
Experimental Autoimmune Encephalomyelitis (EAE)-Induced Elevated Expression of the E1 Isoform of Methyl CpG Binding Protein 2 (MeCP2E1): Implications in Multiple Sclerosis (MS)-Induced Neurological Disability and Associated Myelin Damage

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
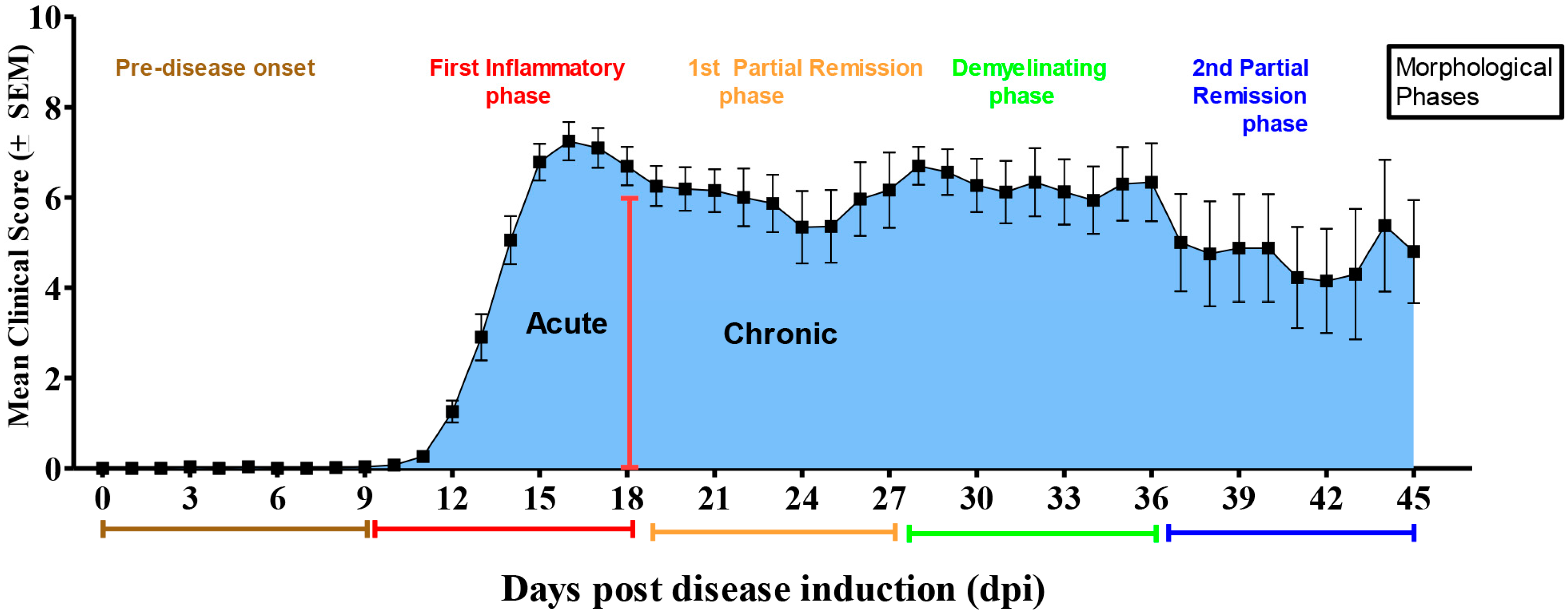
2.1. Neurological Disability Scoring (NDS)
2.2. mRNA Expression of MeCP2E1 and MeCP2E2 Isoforms in the SC
2.3. BDNF Gene and Protein Expression in SC
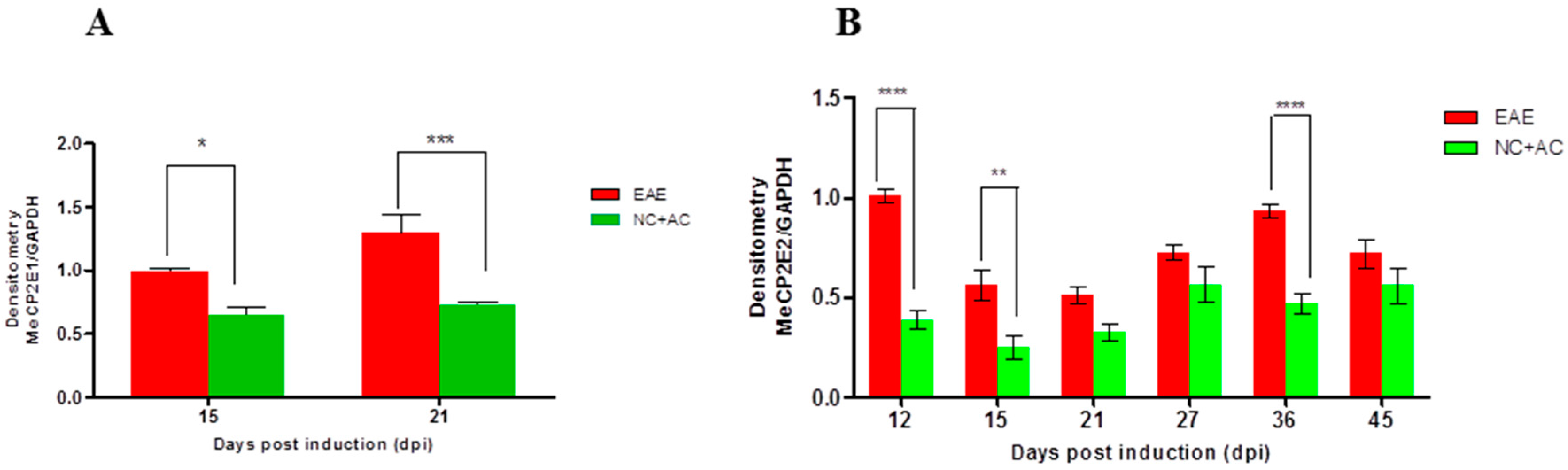
2.4. MeCP2E1 and MeCP2E2 Protein Expression in SC during Acute and Chronic Disease Phase of EAE
2.5. Baseline Protein Expression of MeCP2E1 and MeCP2E2 Isoforms in Spinal Cord (SC) and Dorsal Root Ganglia (DRG) in Naïve Control (NC) Animals
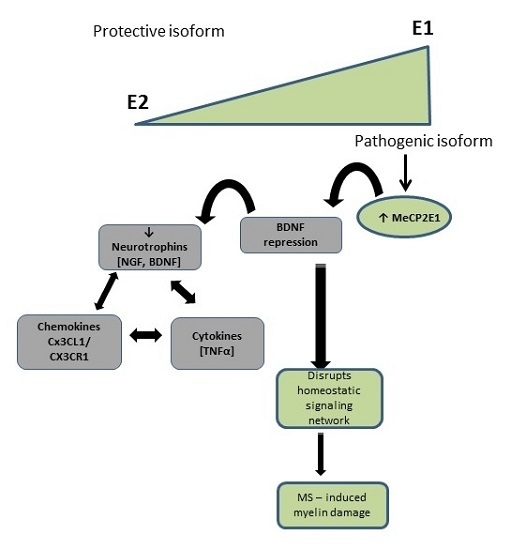
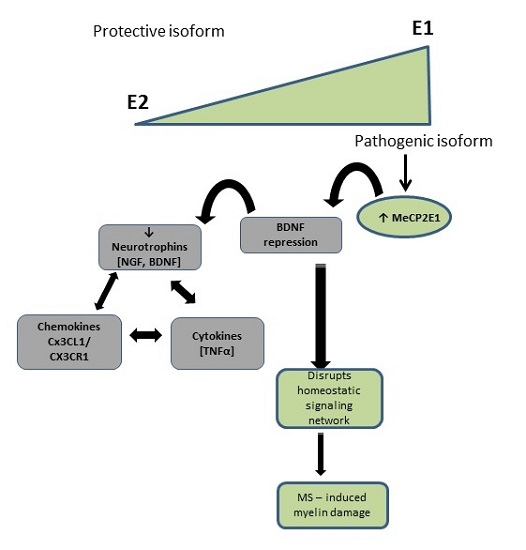
3. Discussion
4. Materials and Methods
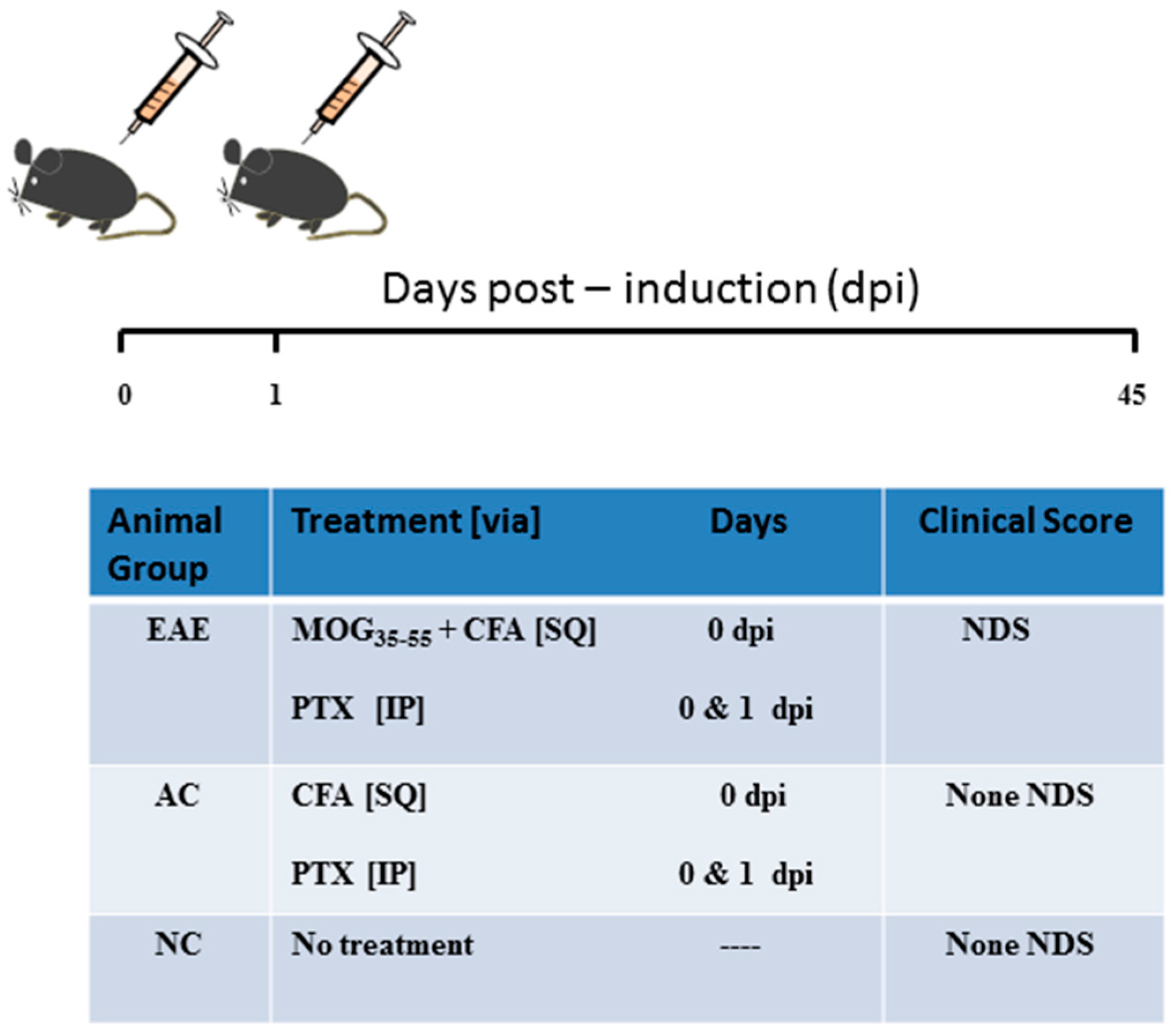
4.1. Induction of EAE
4.2. Gene/Protein Assay
4.3. Quantitative Real Time Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
4.4. Western Blot (WB) Analysis
4.5. Quantitative Enzyme-Linked Immunosorbent Assay (ELISA) Analysis
4.6. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Namaka, M.P.; Ethans, K.; Jensen, B.; Esfahani, F.; Frost, E.E. Multiple sclerosis. In Therapeutic Choices/e-Therapeutic; Canadian Pharmacists Association Publishers, Inc.: Ottawa, ON, Canada, 2011; pp. 309–319. [Google Scholar]
- Lacroix, S.; Hamilton, L.K.; Vaugeois, A.; Beaudoin, S.; Breault-Dugas, C.; Pineau, I.; Levesque, S.A.; Gregoire, C.A.; Fernandes, K.J. Central canal ependymal cells proliferate extensively in response to traumatic spinal cord injury but not demyelinating lesions. PLoS ONE 2014, 9, e85916. [Google Scholar] [CrossRef] [PubMed]
- Gold, R.; Montalban, X. Multiple sclerosis: More pieces of the immunological puzzle. Lancet Neurol. 2012, 11, 9–10. [Google Scholar] [CrossRef]
- Namaka, M.; Turcotte, D.; Leong, C.; Grossberndt, A.; Klassen, D. Multiple sclerosis: Etiology and treatment strategies. Consult. Pharm. 2008, 23, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Acosta, C.M.R.; Cortes, C.; MacPhee, H.; Namaka, M.P. Exploring the role of nerve growth factor in multiple sclerosis: Implications in myelin repair. CNS Neurol. Disord. Drug Targets 2013, 12, 1242–1256. [Google Scholar] [CrossRef] [PubMed]
- Begum, F.; Zhu, W.; Cortes, C.; Macneil, B.; Namaka, M. Elevation of tumor necrosis factor α in dorsal root ganglia and spinal cord is associated with neuroimmune modulation of pain in an animal model of multiple sclerosis. J. Neuroimm. Pharmacol. 2013, 8, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Melanson, M.; Miao, P.; Eisenstat, D.; Gong, Y.; Gu, X.; Au, K.; Zhu, W.; Begum, F.; Frost, E.E.; Namaka, M. Experimental autoimmune encephalomyelitis-induced upregulation of tumor necrosis factor-α in the dorsal root ganglia. Mult. Scler. 