The Role of p16INK4a Pathway in Human Epidermal Stem Cell Self-Renewal, Aging and Cancer

1
Laboratory of Vascular Pathology, Istituto Dermopatico dell'Immacolata, Istituto di Ricovero e Cura a Carattere Scientifico (IDI-IRCCS), Fondazione Luigi Maria Monti (FLMM), via Monti di Creta 104, 00167 Rome, Italy
2
Molecular and Cell Biology Laboratory, Istituto Dermopatico dell'Immacolata, Istituto di Ricovero e Cura a Carattere Scientifico (IDI-IRCCS), Fondazione Luigi Maria Monti (FLMM), via Monti di Creta 104, 00167 Rome, Italy
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(7), 1591; https://doi.org/10.3390/ijms18071591
Submission received: 27 June 2017
/
Revised: 13 July 2017
/
Accepted: 19 July 2017
/
Published: 22 July 2017
(This article belongs to the Special Issue Molecular Research of Epidermal Stem Cells 2017)
Abstract
:The epidermis is a self-renewing tissue. The balance between proliferation and differentiation processes is tightly regulated to ensure the maintenance of the stem cell (SC) population in the epidermis during life. Aging and cancer may be considered related endpoints of accumulating damages within epidermal self-renewing compartment. p16INK4a is a potent inhibitor of the G1/S-phase transition of the cell cycle. p16INK4a governs the processes of SC self-renewal in several tissues and its deregulation may result in aging or tumor development. Keratinocytes are equipped with several epigenetic enzymes and transcription factors that shape the gene expression signatures of different epidermal layers and allow dynamic and coordinated expression changes to finely balance keratinocyte self-renewal and differentiation. These factors converge their activity in the basal layer to repress p16INK4a expression, protecting cells from senescence, and preserving epidermal homeostasis and regeneration. Several stress stimuli may activate p16INK4a expression that orchestrates cell cycle exit and senescence response. In the present review, we discuss the role of p16INK4a regulators in human epidermal SC self-renewal, aging and cancer.
1. Introduction
The epidermis is a self-renewing tissue characterized by several compartmentalized layers of keratinocytes in stages of progressive differentiation by virtue of a temporal and spatial gene regulation: Namely, the basal layer is composed by proliferating keratinocytes; the upper layers are composed by viable and differentiated cells; the horny layer is composed by terminally differentiated cells [1]. Epidermal homeostasis relies on keratinocyte stem cells (SCs) hosted in the basal layer; they provide new cells to replace those lost during tissue turnover or following injury. Indeed, SC properties comprise both the long term self-renewal ability and the differentiation capability [1].
2. Interfollicular Epidermal Stem Cells
Two Distinct Models for Interfollicular Epidermis Self-Renewal Proposed in Mice
The hierarchical model suggests the existence of two distinct proliferative cell compartments consisting of slow-cycling SCs and committed progenitor cells. They are hierarchically arranged and contribute to the homeostasis of discrete regions of epidermis. Indeed, slow-cycling cells give rise to actively cycling progenitors that persist for various periods of time in a transient-amplifying (TA) phase and generate differentiated cells [2]. This view is supported by labelling and genetic lineage-tracing experiments displaying an interfollicular epidermis organization into discrete proliferative units (EPU) from the basal to horny layer. During wound healing, mainly SCs contribute to the repair and long-term regeneration [2,3,4].
The stochastic model suggests that the basal epidermis is composed of a single type of population of actively cycling progenitors functionally equivalent, having 50% probability to differentiate or divide. Thus, they directly generate differentiated spinous cells without entering a TA phase [5,6]. Differences in these studies may arise from the tools used to mark the basal cells or from difference in thickness and gene expression in the epidermis at different body sites.
