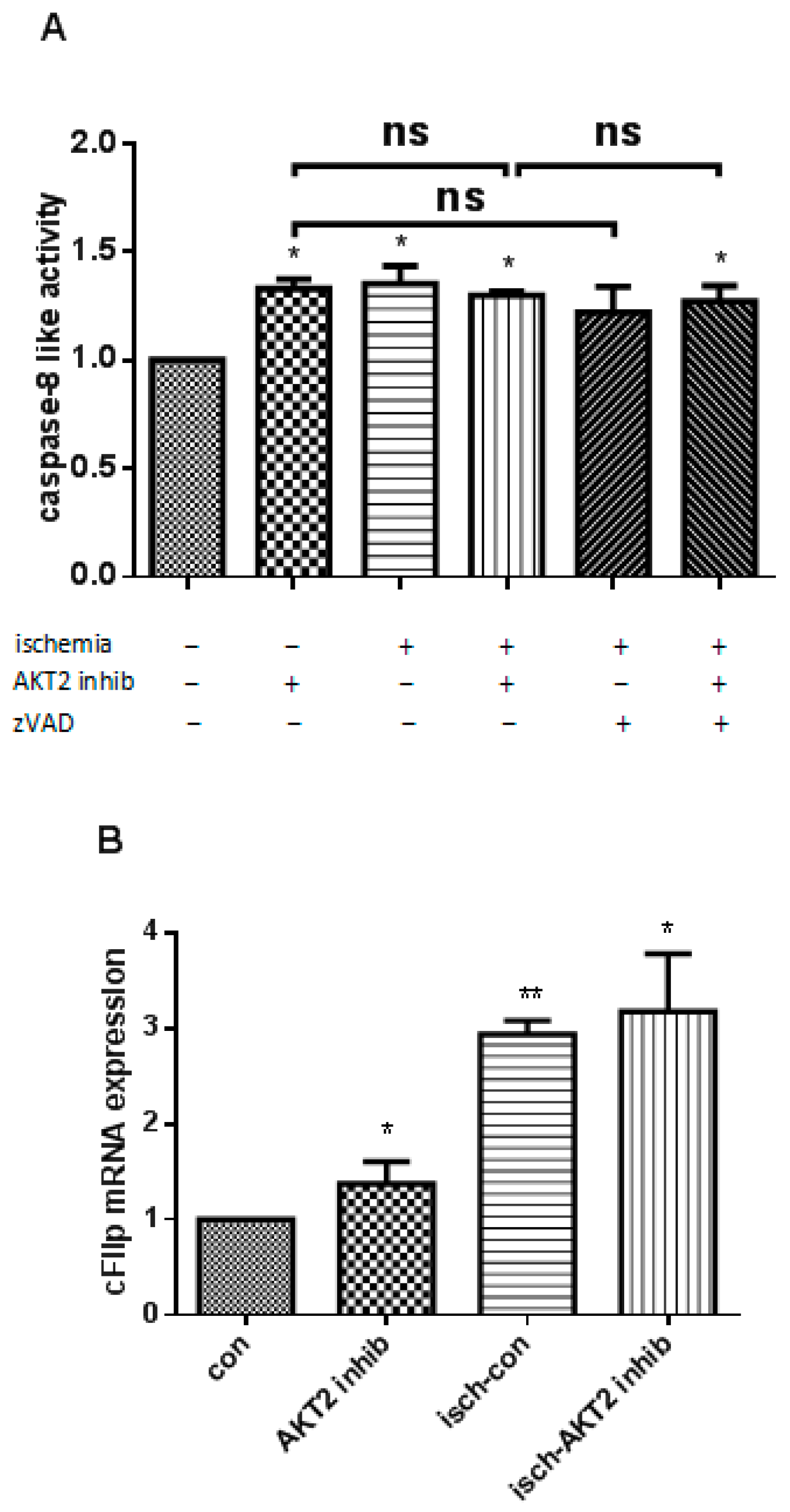
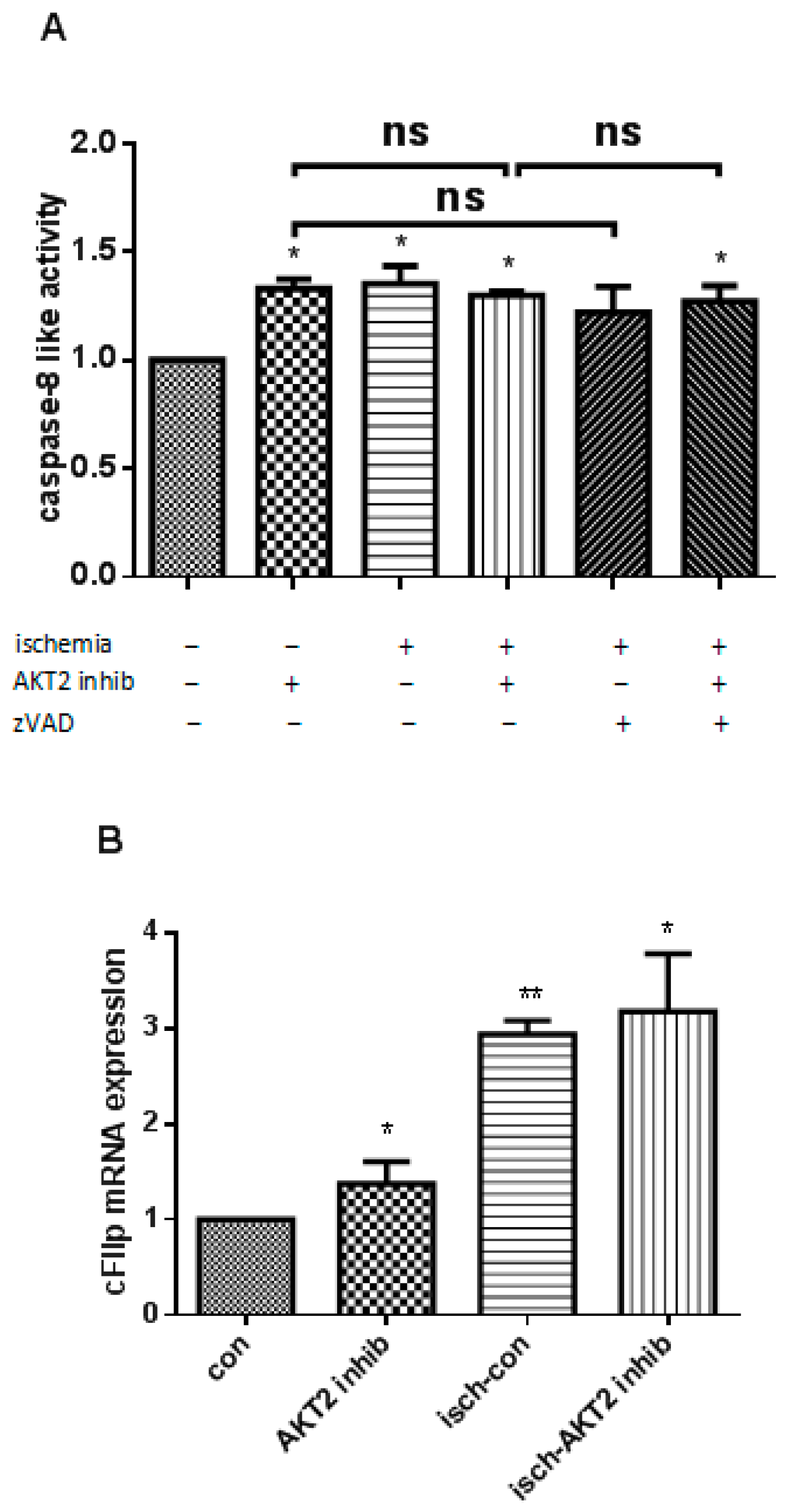
2.3. AKT2 Blockage Diminishes the Activation of Extrinsic Apoptotic Signaling Pathway
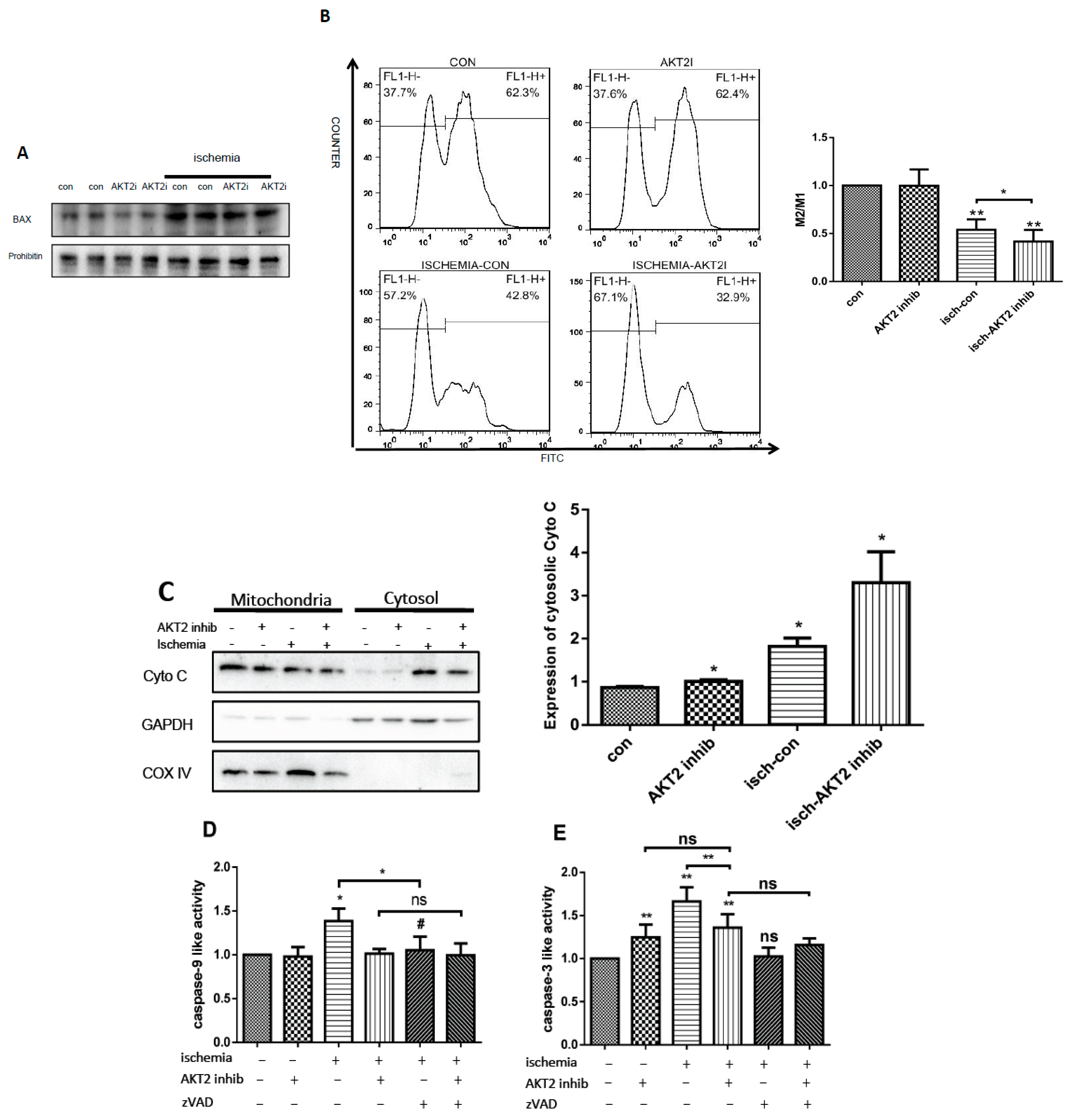
2.4. AKT2 Inhibition Promotes Mitochondrial Membrane Injury Independent of Intrinsic Apoptotic Pathway
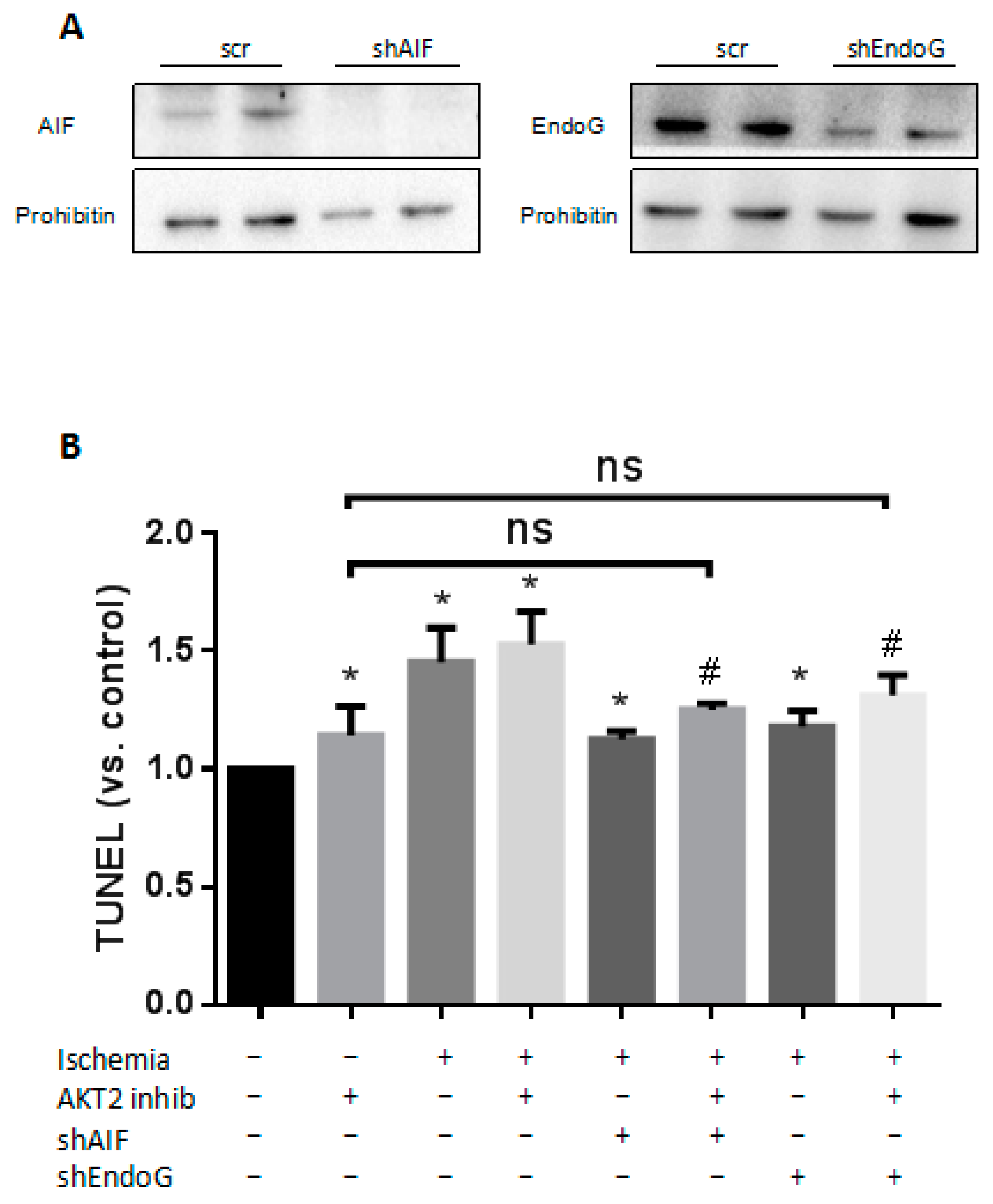
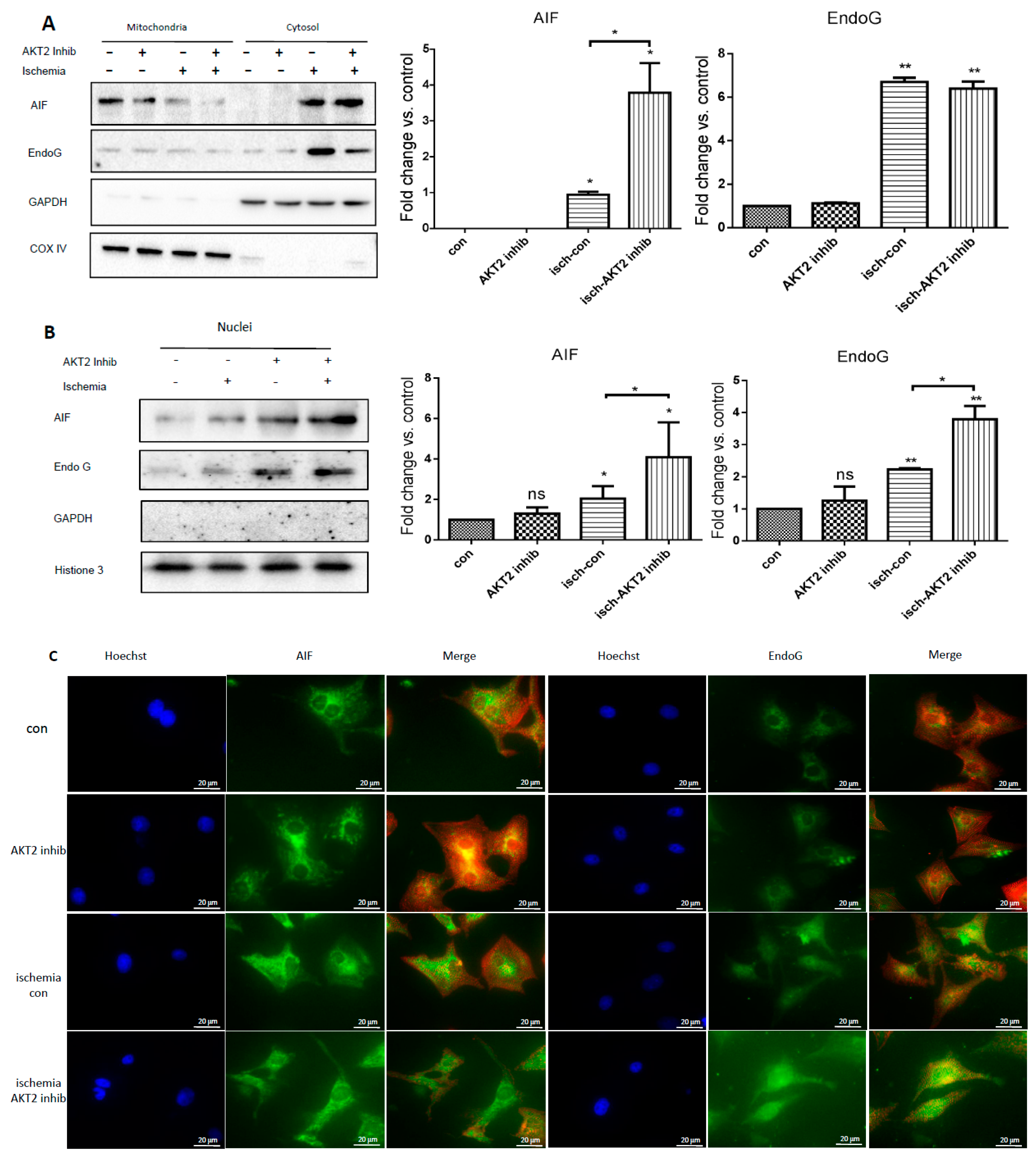
2.5. Cardiac AKT2 Inhibition Facilitates EndoG and AIF Cytosolic and Nucleus Translocation from Mitochondria
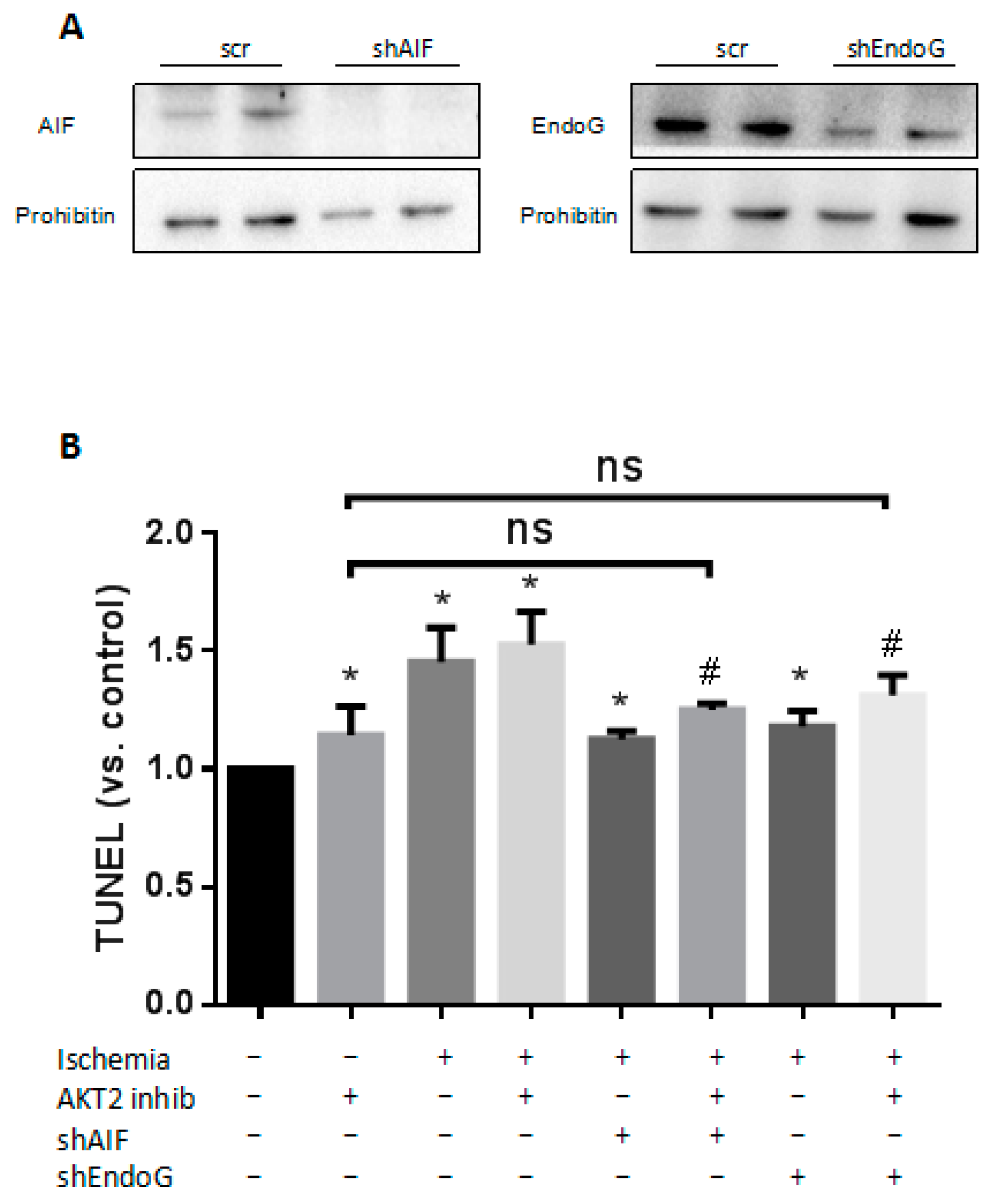
2.6. Both EndoG and AIF Are Responsible for Caspase-Independent Cell Death When AKT2 is Blocked
3. Discussion
4. Materials and Methods
4.1. Cardiomyocyte Culture, Cardiac Fibroblast Culture and Tissue Samples
4.2. Experimental Ischemia
4.3. Histology
4.4. shRNA Knockdown of AKT2, Endo G and AIF
4.5. Chemicals
4.6. RNA Extraction and Real Time Quantitative RT-PCR
4.7. Western Blot and Immunofluorescence
4.8. Measuremennt of MOMP
4.9. Measurement of Apoptosis
4.10. TUNEL Assay
4.11. Enzymatic Caspase Activity Assay
4.12. Mitochondria, Nuclei and Cytosol Extraction
4.13. Statistics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wright, K.M.; Linhoff, M.W.; Potts, P.R.; Deshmukh, M. Decreased apoptosome activity with neuronal differentiation sets the threshold for strict IAP regulation of apoptosis. J. Cell Biol. 2004, 167, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Cryns, V.; Yuan, J. Proteases to die for. Genes Dev. 1998, 12, 1551–1570. [Google Scholar] [CrossRef] [PubMed]