2009, 15, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Frost, E.E.; Begum, F.; Vora, P.; Au, K.; Gong, Y.; Macneil, B.; Pillai, P.; Namaka, M. The role of dorsal root ganglia activation and brain-derived neurotrophic factor in multiple sclerosis. J. Cell Mol. Med. 2012, 16, 1856–1865. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Acosta, C.; MacNeil, B.; Cortes, C.; Intrater, H.; Gong, Y.; Namaka, M. Elevated expression of fractalkine (cx3cl1) and fractalkine receptor (cx3cr1) in the dorsal root ganglia and spinal cord in experimental autoimmune encephalomyelitis: Implications in multiple sclerosis-induced neuropathic pain. BioMed Res. Int. 2013, 2013, 480702. [Google Scholar] [CrossRef] [PubMed]
- Acosta, C.; Cortes, C.; Altaweel, K.; MacPhee, H.; Hoogervorst, B.; Bhullar, H.; MacNeil, B.; Torabi, M.; Burczynski, F.; Namaka, M.P. Immune system induction of nerve growth factor in an animal model of multiple sclerosis: Implications in re-myelination and myelin repair. CNS Neurol. Disord. Drug Targets 2015, 14, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Brian, C.A.; MacNeil, J.; Klonisch, T.; Cortes, C.; Doupe, M.; Gong, Y.; Namaka, M. Spinal cord brain derived neurotrophic factor (BDNF) responsive cells in an experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis (MS): Implications in myelin repair. Res. Immunol. 2014, in press. [Google Scholar] [CrossRef]
- VonDran, M.W.; Singh, H.; Honeywell, J.Z.; Dreyfus, C.F. Levels of BDNF impact oligodendrocyte lineage cells following a cuprizone lesion. J. Neurosci. 2011, 31, 14182–14190. [Google Scholar] [CrossRef] [PubMed]
- Faraco, G.; Cavone, L.; Chiarugi, A. The therapeutic potential of hdac inhibitors in the treatment of multiple sclerosis. Mol. Med. 2011, 17, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Vondran, M.W.; Clinton-Luke, P.; Honeywell, J.Z.; Dreyfus, C.F. BDNF+/− mice exhibit deficits in oligodendrocyte lineage cells of the basal forebrain. Glia 2010, 58, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Linker, R.A.; Lee, D.H.; Demir, S.; Wiese, S.; Kruse, N.; Siglienti, I.; Gerhardt, E.; Neumann, H.; Sendtner, M.; Luhder, F.; et al. Functional role of brain-derived neurotrophic factor in neuroprotective autoimmunity: Therapeutic implications in a model of multiple sclerosis. Brain 2010, 133, 2248–2263. [Google Scholar] [CrossRef] [PubMed]
- Linker, R.; Gold, R.; Luhder, F. Function of neurotrophic factors beyond the nervous system: Inflammation and autoimmune demyelination. Crit. Rev. Immunol. 2009, 29, 43–68. [Google Scholar] [CrossRef] [PubMed]