Mouse and human skin have significant differences in cellular architecture and physiology, making it difficult to apply these concepts to human epidermis. Mouse skin has a dense array of hair follicles, whereas human skin displays larger areas of interfollicular epidermis characterized by downward projections of the epidermal rete ridges. Moreover, the human epidermal compartment is thicker with more cell layers than mouse skin. Indeed, human epidermis turnover occurs every 40–56 days, whereas mouse epidermis turns over every 8–10 days [7]. However, genetically marked xenografts of human skin revealed the presence of long-term columns of labeled cells, resembling EPU [8,9]. The first functional demonstration of the presence of human epidermal SCs was achieved in 1975, when they were maintained in culture and propagated for hundreds of generations without losing stemness [10]. Human keratinocyte cultures are constituted of a heterogeneous population of clonogenic cells endowed with different proliferative potential. They can be classified by clonal analysis [11]: The holoclone is generated from SC and has the highest proliferative capacity; meroclones and paraclones originate from young and old TA-cells, respectively. Clonal analysis remains the most reliable means to discriminate human SC from TA-cells. Indeed, none of the identified cell surface proteins is a keratinocyte SC specific marker even though some are able to capture subpopulations containing SCs [11]. Clonal analyses of several human squamous epithelia have unambiguously shown the existence of both self-renewing cells and non-self-renewing TA cells with different clonogenic capacity [12,13]. Both cell types participate in epithelial regeneration [12,14,15]. Thus, the evidence available for human epidermis is consistent with the hypothesis of the presence of both slow-cycling and activated SCs, as found in the mouse. However, the latter SC type may generate TA cells that sustain the longer turnover time and produce differentiated cells, as proposed for other human tissues [16].
For self-renewal, SC has to enter into the cell cycle and divide, and at least one daughter cell must be maintained in an undifferentiated state. Human clonal evolution from SCs to TA cells is a continuous unidirectional process, is instrumental in building up the epidermal structure since TA cells may generate postmitotic terminally differentiated keratinocytes, which migrate upwards and form suprabasal epidermal layers [11]. The SC division timing and the balance of differentiating vs. proliferating cell production are tightly regulated to ensure the SC population maintenance during life. Indeed, whether SCs are exhausted too quickly or their proliferative potential is reduced following endogenous or exogenous damages, tissue atrophy, and premature aging can arise. On the contrary, some environmental stimuli or genomic instability may promote more frequent SC divisions without appropriate differentiation balance that result in abnormal tissue development and even cancer [1].
Several studies indicate the tumor suppressor p16INK4a as an intrinsic human clonal evolution regulator. Indeed, it increases during in vitro clonal conversion and its forced expression or downregulation impairs this process [17,18,19]. In several tissues p16INK4a governs the processes of SC self-renewal and its deregulation may result in aging or tumour development [20].
3. The p16INK4a Pathway and Its Regulation
3.1. The p16INK4a/pRb Pathway
The SC self-renews, taking advantage of the cell cycle machinery to divide. Mitogenic signals induce the expression of D-type cyclins, which bind to and activate cyclin-dependent kinase 4 (CDK4) or CDK6. These complexes inactivate the pRb proteins through phosphorylation that induces pRb-E2Fs dissociation. The E2F transcription factors promote the G1- to S-phase transition through the transcription of their target genes [21].
p16INK4a is a potent inhibitor of the G1-phase transition of the cell cycle and is considered a tumor suppressor gene. Following several stress stimuli (e.g., DNA damage, oncogenic signals), p16INK4a directly binds CDK4/6, inhibiting its kinase activity and preventing pRb phosphorylation. Thus, pRb remains associated with E2Fs in the cytoplasm, preventing the E2F-mediated transcription and resulting in cell cycle block [21]. The transition from temporary to stable cell cycle arrest, which involves prolonged cyclin inhibition activity by sustained activation of p16INK4a (or p21) pathway, may be considered the initial step of cellular senescence [20]. p16INK4a mediated senescence results in chromatin reorganization or senescence-associated heteochromatin foci (SAHFs) which are related to the repression of genes regulated by E2F1 [22].
The p16INK4a gene (CDKN2A) is contained within the Inhibitor of cdk4/ Alternate Reading Frame (INK4/ARF) locus (Figure 1A), a complex locus critical for proper cell cycle control, on human chromosome 9 p21 [21]. CDKN2A encodes two transcripts for p16INK4a and p14ARF tumor suppressor genes falling on two distinct reading frames. p16INK4a is transcribed from exon 1α and exons 2 and 3, whereas p14ARF is transcribed from exon 1β and exon 2. Different from p16INK4a, p14ARF stabilizes and activates p53. In addition to CDKN2A, the INK4/ARF locus encodes a third tumor suppressor, p15INK4b, which directly blocks the interaction of CDK4/6 with D-type cyclins similarly to p16INK4a, and the long non-coding transcript ANRIL (Anti-sense non-coding RNA in the INK4/ARF Locus), which acts as an epigenetic regulator of the INK4/ARF locus [21,23,24,25].
