
Interleukin 17A Promotes Lymphocytes Adhesion and Induces CCL2 and CXCL1 Release from Brain Endothelial Cells


Abstract
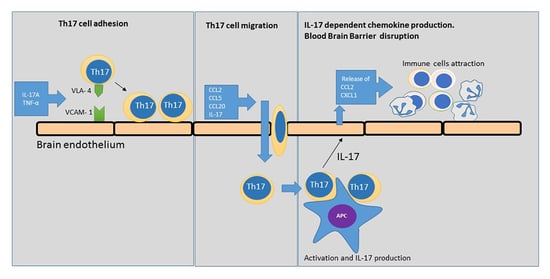
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Th17 Cell Adherence to Brain Endothelium
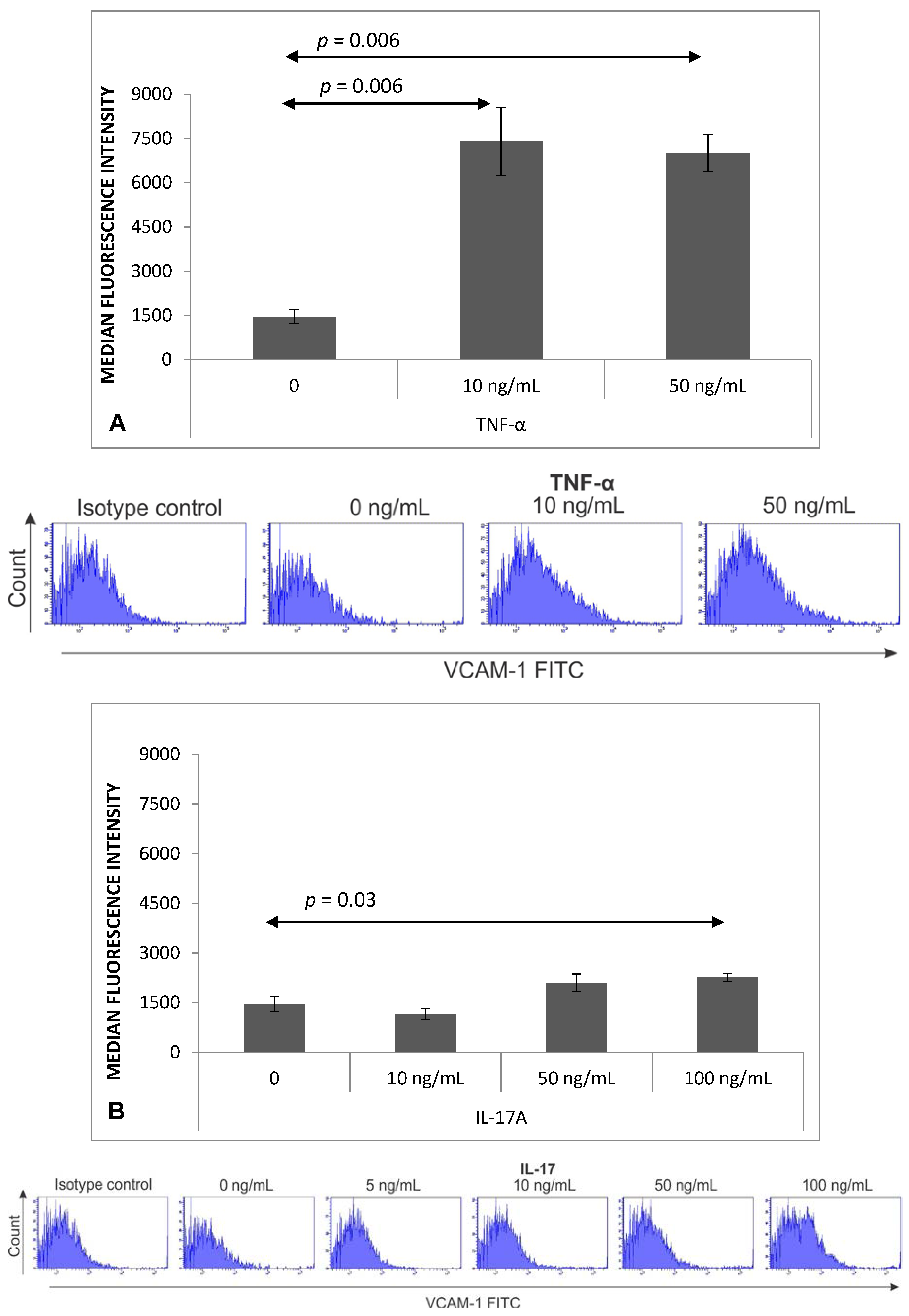
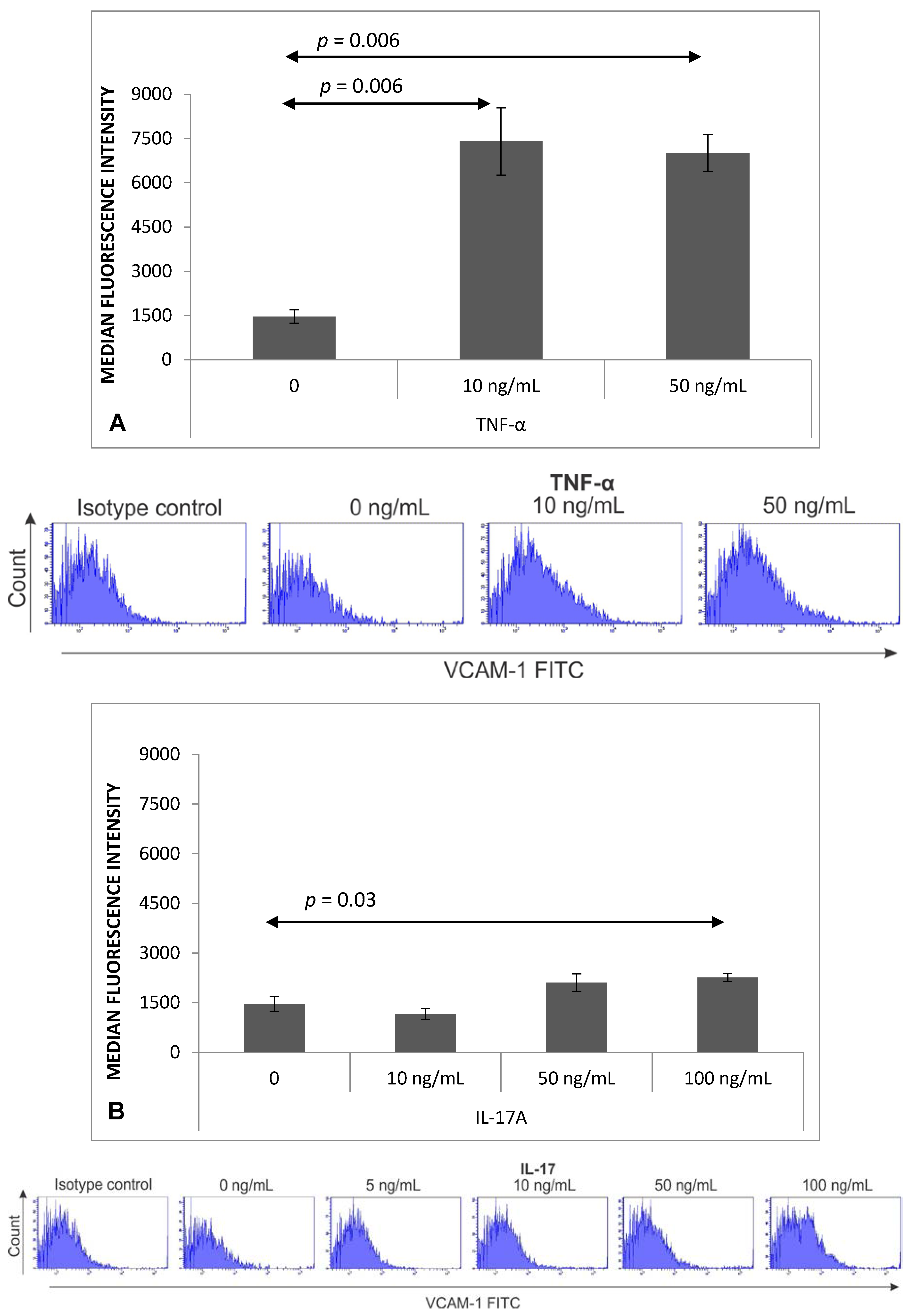
2.2. Expression of VCAM-1 Receptor on Brain Endothelium
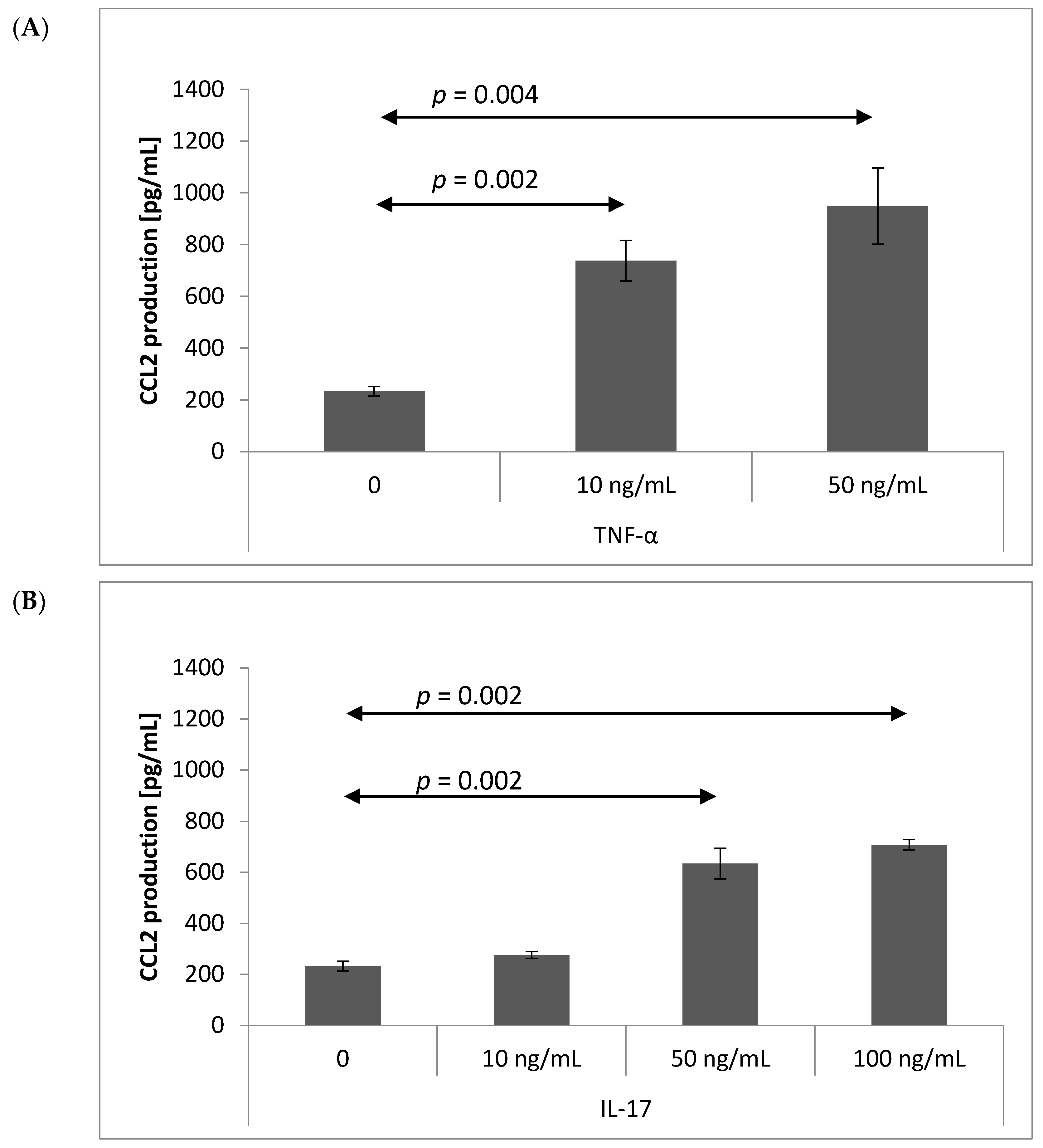
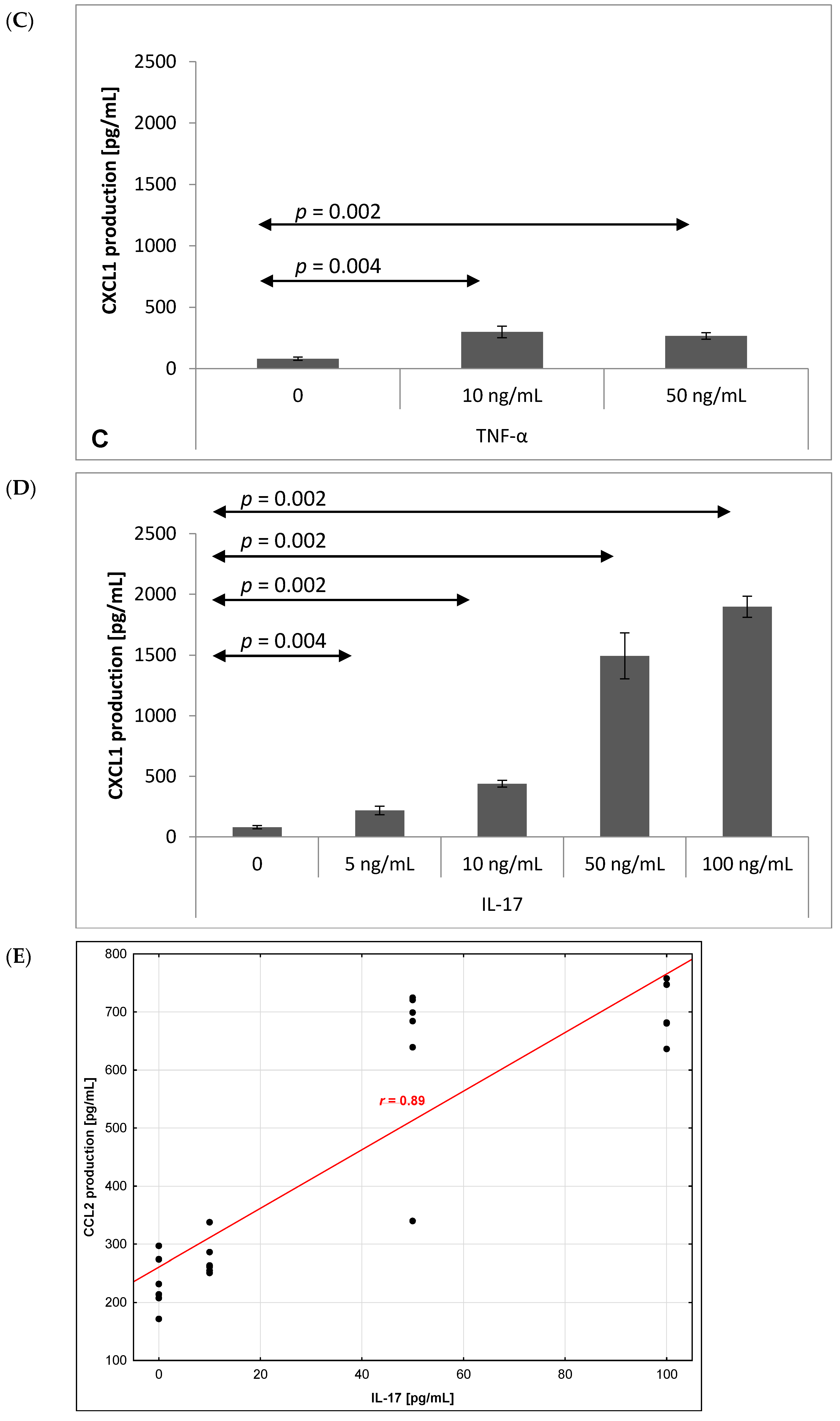
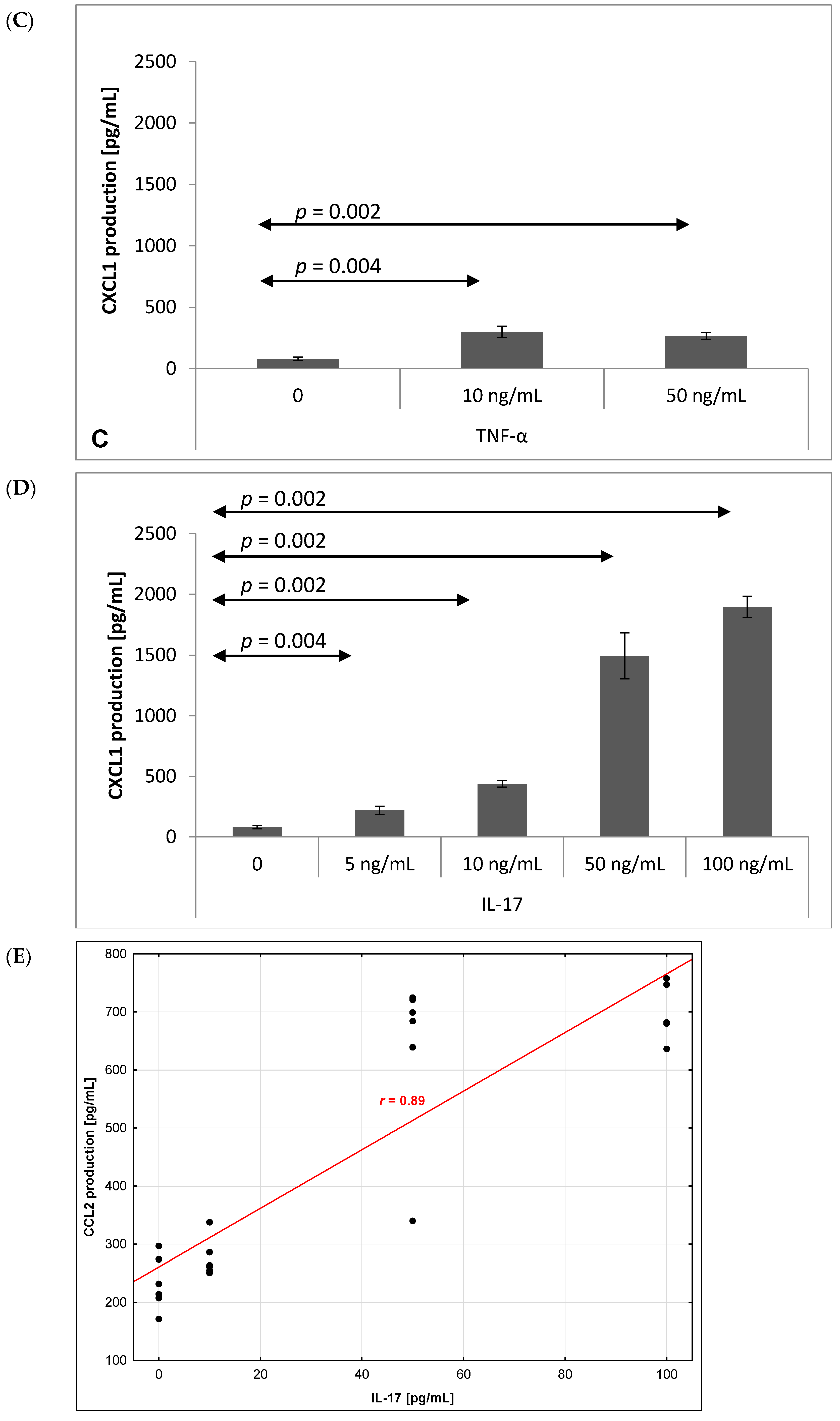
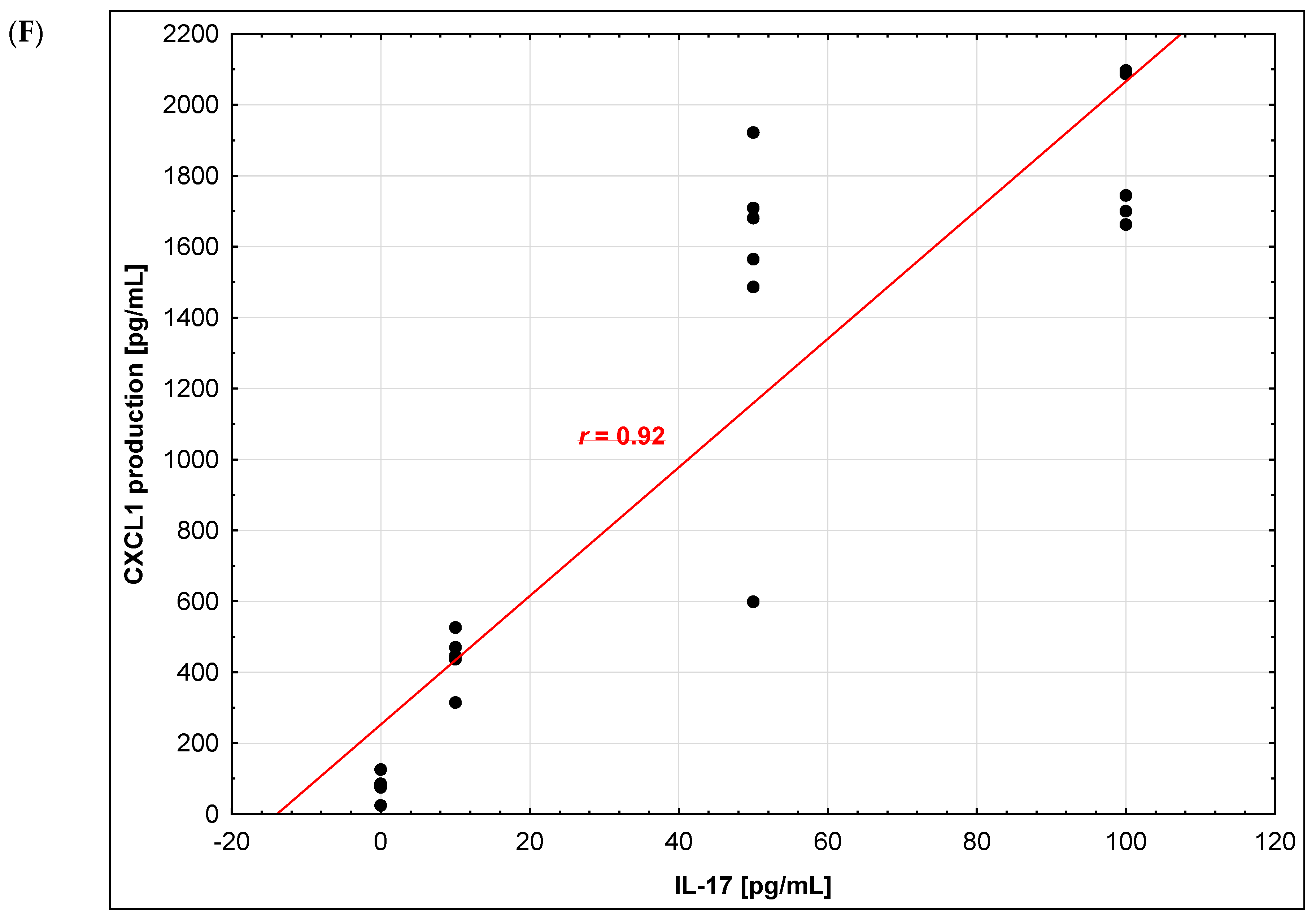
2.3. Chemokine Production by Brain Endothelium
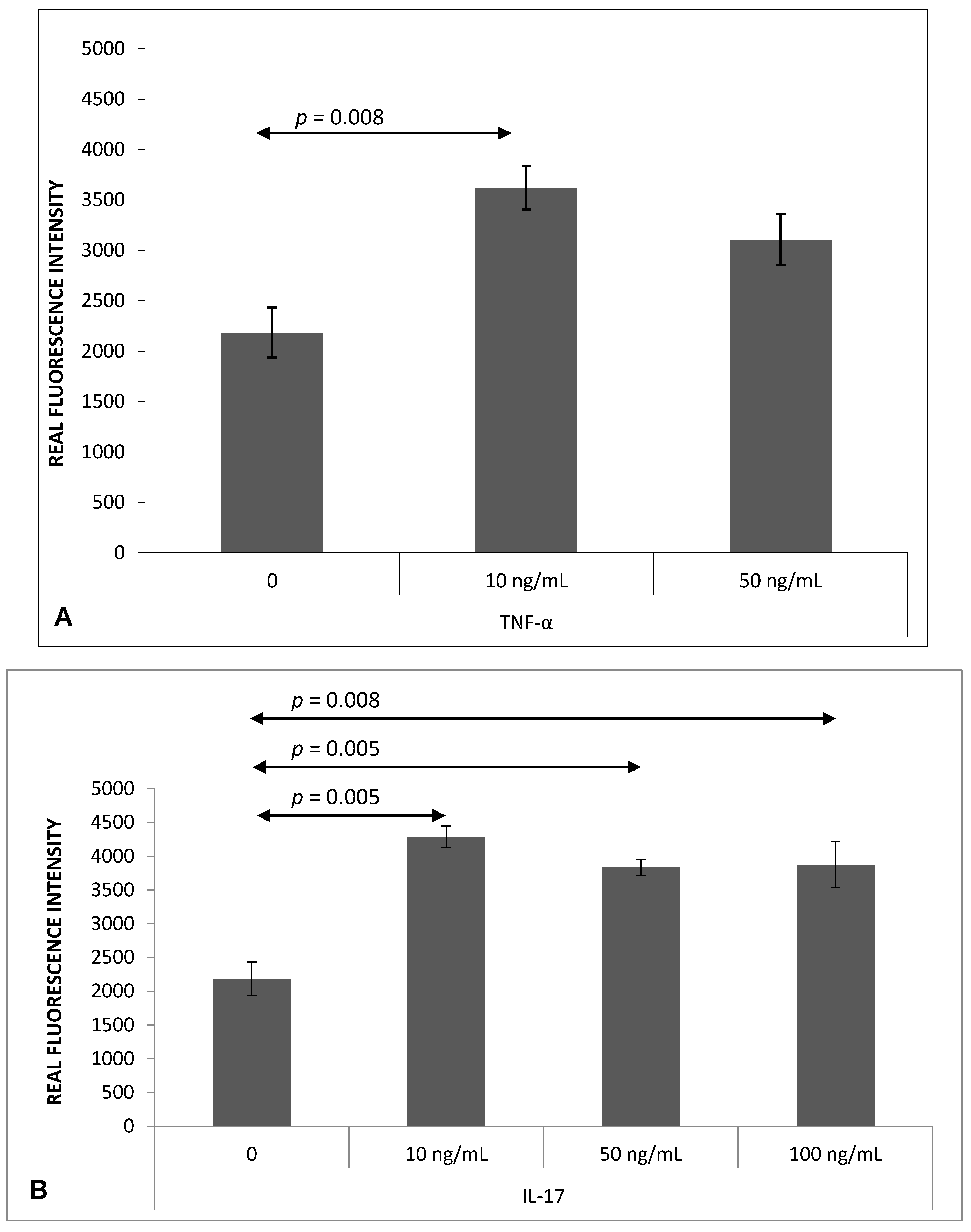
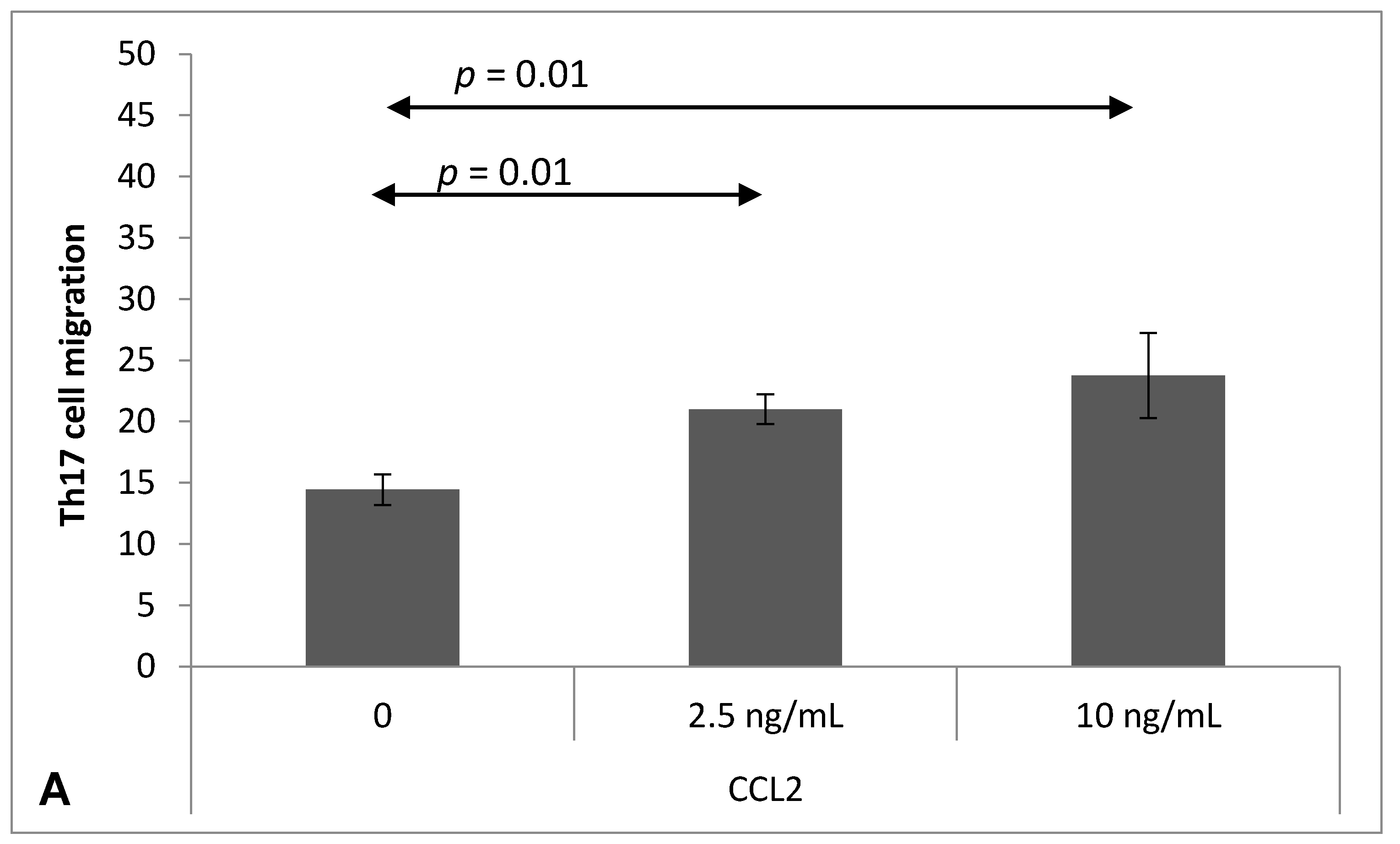
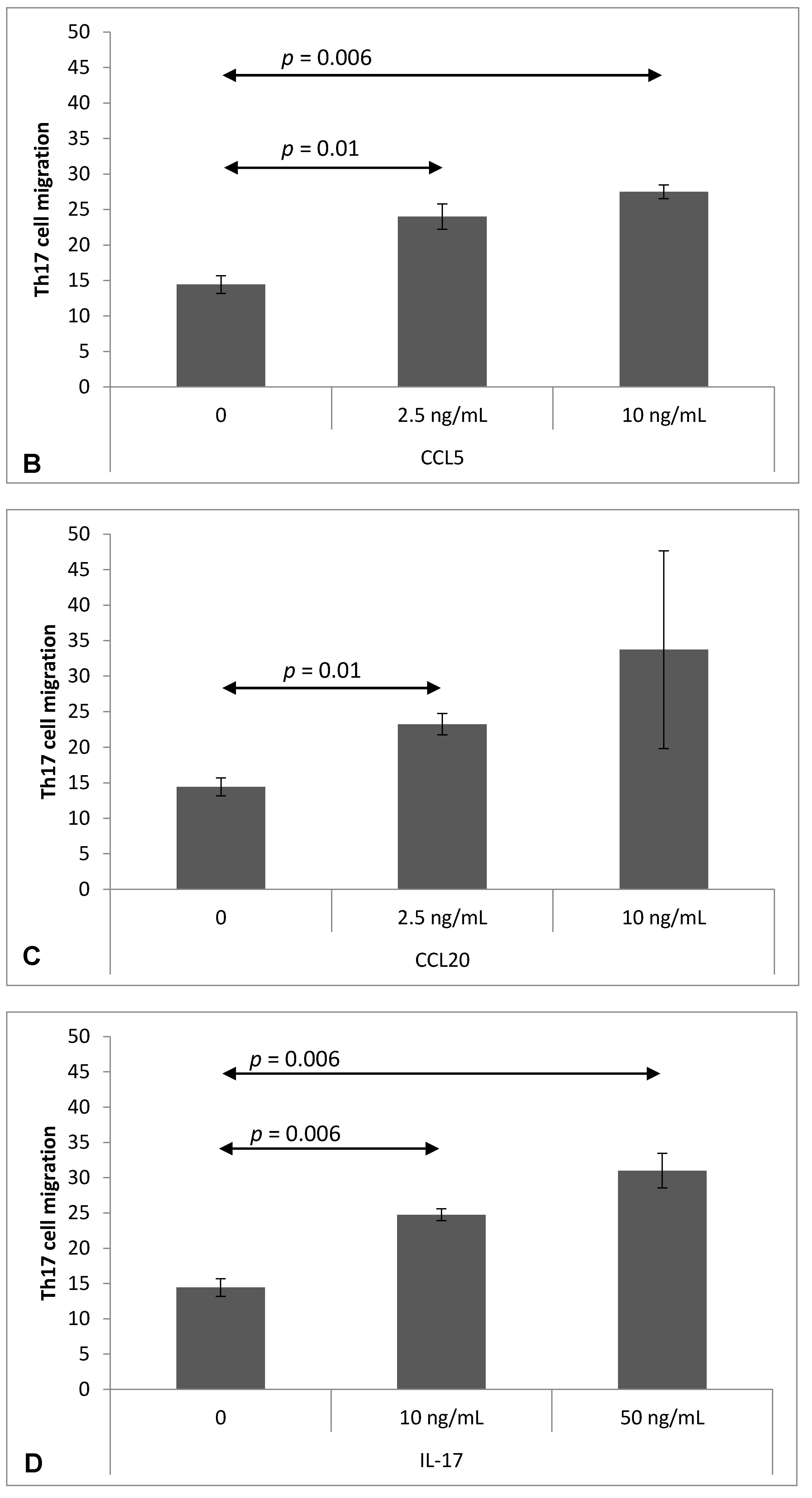
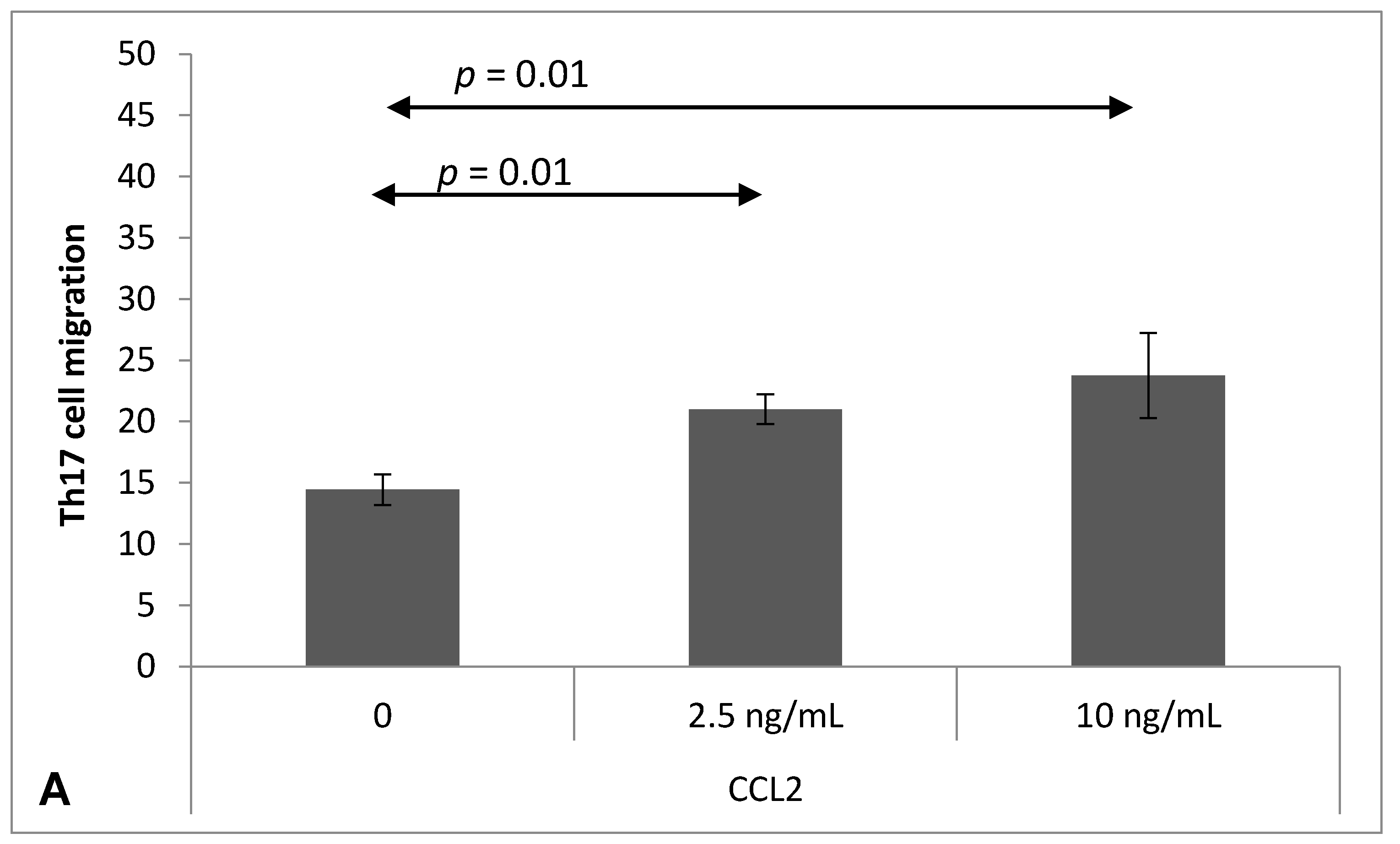
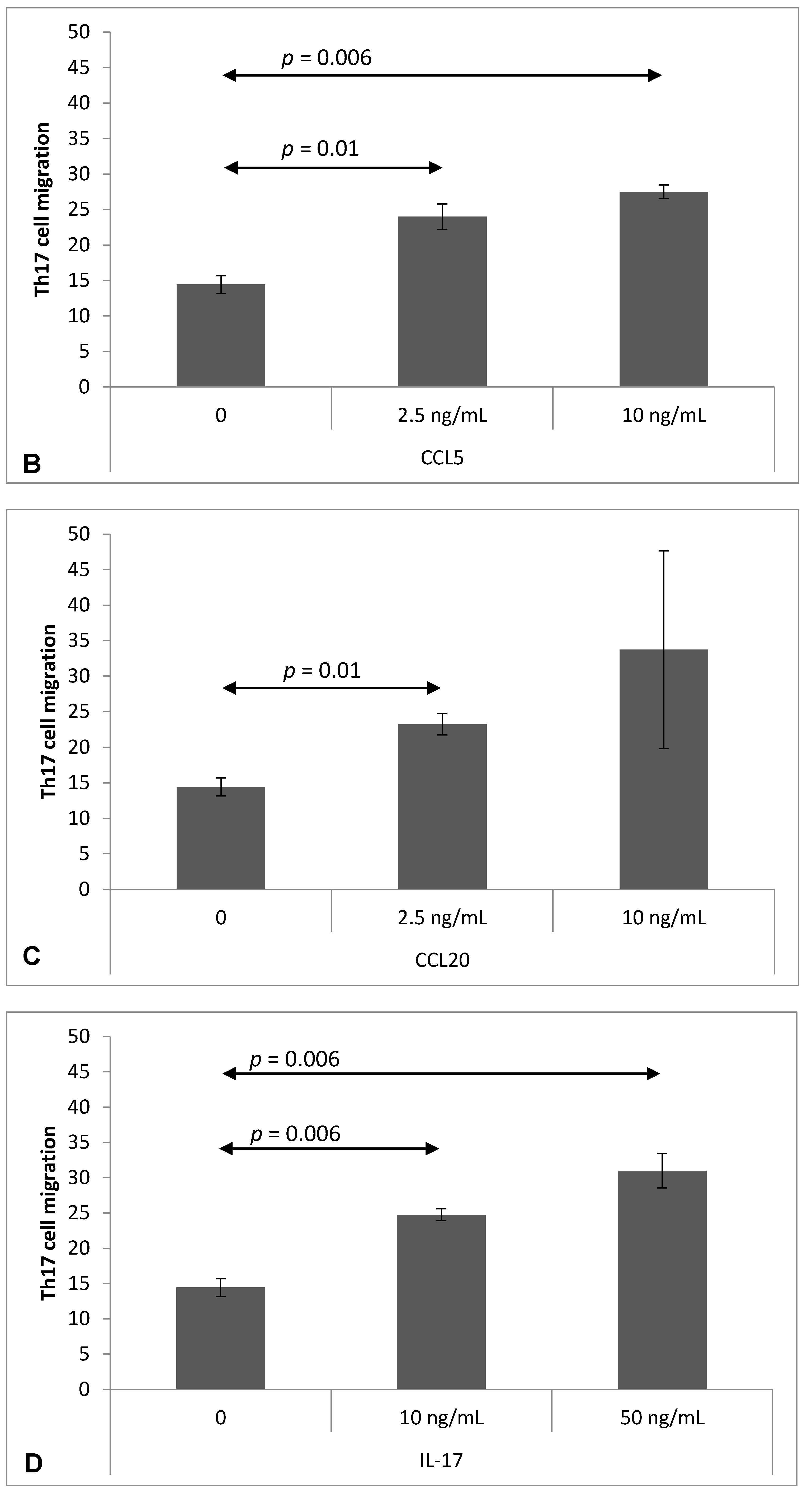
2.4. Chemokine-Induced Transmigration of Th17 Cells through the Brain Endothelium
3. Discussion
4. Materials and Methods
4.1. Mice and Tissue Collection
4.2. Isolation of CD4+ T Cells and Th17 Cell Culture
4.3. Mouse Brain Endothelial Cell (MBEC) Culture
4.4. Analysis of Th17 Cell Adherence to MBEC
4.5. Measurement of Chemokine Level by ELISA
4.6. Flow Cytometry Analysis of VCAM-1 Expression on MBEC
4.7. Analysis of Th17 Cell Migration through the Brain Endothelium
4.8. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gay, F.W.; Drye, T.J.; Dick, G.W.; Esiri, M.M. The application of multifactorial cluster analysis in the staging of plaques in early multiple sclerosis. Identification and characterization of the primary demyelinating lesion. Brain 1997, 120, 1461–1483. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.Q.; Wittmer, S.; Dalton, D.K. Failure to Suppress the Expansion of the Activated CD4 T cell population in interferon γ–deficient mice leads to exacerbation of experimental autoimmune encephalomyelitis. J. Exp. Med. 2000, 192, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Noseworthy, J.H.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G. Multiple sclerosis. N. Engl. J. Med. 2000, 343, 938–952. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Painter, S.L.; Fanslow, W.C.; Ulrich, D.; Macduff, B.M.; Spriggs, M.K.; Armitage, R.J. Human IL-17: A novel cytokine derived from T cells. J. Immunol. 1995, 155, 5483–5486. [Google Scholar] [PubMed]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; Dong, C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.; Closhen, D.; Croxford, A.; White, R.; Kulig, P.; Pietrowski, E.; Bechmann, I.; Becher, B.; Luhmann, H.J.; Waisman, A.; et al. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. 2010, 24, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Kebir, H.; Kreymborg, K.; Ifergan, I.; Dodelet-Devillers, A.; Cayrol, R.; Bernard, M.; Giuliani, F.; Arbour, N.; Becher, B.; Prat, A. Human TH17 lymphocytes promote blood–brain barrier disruption and central nervous system inflammation. Nat. Med. 2007, 13, 1173–1175. [Google Scholar] [CrossRef] [PubMed]
- Ballabh, P.; Braun, A.; Nedergaard, M. The blood–brain barrier: An overview: Structure, regulation, and clinical implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Vestweber, D.; Hallmann, R.; Schulz, M. E- and P-selectin are not involved in the recruitment of inflammatory cells across the blood–brain barrier in experimental autoimmune encephalomyelitis. Blood 1997, 90, 4459–4472. [Google Scholar] [PubMed]