- McTigue, D.M.; Horner, P.J.; Stokes, B.T.; Gage, F.H. Neurotrophin-3 and brain-derived neurotrophic factor induce oligodendrocyte proliferation and myelination of regenerating axons in the contused adult rat spinal cord. J. Neurosci. 1998, 18, 5354–5365. [Google Scholar] [PubMed]
- Van’t Veer, A.; Du, Y.; Fischer, T.Z.; Boetig, D.R.; Wood, M.R.; Dreyfus, C.F. Brain-derived neurotrophic factor effects on oligodendrocyte progenitors of the basal forebrain are mediated through TRKB and the MAP kinase pathway. J. Neurosci. Res. 2009, 87, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Ferner, A.H.; Wong, A.W.; Denham, M.; Kilpatrick, T.J.; Murray, S.S. Extracellular signal-regulated kinase 1/2 signaling promotes oligodendrocyte myelination in vitro. J. Neurochem. 2012, 122, 1167–1180. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.W.; Xiao, J.; Kemper, D.; Kilpatrick, T.J.; Murray, S.S. Oligodendroglial expression of TRKB independently regulates myelination and progenitor cell proliferation. J. Neurosci. 2013, 33, 4947–4957. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Wong, A.W.; Willingham, M.M.; van den Buuse, M.; Kilpatrick, T.J.; Murray, S.S. Brain-derived neurotrophic factor promotes central nervous system myelination via a direct effect upon oligodendrocytes. Neuro-Signals 2010, 18, 186–202. [Google Scholar] [CrossRef] [PubMed]
- Cellerino, A.; Carroll, P.; Thoenen, H.; Barde, Y.-A. Reduced size of retinal ganglion cell axons and hypomyelination in mice lacking brain-derived neurotrophic factor. Mol. Cell. Neurosci. 1997, 9, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Djalali, S.; Holtje, M.; Grosse, G.; Rothe, T.; Stroh, T.; Grosse, J.; Deng, D.R.; Hellweg, R.; Grantyn, R.; Hortnagl, H.; et al. Effects of brain-derived neurotrophic factor (BDNF) on glial cells and serotonergic neurones during development. J. Neurochem. 2005, 92, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Lercher, L.D.; Zhou, R.; Dreyfus, C.F. Mitogen-activated protein kinase pathway mediates effects of brain-derived neurotrophic factor on differentiation of basal forebrain oligodendrocytes. J. Neurosci. Res. 2006, 84, 1692–1702. [Google Scholar] [CrossRef] [PubMed]
- Di Nuzzo, L.; Orlando, R.; Nasca, C.; Nicoletti, F. Molecular pharmacodynamics of new oral drugs used in the treatment of multiple sclerosis. Drug Design Dev. Ther. 2014, 8, 555–568. [Google Scholar]
- Arnon, R.; Aharoni, R. Neuroprotection and neurogeneration in MS and its animal model EAE effected by glatiramer acetate. J. Neural. Transm. 2009, 116, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- La Mantia, L.; Munari, L.M.; Lovati, R. Glatiramer acetate for multiple sclerosis. Cochrane Database Syst. Rev. 2010, 5, CD004678. [Google Scholar]