3.2. The p16INK4a Expression Regulation
To maintain tissue homeostasis, the ability of p16INK4a to inhibit cellular proliferation must be tightly controlled. To this aim, the regulation of p16INK4a expression is complex and involves multiple transcription factors and a finely tuned epigenetic control [24].
3.2.1. Epigenetic Regulation
Epigenetic regulators have specific enzymatic activities, which modify DNA accessibility and chromatin structure, and, in turn, control gene expression (Figure 1A).
DNA methyltransferase (DNMT) and ten-eleven translocation (TET) family enzymes—Conversion of cytosine DNA bases to 5-methyl-cytosine (5mC) is one of the best-characterized epigenetic modifications, which occurs predominantly in CpG islands, and is mostly associated with transcriptional repression. Indeed, 5mC may inhibit transcription by preventing the transcription factor binding to DNA or it may recruit methyl-DNA-binding proteins that facilitate the assembling of chromatin repressor complexes [26]. However, the impact of the DNA methylation on transcription is quite nuanced. Dynamic DNA methylation depends on the interplay between DNMT and TET enzymes. Three DNMT enzymes (DNMT1, DNMT3A, and DNMT3B) catalyze the promoter DNA methylation of several genes codifying proteins able to block cell cycle progression, such as p16INK4a [27]. Specifically, DNMT1 copies the pattern of methyl marks from the parent strand to the daughter strand after cell division, whereas DNMT3A and DNMT3B catalyze de novo DNA methylation. Although no mechanism of direct demethylation has been identified, TET family enzymes may oxidize 5mC to 5-Hydroxymethylcytosine (5-hmC) that is not recognized by DNMT1 and, in turn, DNA methylation is lost during replication [28,29].
Polycomb group (PcG) protein and Jumanji protein families—In mammals, PcG proteins belong to two classes of complexes, Polycomb Repressor Complex 1 and 2 (PRC1 and PRC2), which operate a transcriptional repression via histone modifications and chromatin compaction. PRC2 functions as an initiator of transcription repression and PRC1 functions as a repressor maintenance complex. These complexes are critical for the homeostatic regulation of the INK4/ARF transcription. When they repress the transcription, SCs may enter in the cell cycle [30,31]. The PRC2 complex is composed of methyltransferase subunits Ezh1 and Ezh2, which mediates histone H3 lysine 27 trimethylation (H3K27me3), EED, Suppressor of zeste 12 homolog (Suz12), and Retinoblastoma-associated protein (RBA) p46/48. The H3K27me3 mark mediated by PRC2 is a pre-requisite for PRC1 binding to the chromatin [30]. Multiple variants of canonical PRC1 complex exist and can bind to INK4/ARF locus, likely carrying out different biochemical functions. Moreover, the subunit composition is cell type specific [32]. Canonical PRC1 is formed by four different ortholog proteins, CBX (polycomb), PCGF (polycomb group factor), HPH (human polyhomeotic homolog), and the E3-ligase protein (RING) that catalyzes histone H2A monoubiquitylation (H2AK119ub1). This modification prevents RNA polymerase II elongation and promotes chromatin compaction and silencing [32]. PRC1 complexes contain a single representative of the CBX and PCGF subunits. However, these proteins have numerous orthologs, such as five CBX (CBX2, 4, 6, 7, 8), six PCGF (PCGF1, 2, 3, 4, 5, 6), three HPH (HPH1, 2, 3), and two RING proteins (RING1, 2), and can assemble diverse compositions of the PRC1 complex. Different CBX proteins have a distinct pattern of chromatin binding, suggesting that the CBX unit confers specificity to the complex. Indeed, the CBX subunit of canonical PRC1 complex recognizes H3K27me3 via its chromodomain and enables PRC1 recruitment to its target genes [29,30,32]. This results in long-term reversible suppression of INK4/ARF genes. The recruitment of PRC1 and PRC2 to the INK4/ARF locus is mediated by the long noncoding RNA, ANRIL. ANRIL binds SUZ12 subunit of PRC2 to induce the Ezh2-mediated H3K27me3 and consequent silencing of the INK4/ARF locus. ANRIL binds the CBX7 subunit of PRC1, allowing the recognition of H3K27me3 necessary for the H2AK119ub1 and maintenance of silencing. Therefore, ANRIL transcription modulation affects the repressing ability of PcG proteins [33,34].