- Sikorski, E.E.; Hallmann, R.; Berg, E.L.; Butcher, E.C. The Peyer’s patch high endothelial receptor for lymphocytes, the mucosal vascular addressin, is induced on a murine endothelial cell line by tumor necrosis factor-α and IL-1. J. Immunol. 1993, 151, 5239–5250. [Google Scholar] [PubMed]
- Alvarez, J.I.; Cayrol, R.; Prat, A. Disruption of central nervous system barriers in multiple sclerosis. Biochim. Biophys. Acta 2010, 1812, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, C.; Agardh, C.-D.; Zetterqvist, A.V.; Nilsson, J.; Agardh, E.; Gomez, M.F. Vascular cellular adhesion molecule-1 (VCAM-1) expression in mice retinal vessels is affected by both hyperglycemia and hyperlipidemia. PLoS ONE 2010, 5, e12699. [Google Scholar] [CrossRef] [PubMed]
- Schneider-Hohendorf, T.; Rossaint, J.; Mohan, H.; Böning, D.; Breuer, J.; Kuhlmann, T.; Gross, C.C.; Flanagan, K.; Sorokin, L.; Vestweber, D.; et al. VLA-4 blockade promotes differential routes into human CNS involving PSGL-1 rolling of T cells and MCAM-adhesion of TH17 cells. J. Exp. Med. 2014, 211, 1833–1846. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Heink, S.; Petermann, F.; Srivastava, R.; Claussen, M.C.; Hemmer, B.; Korn, T. Th17 lymphocytes traffic to the central nervous system independently of α4 integrin expression during EAE. J. Exp. Med. 2011, 208, 2465–2476. [Google Scholar] [CrossRef] [PubMed]
- Coisne, C.; Mao, W.; Engelhardt, B. Cutting Edge: Natalizumab blocks adhesion but not initial contact of human T cells to the blood–brain barrier in vivo in an animal model of multiple sclerosis. J. Immunol. 2009, 182, 5909–5913. [Google Scholar] [CrossRef] [PubMed]