- Kala, M.; Miravalle, A.; Vollmer, T. Recent insights into the mechanism of action of glatiramer acetate. J. Neuroimmunol. 2011, 235, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Comi, G.; Pulizzi, A.; Rovaris, M.; Abramsky, O.; Arbizu, T.; Boiko, A.; Gold, R.; Havrdova, E.; Komoly, S.; Selmaj, K.; et al. Effect of laquinimod on MRI-monitored disease activity in patients with relapsing-remitting multiple sclerosis: A multicentre, randomised, double-blind, placebo-controlled phase IIB study. Lancet 2008, 371, 2085–2092. [Google Scholar] [CrossRef]
- Thone, J.; Ellrichmann, G.; Seubert, S.; Peruga, I.; Lee, D.H.; Conrad, R.; Hayardeny, L.; Comi, G.; Wiese, S.; Linker, R.A.; et al. Modulation of autoimmune demyelination by laquinimod via induction of brain-derived neurotrophic factor. Am. J. Pathol. 2012, 180, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Meehan, R.R.; Lewis, J.D.; McKay, S.; Kleiner, E.L.; Bird, A.P. Identification of a mammalian protein that binds specifically to DNA containing methylated CPGs. Cell 1989, 58, 499–507. [Google Scholar] [CrossRef]
- Zachariah, R.M.; Olson, C.O.; Ezeonwuka, C.; Rastegar, M. Novel MECP2 isoform-specific antibody reveals the endogenous MECP2e1 expression in murine brain, primary neurons and astrocytes. PLoS ONE 2012, 7, e49763. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.D.; Meehan, R.R.; Henzel, W.J.; Maurer-Fogy, I.; Jeppesen, P.; Klein, F.; Bird, A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 1992, 69, 905–914. [Google Scholar] [CrossRef]
- Nan, X.; Campoy, F.J.; Bird, A. MECP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 1997, 88, 471–481. [Google Scholar] [CrossRef]
- Chen, W.G.; Chang, Q.; Lin, Y.; Meissner, A.; West, A.E.; Griffith, E.C.; Jaenisch, R.; Greenberg, M.E. Derepression of bdnf transcription involves calcium-dependent phosphorylation of MECP2. Science 2003, 302, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Khare, G.; Dani, V.; Nelson, S.; Jaenisch, R. The disease progression of MECP2 mutant mice is affected by the level of bdnf expression. Neuron 2006, 49, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef] [PubMed]
- Wade, P.A.; Jones, P.L.; Vermaak, D.; Veenstra, G.J.; Imhof, A.; Sera, T.; Tse, C.; Ge, H.; Shi, Y.B.; Hansen, J.C.; et al. Histone deacetylase directs the dominant silencing of transcription in chromatin: Association with MECP2 and the MI-2 chromodomain SWI/SNF ATPase. Cold Spring Harb. Symp. Quant. Biol. 1998, 63, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.L.; Veenstra, G.J.; Wade, P.A.; Vermaak, D.; Kass, S.U.; Landsberger, N.; Strouboulis, J.; Wolffe, A.P. Methylated DNA and MECP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998, 19, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Abuhatzira, L.; Makedonski, K.; Kaufman, Y.; Razin, A.; Shemer, R. MECP2 deficiency in the brain decreases bdnf levels by rest/corest-mediated repression and increases TRKB production. Epigenetics 2007, 2, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chan, S.-A.; Ogier, M.; Hellard, D.; Wang, Q.; Smith, C.; Katz, D.M. Dysregulation of brain-derived neurotrophic factor expression and neurosecretory function in MECP2 null mice. J. Neurosci. 2006, 26, 10911–10915. [Google Scholar] [CrossRef] [PubMed]