- Regula, K.M.; Kirshenbaum, L.A. Apoptosis of ventricular myocytes: A means to an end. J. Mol. Cell. Cardiol. 2005, 38, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Cain, K.; Bratton, S.B.; Cohen, G.M. The Apaf-1 apoptosome: A large caspase-activating complex. Biochimie 2002, 84, 203–214. [Google Scholar] [CrossRef]
- Lopez-Neblina, F.; Toledo, A.H.; Toledo-Pereyra, L.H. Molecular biology of apoptosis in ischemia and reperfusion. J. Investig. Surg. 2005, 18, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309. [Google Scholar] [CrossRef] [PubMed]
- David, K.K.; Sasaki, M.; Yu, S.W.; Dawson, T.M.; Dawson, V.L. EndoG is dispensable in embryogenesis and apoptosis. Cell. Death Differ. 2006, 13, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Daugas, E.; Susin, S.A.; Zamzami, N.; Ferri, K.F.; Irinopoulou, T.; Larochette, N.; Prévost, M.C.; Leber, B.; Andrews, D.; Penninger, J.; et al. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 2000, 14, 729–739. [Google Scholar] [PubMed]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [PubMed]
- Susin, S.A.; Daugas, E.; Ravagnan, L.; Samejima, K.; Zamzami, N.; Loeffler, M.; Costantini, P.; Ferri, K.F.; Irinopoulou, T.; Prévost, M.C.; et al. Two distinct pathways leading to nuclear apoptosis. J. Exp. Med. 2000, 192, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Bahi, N.; Zhang, J.; Llovera, M.; Ballester, M.; Comella, J.X.; Sanchis, D. Switch from caspase-dependent to caspase-independent death during heart development essential role of endonuclease G in ischemia-induced DNA processing of differentiated cardiomyocytes. J. Biol. Chem. 2006, 281, 22943–22952. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Brunet, A.; Greenberg, M.E. Cellular survival: A play in three Akts. Genes Dev. 1999, 13, 2905–2927. [Google Scholar] [CrossRef] [PubMed]
- DeBosch, B.; Sambandam, N.; Weinheimer, C.; Courtois, M.; Muslin, A.J. Akt2 regulates cardiac metabolism and cardiomyocyte survival. J. Biol. Chem. 2006, 281, 32841–32851. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Rosenzweig, A. Convergent signal transduction pathways controlling cardiomyocyte survival and function: The role of PI 3-kinase and Akt. J. Mol. Cell. Cardiol. 2005, 38, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Muslin, A.J. Akt2: A critical regulator of cardiomyocyte survival and metabolism. Pediatr. Cardiol. 2011, 32, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Etzion, S.; Etzion, Y.; DeBosch, B.; Crawford, P.A.; Muslin, A.J. Akt2 deficiency promotes cardiac induction of Rab4a and myocardial β-adrenergic hypersensitivity. J. Mol. Cell. Cardiol. 2010, 49, 931–940. [Google Scholar] [CrossRef] [PubMed]
- Linares-Palomino, J.; Husainy, M.A.; Lai, V.K.; Dickenson, J.M.; Galiñanes, M. Selective blockade of protein kinase B protects the rat and human myocardium against ischaemic injury. J. Physiol. 2010, 588, 2173–2191. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Deng, W.; Chen, Y.; Fan, W.; Baldwin, K.M.; Jope, R.S.; Wallace, D.C.; Wang, P.H. Impaired translocation and activation of mitochondrial Akt1 mitigated mitochondrial oxidative phosphorylation Complex V activity in diabetic myocardium. J. Mol. Cell. Cardiol. 