- Chigaev, A.; Wu, Y.; Williams, D.B.; Smagley, Y.; Sklar, L.A. Discovery of very late antigen-4 (VLA-4, α4β1 integrin) allosteric antagonists. J. Biol. Chem. 2010, 286, 5455–5463. [Google Scholar] [CrossRef] [PubMed]
- Antezana, A.; Sigal, S.; Herbert, J.; Kister, I. Natalizumab-induced hepatic injury: A case report and review of literature. Mult. Scler. Relat. Disord. 2015, 4, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Bloomgren, G.; Richman, S.; Hotermans, C.; Subramanyam, M.; Goelz, S.; Natarajan, A.; Lee, S.; Plavina, T.; Scanlon, J.V.; Sandrock, A.; et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N. Engl. J. Med. 2012, 366, 1870–1880. [Google Scholar] [CrossRef] [PubMed]
- Ifergan, I.; Kébir, H.; Bernard, M.; Wosik, K.; Dodelet-Devillers, A.; Cayrol, R.; Arbour, N.; Prat, A. The blood–brain barrier induces differentiation of migrating monocytes into Th17-polarizing dendritic cells. Brain 2008, 131, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Steiner, O.; Coisne, C.; Cecchelli, R.; Boscacci, R.; Deutsch, U.; Engelhardt, B.; Lyck, R. Differential roles for endothelial ICAM-1, ICAM-2, and VCAM-1 in shear-resistant t cell arrest, polarization, and directed crawling on blood–brain barrier endothelium. J. Immunol. 2010, 185, 4846–4855. [Google Scholar] [CrossRef] [PubMed]
- Semple, B.D.; Kossmann, T.; Morganti-Kossmann, M.C. Role of chemokines in CNS health and pathology: A focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J. Cereb. Blood Flow Metab. 2010, 30, 459–473. [Google Scholar] [CrossRef] [PubMed]