- Ogier, M.; Wang, H.; Hong, E.; Wang, Q.; Greenberg, M.E.; Katz, D.M. Brain-derived neurotrophic factor expression and respiratory function improve after ampakine treatment in a mouse model of rett syndrome. J. Neurosci. 2007, 27, 10912–10917. [Google Scholar] [CrossRef] [PubMed]
- Deng, V.; Matagne, V.; Banine, F.; Frerking, M.; Ohliger, P.; Budden, S.; Pevsner, J.; Dissen, G.A.; Sherman, L.S.; Ojeda, S.R. FXYD1 is an MECP2 target gene overexpressed in the brains of rett syndrome patients and MECP2-null mice. Hum. Mol. Genet. 2007, 16, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Aid, T.; Kazantseva, A.; Piirsoo, M.; Palm, K.; Timmusk, T. Mouse and rat BDNF gene structure and expression revisited. J. Neurosci. Res. 2007, 85, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.; Qin, J.; Zoghbi, H.Y. MECP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Mnatzakanian, G.N.; Lohi, H.; Munteanu, I.; Alfred, S.E.; Yamada, T.; MacLeod, P.J.; Jones, J.R.; Scherer, S.W.; Schanen, N.C.; Friez, M.J.; et al. A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to rett syndrome. Nat. Genet. 2004, 36, 339–341. [Google Scholar] [CrossRef] [PubMed]
- Kriaucionis, S.; Bird, A. The major form of MECP2 has a novel N-terminus generated by alternative splicing. Nucleic Acids Res. 2004, 32, 1818–1823. [Google Scholar] [CrossRef] [PubMed]
- Dastidar, S.G.; Bardai, F.H.; Ma, C.; Price, V.; Rawat, V.; Verma, P.; Narayanan, V.; D’Mello, S.R. Isoform-specific toxicity of MECP2 in postmitotic neurons: Suppression of neurotoxicity by FOXG1. J. Neurosci. 2012, 32, 2846–2855. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, V.R.; Zachariah, R.M.; Rastegar, M. Decitabine alters the expression of MECP2 isoforms via dynamic DNA methylation at the MECP2 regulatory elements in neural stem cells. Mol. Autism 2013, 4, 46. [Google Scholar] [CrossRef] [PubMed]
- KhorshidAhmad, T.; Acosta, C.; Cortes, C.; Lakowski, T.M.; Gangadaran, S.; Namaka, M. Transcriptional regulation of brain-derived neurotrophic factor (BDNF) by methyl CPG binding protein 2 (MECP2): A novel mechanism for re-myelination and/or myelin repair involved in the treatment of multiple sclerosis (MS). Mol. Neurobiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Vora, P.; Mina, R.; Namaka, M.; Frost, E.E. A novel transcriptional regulator of myelin gene expression: Implications for neurodevelopmental disorders. Neuroreport 2010, 21, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hong, E.J.; Cohen, S.; Zhao, W.N.; Ho, H.Y.; Schmidt, L.; Chen, W.G.; Lin, Y.; Savner, E.; Griffith, E.C.; et