2013, 59, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Mayorga, M.; Bahi, N.; Ballester, M.; Comella, J.X.; Sanchis, D. Bcl-2 is a key factor for cardiac fibroblast resistance to programmed cell death. J. Biol. Chem. 2004, 279, 34882–34889. [Google Scholar] [CrossRef] [PubMed]
- Yeh, W.C.; Itie, A.; Elia, A.J.; Ng, M.; Shu, H.B.; Wakeham, A.; Mirtsos, C.; Suzuki, N.; Bonnard, M.; Goeddel, D.V.; Mak, T.W. Requirement for Casper (c-FLIP) in regulation of death receptor–induced apoptosis and embryonic development. Immunity 2000, 12, 633–642. [Google Scholar] [CrossRef]
- Chin, Y.R.; Yuan, X.; Balk, S.P.; Toker, A. PTEN-deficient tumors depend on AKT2 for maintenance and survival. Cancer Discov. 2014, 4, 942–955. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.Q.; Feldman, R.I.; Sussman, G.E.; Coppola, D.; Nicosia, S.V.; Cheng, J.Q. Akt2 Inhibition of Cisplatin-induced JNK/p38 and Bax Activation by Phosphorylation of ASK1 Implication of AKT2 in Chemoresistance. J. Biol. Chem. 2003, 278, 23432–23440. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, N.J.; Whyte, M.K.; Gilbert, C.S.; Evan, G.I. Inhibition of Ced-3/ICE-related proteases does not prevent cell death induced by oncogenes, DNA damage, or the Bcl-2 homologue Bak. J. Cell Biol. 1997, 136, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Chao, D.T.; Korsmeyer, S.J. BAX-induced cell death may not require interleukin 1 beta-converting enzyme-like proteases. Proc. Natl. Acad. Sci. USA 1996, 93, 14559–14563. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Gaume, B.; Karbowski, M.; Sharpe, J.C.; Cecconi, F.; Youle, R.J. Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak-mediated permeabilization. EMBO J. 2003, 22, 4385–4399. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Parone, P.; Martinou, J.C.; Antonsson, B.; Estaquier, J.; Ameisen, J.C. Mitochondrial release of apoptosis-inducing factor occurs downstream of cytochrome c release in response to several proapoptotic stimuli. J. Cell Biol. 2002, 159, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.Y.; Ribecco, M.; Pandey, S.; Walker, P.R.; Sikorska, M. Apoptosis-related Functional Features of the DNaseI-like Family of Nucleases. Ann. N. Y. Acad. Sci. 1999, 887, 60–76. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.B.; Toledo-Pereyra, L.H. Caspase-independent programmed cell death following ischemic stroke. J. Investig. Surg. 2008, 21, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Cregan, S.P.; Fortin, A.; MacLaurin, J.G.; Callaghan, S.M.; Cecconi, F.; Yu, S.W.; Dawson, T.M.; Dawson, V.L.; Park, D.S.; Kroemer, G.; et al. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J. Cell Biol. 2002, 158, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sun, S.; Zhou, J.; Liu, J.; Lv, J.H.; Yu, X.Q.; Li, C.; Gong, L.; Yan, Q.; Deng, M.; et al. Knockdown of Akt1 promotes Akt2 upregulation and resistance to oxidative-stress-induced apoptosis through control of multiple signaling pathways. Antioxid. Redox Signal. 