- Mardiguian, S.; Serres, S.; Ladds, E.; Campbell, S.J.; Wilainam, P.; McFadyen, C.; McAteer, M.; Choudhury, R.P.; Smith, P.; Saunders, F.; et al. Anti–IL-17A treatment reduces clinical score and VCAM-1 expression detected by in vivo magnetic resonance imaging in chronic relapsing EAE ABH mice. Am. J. Pathol. 2013, 182, 2071–2081. [Google Scholar] [CrossRef] [PubMed]
- Glabinski, A.; Mirecka, M.; Pokoca, L. Tumor necrosis factor α but not lymphotoxin is overproduced by blood mononuclear cells in multiple sclerosis. Acta Neurol. Scand. 1995, 91, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Schnyder, B.; Schnyder-Candrian, S.; Pansky, A.; Schmitz, M.L.; Heim, M.; Ryffel, B.; Moser, R. IL-17 reduces TNF-induced Rantes and VCAM-1 expression. Cytokine 2005, 31, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Griffin, G.K.; Newton, G.; Tarrio, M.L.; Bu, D.X.; Maganto-Garcia, E.; Azcutia, V.; Alcaide, P.; Grabie, N.; Luscinskas, F.W.; Croce, K.J.; et al. IL-17 and TNFα sustain neutrophil recruitment during inflammation through synergistic effects on endothelial activation. J. Immunol. 2012, 188, 6287–6299. [Google Scholar] [CrossRef] [PubMed]
- Leonard, E.J.; Skeel, A.; Yoshimura, T. Biological aspects of monocyte chemoattractant protein-1 (MCP-1). Adv. Exp. Med. Biol. 1991, 305, 57–64. [Google Scholar] [PubMed]
- Mantovani, A.; Sozzani, S.; Bottazzi, B.; Peri, G.; Sciacca, F.L.; Locati, M.; Colotta, F. Monocyte chemotactic protein-1 (MCP-1): Signal transduction and involvement in the regulation of macrophage traffic in normal and neoplastic tissues. Adv. Exp. Med. Biol. 1993, 351, 47–54. [Google Scholar] [PubMed]