al. Brain-specific phosphorylation of MECP2 regulates activity-dependent BDNF transcription, dendritic growth, and spine maturation. Neuron 2006, 52, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Rousseaud, A.; Delepine, C.; Nectoux, J.; Billuart, P.; Bienvenu, T. Differential expression and regulation of brain-derived neurotrophic factor (BDNF) mrna isoforms in brain cells from MECP2 mouse model. J. Mol. Neurosci. 2015, 56, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Tochiki, K.K.; Cunningham, J.; Hunt, S.P.; Geranton, S.M. The expression of spinal methyl-CPG-binding protein 2, DNA methyltransferases and histone deacetylases is modulated in persistent pain states. Mol. Pain 2012, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Huang, M.; Cao, Z.; Qi, J.; Qiu, Z.; Chiang, L.Y. MECP2 plays an analgesic role in pain transmission through regulating creb/miR-132 pathway. Mol. Pain 2015, 11, 19. [Google Scholar] [CrossRef] [PubMed]
- Pedre, X.; Mastronardi, F.; Bruck, W.; Lopez-Rodas, G.; Kuhlmann, T.; Casaccia, P. Changed histone acetylation patterns in normal-appearing white matter and early multiple sclerosis lesions. J. Neurosci. 2011, 31, 3435–3445. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Lee, S.M.; Gao, B.; Zhang, J.; Fang, D. Histone deacetylase sirtuin 1 deacetylates IRF1 protein and programs dendritic cells to control TH17 protein differentiation during autoimmune inflammation. J. Biol. Chem. 2013, 288, 37256–37266. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Singh, J.; Pillai, P.P.; Frost, E.E. Involvement of MECP2 in regulation of myelin-related gene expression in cultured rat oligodendrocytes. J. Mol. Neurosci. 2015, 57, 176–184. [Google Scholar] [CrossRef] [PubMed]
- De Felice, C.; Della Ragione, F.; Signorini, C.; Leoncini, S.; Pecorelli, A.; Ciccoli, L.; Scalabri, F.; Marracino, F.; Madonna, M.; Belmonte, G.; et al. Oxidative brain damage in MECP2-mutant murine models of rett syndrome. Neurobiol. Dis. 2014, 68, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Ben-Zeev, B.; Aharoni, R.; Nissenkorn, A.; Arnon, R. Glatiramer acetate (GA, copolymer-1) an hypothetical treatment option for rett syndrome. Med. Hypotheses 2011, 76, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Costa, O.; Divoux, D.; Ischenko, A.; Tron, F.; Fontaine, M. Optimization of an animal model of experimental autoimmune encephalomyelitis achieved with a multiple MOG(35–55)peptide in c57bl6/j strain of mice. J. Autoimmun. 2003, 20, 51–61. [Google Scholar] [CrossRef]
- Namaka, M.; Kapoor, S.; Simms, L.; Leong, C.; Grossberndt, A.; Prout, M.; Frost, E.; Esfahani, F.; Gomori, A.; Mulvey, M.R. Molecular mimicry and multiple sclerosis. Neural Regen Res. 2011, 6, 1322–1333. [Google Scholar]