2011, 15, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Mikhalkova, D.; Gao, E.; Zhang, J.; Myers, V.; Zincarelli, C.; Lei, Y.; Song, J.; Koch, W.J.; Peppel, K.; et al. Myocardial injury after ischemia-reperfusion in mice deficient in Akt2 is associated with increased cardiac macrophage density. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1932–H1940. [Google Scholar] [CrossRef] [PubMed]
- Shiojima, I.; Walsh, K. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev. 2006, 20, 3347–3365. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Chua, C.C.; Ho, Y.S.; Hamdy, R.C.; Chua, B.H. Overexpression of Bcl-2 attenuates apoptosis and protects against myocardial I/R injury in transgenic mice. Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H2313–H2320. [Google Scholar] [PubMed]
- Huss, J.M.; Kelly, D.P. Mitochondrial energy metabolism in heart failure: A question of balance. J. Clin. Investig. 2005, 115, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Zamzami, N.; Susin, S.A.; Marchetti, P.; Hirsch, T.; Gómez-Monterrey, I.; Castedo, M.; Kroemer, G. Mitochondrial control of nuclear apoptosis. J. Exp. Med. 1996, 183, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Dallaporta, B.; Resche-Rigon, M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 1998, 60, 619–642. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.T.; Chiang, M.C.; Tasi, C.Y.; Kuo, C.H.; Shyu, W.C.; Kao, C.L.; Huang, C.Y.; Lee, S.D. Cardiac Fas-Dependent and Mitochondria-Dependent Apoptotic Pathways in a Transgenic Mouse Model of Huntington’s Disease. Cardiovasc. Toxicol. 