- Paul, D.; Ge, S.; Lemire, Y.; Jellison, E.R.; Serwanski, D.R.; Ruddle, N.H.; Pachter, J.S. Cell-selective knockout and 3D confocal image analysis reveals separate roles for astrocyte-and endothelial-derived CCL2 in neuroinflammation. J. Neuroinflamm. 2014, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Izikson, L.; Klein, R.S.; Luster, A.D.; Weiner, H.L. Targeting monocyte recruitment in CNS autoimmune disease. Clin. Immunol. 2002, 103, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.J.; Ransohoff, R.M. The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Semin. Immunol. 2003, 15, 23–32. [Google Scholar] [CrossRef]
- Conductier, G.; Blondeau, N.; Guyon, A.; Nahon, J.L.; Rovère, C. The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J. Neuroimmunol. 2010, 224, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Glabinski, A.R.; Bielecki, B.; Kolodziejski, P.; Han, Y.; Selmaj, K.; Ransohoff, R.M. TNF-α microinjection upregulates chemokines and chemokine receptors in the central nervous system without inducing leukocyte infiltration. J. Interferon Cytokine Res. 2003, 23, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Pachter, J.S. Monocyte chemoattractant protein-1 alters expression of tight junction-associated proteins in brain microvascular endothelial cells. Microvasc. Res. 2004, 67, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Stamatovic, S.M.; Keep, R.F.; Kunkel, S.L.; Andjelkovic, A.V. Potential role of MCP-1 in endothelial cell tight junction ‘opening’: Signaling via Rho and Rho kinase. J. Cell Sci. 2003, 116, 4615–4628. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Tsirka, S.E. Truncation of monocyte chemoattractant protein 1 by plasmin promotes blood–brain barrier disruption. J. Cell Sci. 2011, 124, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Novotny, M.; Bulek, K.; Liu, C.; Li, X.; Hamilton, T. Interleukin 17 treatment prolongs CXCL1 mRNA half-life via TRAF5 and the splicing regulatory factor SF2/ASF. Nat. Immunol. 