On the contrary, in the presence of stress signals, p16INK4a expression must be induced to prevent uncontrolled cell cycle progression. The expression of H3K27me3 demethylases of the Jumanji family proteins (JMJD3 and UTX) increases following oncogenic stimuli and removes the repressive histone marks from the p16INK4a promoter [29]. Jun dimerization protein 2 (JDP2) binds to H3K27, masking it from the actions of PRC2. Thus, JDP2 and JMJD3 work in concert to maintain a permissive state of p16INK4a expression. In this setting, PRC1 is not able to recognize the unmethylated H3K27, the chromatin in the region surrounding p16INK4a promoter becomes decondensed and accessible for transcription factor activity [29]. In addition, a cell cycle dysregulation may restrain the PRC2 activity since p53 activation represses EZH2 expression through pRb-mediated inhibition of E2F1 activity [31]. Thus, the homeostatic regulation of INK4/ARF transcription relies on the interplay balance between PRC complexes and histone demethylases.
The histone methyltransferase enzymes—The enzyme SET domain containing (lysine methyltransferase) 8 (Setd8)/lysine methyltransferase 5A (KMT5a) catalyzes mono-methylation of histone H4 at lysine 20 (H4K20me1) that characterizes transcriptionally active genes. Indeed, inhibition of Setd8 induces the reduction of H4K20me1 at several loci, such as INK4a/ARF, and, in turn, increases p16INK4a expression [35].
The enzyme, histone-lysine N-methyltransferase 2A (KMT2A) or myeloid/lymphoid or mixed-lineage leukemia 1 (MLL1), trimethylates the histone H3 at lysine 4 (H3K4me3), a mark associated with transcriptional activation. MLL1 and the E3 ubiquitin ligase DDB1-CUL4-ROC1 complex directly bind to the INK4a/ARF locus and mediate p16INK4a induction during replicative and oncogene-induced senescence, counteracting PRC-mediated repression [36].
Histone acetyltransferases (HATs) and histone deacetylases (HDACs)—Histone acetylation is a dynamic process controlled by two large families of enzymes with opposed activities: HATs and HDACs. HATs catalyze the acetylation of ε-amino groups of lysine residues within histone tails, facilitating chromatin relaxing and subsequent gene transactivation. The p16INK4a regulation mediated by the HAT p300 mainly depends on availability of several transcriptional co-factors. Indeed, the transcription factors SP1 and HMG box-containing protein 1 (HBP1), recruit p300 to the p16INK4a promoter. Both chromatin acetylation and HBP1 foster chromatin decondensation and subsequent gene transactivation [37]. p300 is also able to target the Myb-related protein B (B-Myb) promoter that encodes a p16INK4a repressor [24]. HDACs remove the acetyl groups and allow a tighter wrap of the histone to DNA that results in gene silencing. All HDACs have been reported to bind and repress transcription from the p16INK4a promoter. Most of these interactions depend on activity of transcription factors such as Lymphoid Specific Helicase (LSH), HLX1, ZBP-89, and YY1. For instance, HDAC3 and HDAC4 are recruited to p16INK4a promoter by YY1 to repress its expression. HDAC2 may also directly bind the p16INK4a promoter [38,39].
Chromatin remodelers—The multisubunit SWI/SNF family complexes contain SWI2/SNF2-like ATPases that hydrolize ATP to disrupt histone-DNA interactions within the nucleosome. This structural alteration results in an open chromatin state suitable for transcription machinery access [40]. As a subunit of the SWI/SNF complex physically interacts with JunB, the SWI/SNF complex may be directly recruited to the p16INK4a promoter to activate transcription. Moreover, SWI/SNF displaces PcG silencing complexes from p16INK4a promoter and reduces its DNA methylation [41]. Among SWI/SNF family, BRG/BRM-associated factor (BAF) complexes consist of one ATPase catalytic component (BRG1 or BRM) and several regulatory subunits called BAFs. BRG1 binds to p16INK4a and p16INK4a/BRG1 interaction negatively modulates the chromatin remodelling activity of BRG1 itself. However, BRG1 is not required for p16INK4a-induced cell cycle inhibition. Taken together, these findings indicate a putative feedback loop between p16INK4a and the SWI/SNF complex [42].