- Berard, J.L.; Wolak, K.; Fournier, S.; David, S. Characterization of relapsing-remitting and chronic forms of experimental autoimmune encephalomyelitis in c57bl/6 mice. Glia 2010, 58, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shachar, S.; Chahrour, M.; Thaller, C.; Shaw, C.A.; Zoghbi, Y.H. Mouse models of MECP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum. Mol. Genet. 2009, 18, 2431–2442. [Google Scholar] [CrossRef] [PubMed]
- Dragich, J.M.; Kim, Y.H.; Arnold, A.P.; Schanen, N.C. Differential distribution of the MECP2 splice variants in the postnatal mouse brain. J. Comparative Neurol. 2007, 501, 526–542. [Google Scholar] [CrossRef] [PubMed]
- Saunders, C.J.; Minassian, B.E.; Chow, E.W.; Zhao, W.; Vincent, J.B. Novel exon 1 mutations in MECP2 implicate isoform MECP2_e1 in classical rett syndrome. Am. J. Med. Genet. Part A 2009, 149A, 1019–1023. [Google Scholar] [CrossRef] [PubMed]
- Fichou, Y.; Nectoux, J.; Bahi-Buisson, N.; Rosas-Vargas, H.; Girard, B.; Chelly, J.; Bienvenu, T. The first missense mutation causing rett syndrome specifically affecting the MECP2_e1 isoform. Neurogenetics 2009, 10, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Orlic-Milacic, M.; Kaufman, L.; Mikhailov, A.; Cheung, A.Y.; Mahmood, H.; Ellis, J.; Gianakopoulos, P.J.; Minassian, B.A.; Vincent, J.B. Over-expression of either MECP2_e1 or MECP2_e2 in neuronally differentiated cells results in different patterns of gene expression. PLoS ONE 2014, 9, e91742. [Google Scholar] [CrossRef] [PubMed]
- Meinnel, T.; Peynot, P.; Giglione, C. Processed N-termini of mature proteins in higher eukaryotes and their major contribution to dynamic proteomics. Biochimie 2005, 87, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Del Gaudio, D.; Fang, P.; Scaglia, F.; Ward, P.A.; Craigen, W.J.; Glaze, D.G.; Neul, J.L.; Patel, A.; Lee, J.A.; Irons, M.; et al. Increased MECP2 gene copy number as the result of genomic duplication in neurodevelopmentally delayed males. Genet. Med. 2006, 8, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Van Esch, H.; Bauters, M.; Ignatius, J.; Jansen, M.; Raynaud, M.; Hollanders, K.; Lugtenberg, D.; Bienvenu, T.; Jensen, L.R.; Gecz, J.; et al. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am. J. Human Genet. 2005, 77, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Chatrchyan, S.; Khachatryan, V.; Sirunyan, A.M.; Tumasyan, A.; Adam, W.; Bergauer, T.; Dragicevic, M.; Ero, J.; Fabjan, C.; Friedl, M.; et al. Evidence of b-Jet quenching in PbPb collisions at radical(s[NN]) = 2.76 TeV. Phys. Rev. Lett. 2014, 113, 132301. [Google Scholar] [CrossRef] [PubMed]
- Mendel, I.; Kerlero de Rosbo, N.; Ben-Nun, A. A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2B mice: Fine specificity and T cell receptor V β expression of encephalitogenic T cells. Eur. J. Immunol. 1995, 25, 1951–1959. [Google Scholar] [CrossRef] [PubMed]