2011, 12, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Miyake, M.; Goodison, S.; Urquidi, V.; Gomes Giacoia, E.; Rosser, C.J. Expression of CXCL1 in human endothelial cells induces angiogenesis through the CXCR2 receptor and the ERK1/2 and EGF pathways. Lab. Investig. 2013, 93, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Stoler-Barak, L.; Barzilai, S.; Zauberman, A.; Alon, R. Transendothelial migration of effector T cells across inflamed endothelial barriers does not require heparan sulfate proteoglycans. Int. Immunol. 2014, 26, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Wojkowska, D.W.; Szpakowski, P.; Ksiazek-Winiarek, D.; Leszczynski, M.; Glabinski, A. Interactions between neutrophils, Th17 cells, and chemokines during the initiation of experimental model of multiple sclerosis. Mediat. Inflamm. 2014, 2014, 590409. [Google Scholar] [CrossRef] [PubMed]
- Carlson, T.; Kroenke, M.; Rao, P.; Lane, T.E.; Segal, B. The Th17–ELR+ CXC chemokine pathway is essential for the development of central nervous system autoimmune disease. J. Exp. Med. 2008, 205, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Erbel, C.; Chen, L.; Bea, F.; Wangler, S.; Celik, S.; Lasitschka, F.; Wang, Y.; Böckler, D.; Katus, H.A.; Dengler, T.J. Inhibition of IL-17A Attenuates Atherosclerotic Lesion Development in ApoE-Deficient Mice. J. Immunol. 2009, 183, 8167–8175. [Google Scholar] [CrossRef] [PubMed]