2016, 16, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Eskes, R.; Antonsson, B.; Osen-Sand, A.; Montessuit, S.; Richter, C.; Sadoul, R.; Mazzei, G.; Nichols, A.; Martinou, J.C. Bax-induced cytochrome C release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+ ions. J. Cell Biol. 1998, 143, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Ohno, M.; Takemura, G.; Ohno, A.; Misao, J.; Hayakawa, Y.; Minatoguchi, S.; Fujiwara, T.; Fujiwara, H. “Apoptotic” Myocytes in Infarct Area in Rabbit Hearts May Be Oncotic Myocytes with DNA Fragmentation Analysis by Immunogold Electron Microscopy Combined With In Situ Nick End-Labeling. Circulation 1998, 98, 1422–1430. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Blömer, U.; Gage, F.H.; Trono, D.; Verma, I.M. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. USA 1996, 93, 11382–11388. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Oligonucleotides Sequences (5′→3′) |
|---|---|
| Endo G | gatccccGGAACAACCTTGAGAAGTAttcaagagaTACTTCTCAAGGTTGTTCCttttta |
| agcttaaaaaGGAACAACCTTGAGAAGTAtctcttgaaTACTTCTCAAGGTTGTTCCggg | |
| AIF | gatccccGCATGCTTCTATGATATAATTCAAGAGATTATATCATAGAAGCATGCttttta |
| agcttaaaaaGCATGCTTCTATGATATAATCTCTTGAATTATATCATAGAAGCATGCggg | |
| Scrambled | gatccccGAATGCTAAGATGTCTAATttcaagagaATTAGACATCTTAGCATTCttttta |
| agcttaaaaaGAATGCTAAGATGTCTAATtctcttgaaATTAGACATCTTAGCATTCggg |
| Antigen | Provider | Catalogue Number | Application |
|---|---|---|---|
| AKT2 | Cell Signaling | 3063 | WB |
| P-AKT2 | Cell Signaling | 8599 | WB |
| Bax | Cell Signaling | 2772 | WB |