LSH, a member of the SNF2/helicase family, regulates DNA methylation and transcriptional silencing [43]. LSH operates by recruiting DNMTs (Myant and Stancheva, 2008), or through interaction with members of the PRC1 and HDACs [44]. Specifically, LSH represses p16INK4a expression by recruiting HDAC1 that deacetylates the H3 histone to establish a repressive chromatin structure at the p16INK4a promoter and delays cell senescence [45].
Nuclear architecture remodelers—The specialized AT-rich binding protein 1 (SATB1) is a genome organizer that interacts with AT-rich sequences and recruits chromatin-remodeling enzymes to regulate chromatin structure and gene expression [46]. Thus, depending on its posttranslational modifications, SATB1 activates or represses multiple genes. SATB1 may interact with pRB/E2F1 complex in gene regulation of p16INK4a promoter [47].
CCCTC-motif binding factor (CTCF)—CTCF is a zinc finger transcription factor that binds the INK4/ARF locus, regulating both chromatin compaction and gene expression. CTCF interaction with the chromosomal boundary is important for p16INK4a expression maintenance [48]. Indeed, deletion of CTCF resulted in hypermethylation of the p16INK4a promoter and, in turn, p16INK4a downregulation. Furthermore, CTCF inhibits epigenetic silencing through chromatin remodeling of the p16INK4a locus [49,50,51].
3.2.2. Transcriptional Regulation
The presence of several cell cycle genes grouped within the locus INK4a/ARF allow an overall regulation by the same chromatin remodelling event(s) following specific stimuli. However, a permissive chromatin state is necessary but not sufficient for gene expression. Indeed, transcription requires the binding of activation factors to specific enhancer and promoter elements and the subsequent recruitment of RNA polymerase. Transcription factors model the precise gene expression pattern required for specific functions, binding cooperatively to DNA in the form of cell-type-specific multiprotein complexes [24,25]. Transcriptional regulation of the p16INK4a gene is finely controlled by several transcription factor antagonistic pathways (Figure 1B).
Ets-binding site-mediated regulation—The E-box binding transcription factors E-26 transformation-specific (ETS) proteins (Ets1 and Ets2) are activated by the RAS-MAP kinase pathway following dangerous stimuli. Ets 1 and Ets 2 bind to p16INK4a gene and increase its transcription, leading to cellular senescence in fibroblasts [52]. To suppress p16INK4a overexpression, the Inhibitor of DNA Binding 1 (ID1), a helix loop helix (HLH) transcription factor, binds Ets transcription factors and inhibits their activity [52]. Thus, the balance between Ets1/2 and ID1 seems to act as a sensor of aberrant growth signals.
E-box-mediated regulation—E47 is a basic helix-loop-helix (bHLH) protein that homo- and heterodimerizes with other HLH proteins and binds two consensus sequences in the E-box element to activate p16INK4a expression. ID1 may influence the transactivation activity of E47 acting as dominant negative following heterodimerization. TWIST1 inhibits p16INK4a transcription by decreasing E47 expression [53]. Myc is an E-box-binding transcription factor that binds the p16INK4a promoter to upregulate its expression in human cells [54].
Y box-mediated regulation—Y box-binding protein 1 (YB-1) is a transcription factor that represses the p16INK4a transcription through direct association with its promoter region, resulting in the cell growth promotion and cellular senescence prevention [55].
Sp1-binding site-mediated regulation—The GC-rich region within the p16INK4a promoter contains at least five putative GC boxes that represent the putative binding target sites for Sp transcription factors [53,56,57]. A positive transcription regulatory element in the p16INK4a promoter harbors a GC box for Sp1 binding [56] that is enhanced during cellular senescence mainly due to an increase in Sp1 binding affinity. Sp1 positively regulates p16INK4a transcription by direct binding to DNA and recruiting P300 to the p16INK4a promoter [57].