- Williams, I.R. CCR6 and CCL20: Partners in intestinal immunity and lymphorganogenesis. Ann. N. Y. Acad. Sci. 2006, 1072, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.P.; Zhang, H.H.; Foley, J.F.; Hedrick, M.N.; Farber, J.M. Human T cells that are able to produce IL-17 express the chemokine receptor CCR6. J. Immunol. 2008, 180, 214–221. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Yang, X.O.; Chung, Y.; Fukunaga, A.; Nurieva, R.; Pappu, B.; Martin-Orozco, N.; Kang, H.S.; Ma, L.; Panopoulos, A.D.; et al. CCR6 regulates the migration of inflammatory and regulatory T cells. J. Immunol. 2008, 181, 8391–8401. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wojkowska, D.W.; Szpakowski, P.; Glabinski, A. Interleukin 17A Promotes Lymphocytes Adhesion and Induces CCL2 and CXCL1 Release from Brain Endothelial Cells. Int. J. Mol. Sci. 2017, 18, 1000. https://doi.org/10.3390/ijms18051000
Wojkowska DW, Szpakowski P, Glabinski A. Interleukin 17A Promotes Lymphocytes Adhesion and Induces CCL2 and CXCL1 Release from Brain Endothelial Cells. International Journal of Molecular Sciences. 2017; 18(5):1000. https://doi.org/10.3390/ijms18051000
Chicago/Turabian StyleWojkowska, Dagmara Weronika, Piotr Szpakowski, and Andrzej Glabinski. 2017. "Interleukin 17A Promotes Lymphocytes Adhesion and Induces CCL2 and CXCL1 Release from Brain Endothelial Cells" International Journal of Molecular Sciences 18, no. 5: 1000. https://doi.org/10.3390/ijms18051000
APA StyleWojkowska, D. W., Szpakowski, P., & Glabinski, A. (2017). Interleukin 17A Promotes Lymphocytes Adhesion and Induces CCL2 and CXCL1 Release from Brain Endothelial Cells. International Journal of Molecular Sciences, 18(5), 1000. https://doi.org/10.3390/ijms18051000