- Marcondes, M.C.; Furtado, G.C.; Wensky, A.; Curotto de Lafaille, M.A.; Fox, H.S.; Lafaille, J.J. Immune regulatory mechanisms influence early pathology in spinal cord injury and in spontaneous autoimmune encephalomyelitis. Am. J. Pathol. 2005, 166, 1749–1760. [Google Scholar] [CrossRef]
- Orton, S.M.; Herrera, B.M.; Yee, I.M.; Valdar, W.; Ramagopalan, S.V.; Sadovnick, A.D.; Ebers, G.C. Canadian Collaborative Study Group. Sex ratio of multiple sclerosis in canada: A longitudinal study. Lancet Neurol. 2006, 5, 932–936. [Google Scholar] [CrossRef]
- Rounds, W.H.; Salinas, E.A.; Wilks, T.B., 2nd; Levin, M.K.; Ligocki, A.J.; Ionete, C.; Pardo-Villamizar, C.; Vernino, S.; Greenberg, B.M.; Bigwood, D.W.; et al. Msprecise: A molecular diagnostic test for multiple sclerosis using next generation sequencing. Gene 2015, 572, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Crawford, M.P.; Ortega, S.B.; Karandikar, N.J. Multiparameter flow cytometric assays to quantify effector and regulatory T-cell function in multiple sclerosis. J. Mult. Scler. 2015, 2. [Google Scholar] [CrossRef]
- Zachariah, R.M.; Rastegar, M. Linking epigenetics to human disease and rett syndrome: The emerging novel and challenging concepts in MECP2 research. Neural Plast. 2012, 2012, 415825. [Google Scholar] [CrossRef] [PubMed]







© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khorshid Ahmad, T.; Zhou, T.; AlTaweel, K.; Cortes, C.; Lillico, R.; Lakowski, T.M.; Gozda, K.; Namaka, M.P. Experimental Autoimmune Encephalomyelitis (EAE)-Induced Elevated Expression of the E1 Isoform of Methyl CpG Binding Protein 2 (MeCP2E1): Implications in Multiple Sclerosis (MS)-Induced Neurological Disability and Associated Myelin Damage. Int. J. Mol. Sci. 2017, 18, 1254. https://doi.org/10.3390/ijms18061254
Khorshid Ahmad T, Zhou T, AlTaweel K, Cortes C, Lillico R, Lakowski TM, Gozda K, Namaka MP. Experimental Autoimmune Encephalomyelitis (EAE)-Induced Elevated Expression of the E1 Isoform of Methyl CpG Binding Protein 2 (MeCP2E1): Implications in Multiple Sclerosis (MS)-Induced Neurological Disability and Associated Myelin Damage. International Journal of Molecular Sciences. 2017; 18(6):1254. https://doi.org/10.3390/ijms18061254
Chicago/Turabian StyleKhorshid Ahmad, Tina, Ting Zhou, Khaled AlTaweel, Claudia Cortes, Ryan Lillico, Ted Martin Lakowski, Kiana Gozda, and Michael Peter Namaka. 2017. "Experimental Autoimmune Encephalomyelitis (EAE)-Induced Elevated Expression of the E1 Isoform of Methyl CpG Binding Protein 2 (MeCP2E1): Implications in Multiple Sclerosis (MS)-Induced Neurological Disability and Associated Myelin Damage" International Journal of Molecular Sciences 18, no. 6: 1254. https://doi.org/10.3390/ijms18061254