| Cytochrome C | Cell Signaling | 4272 | WB |
| AIF | Cell Signaling | 5318 | WB |
| Endonuclease G | Abcam | Ab76122 | WB |
| Bcl-2 | Wanleibio | WL01556 | WB |
| Cleaved caspase-3 | Cell Signaling | 9661 | WB |
| Caspase-8 | Cell Signaling | 9746 | WB |
| COXIV | Cell Signaling | 4850 | WB |
| Histon 3 | Ruiying Biological | RLM3038 | WB |
| Prohibitin | NeoMarkers | 292P501F | WB |
| GAPDH | BBI Life Sciences | D110016-0200 | WB |
| α-actinin | SIGMA | A7811 | IF |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, S.; Zhao, X.; Xu, H.; Chen, F.; Xu, Y.; Li, Z.; Sanchis, D.; Jin, L.; Zhang, Y.; Ye, J. AKT2 Blocks Nucleus Translocation of Apoptosis-Inducing Factor (AIF) and Endonuclease G (EndoG) While Promoting Caspase Activation during Cardiac Ischemia. Int. J. Mol. Sci. 2017, 18, 565. https://doi.org/10.3390/ijms18030565
Yang S, Zhao X, Xu H, Chen F, Xu Y, Li Z, Sanchis D, Jin L, Zhang Y, Ye J. AKT2 Blocks Nucleus Translocation of Apoptosis-Inducing Factor (AIF) and Endonuclease G (EndoG) While Promoting Caspase Activation during Cardiac Ischemia. International Journal of Molecular Sciences. 2017; 18(3):565. https://doi.org/10.3390/ijms18030565
Chicago/Turabian StyleYang, Shuai, Xinmei Zhao, Hui Xu, Fan Chen, Yitao Xu, Zhe Li, Daniel Sanchis, Liang Jin, Yubin Zhang, and Junmei Ye. 2017. "AKT2 Blocks Nucleus Translocation of Apoptosis-Inducing Factor (AIF) and Endonuclease G (EndoG) While Promoting Caspase Activation during Cardiac Ischemia" International Journal of Molecular Sciences 18, no. 3: 565. https://doi.org/10.3390/ijms18030565