HBP1-binding site-mediated regulation—p16INK4a promoter contains a HBP1 binding site. This is a downstream effector in the Ras/MAPK signaling pathway positively regulating p16INK4a transcription by direct binding to DNA and recruiting P300 to its promoter [37]. Of note, HBP1 also represses the DNMT1 gene, resulting in both p16INK4a-specific and global DNA hypomethylation changes [58].
INK4a transcription silence element (ITSE)-mediated regulation—The p16INK4a promoter harbors a negative regulatory element that contains a binding site for B-Myb, a transcription factor involved in the regulation of cell survival, proliferation, and differentiation [59].
Ap1 site-mediated regulation—AP1 proteins are homodimers and heterodimers composed of basic region-leucine zipper (bZIP) proteins, including Jun proteins (c-Jun, JunB, JunD), Fos proteins (c-Fos, FosB, Fra-1 and Fra-2), Jun dimerization partners (JDP1 and JDP2), and the closely related activating transcription factors (ATF2, LRF1/ATF3, and B-ATF). JunB activates p16INK4a transcription by binding to three identified AP1-like sites within the p16INK4a promoter. Conversely, c-Jun acts as a JunB antagonist, downregulating p16INK4a expression. Interestingly, p16INK4a is able to bind to JNKs and inhibit c-Jun phosphorylation and AP-1 activity [24,60].
Peroxisome proliferator response element (PPRE)—Peroxisome proliferator-activated receptor alpha (PPARα) specifically binds to the canonical PPRE region of the p16INK4a promoter and interacts with Sp1, enhancing p16INK4a expression. PPARγ phosphorylation represses its transactivation function [61].
4. p16INK4a and Epidermal Homeostasis
A key requirement for SC maintenance throughout life is the p16INK4a repression [20]. Indeed, its increase may be considered a barrier to SC pluripotency. For instance, during reprogramming of induced pluripotent SCs (iPS), the silencing of INK4/ARF gene transcription is associated with SC marker induction [25].
4.1. The p16INK4a/pRb Pathway in Epidermal Homeostasis
p16INK4a expression is undetectable during embryogenesis and in young tissues. In human epidermis, p16INK4a is undetectable in primary human keratinocyte cultures from young donors [17,18,19]. p16INK4a is a key regulator of clonal conversion and its inactivation in epidermal SCs is necessary for maintaining their stemness [18]. When p16INK4a is repressed, CDK4-cyclin D1complex is in the active state and allows E2F-mediated transcription of gene involved in cell cycle progression through inhibition of pRb. Indeed, the CDK4-cyclin D1complex regulates keratinocyte proliferation, but is not implicated in calcium-induced keratinocyte differentiation [62,63,64]. Specifically, E2F1 maintains primary human keratinocytes in a proliferative and undifferentiated state. During differentiation, E2F1 levels decrease. Moreover, E2Fs become upregulated after wounding. Specifically, E2F1 and E2F2 are expressed in all layers of proliferating keratinocytes and in migrating cells at the wound margin [63]. Activation of E2F transcription results in increased transcription of DNMT1 that, in turn, methylates p16INK4a promoter [23].
INK4/ARF locus activation plays a limited role in skin development and homeostasis [65]. However, deregulation of epigenetic pathways regulating epidermal homeostasis, induces p16INK4a expression [66,67,68,69,70,71]. Thus, p16INK4a may be considered a master sensor of aberrant chromatin status that rapidly drives cell cycle exit, inhibiting CDK4-cyclin D1 complex activity [72]. The pRb and E2F family members modulate genes involved either in cell cycle exit and differentiation. The three pRb family proteins are expressed in human epidermis. Specifically, pRb and p107 proteins are expressed in all layers, whereas p130 is restricted to suprabasal keratinocytes, suggesting a different coordinated role during epidermal differentiation. pRb in conjunction with p107 plays a central role in regulating epidermal homeostasis. pRb is essential for the maintenance of the terminally differentiated state, preventing cell cycle re-entry, and p107 compensates for the effects of Rb loss [73]. Rb proteins modulate the epidermal homeostasis by differential interaction with E2Fs. Indeed, E2F family members are divided into two groups: activators (E2F1-E2F3a) and repressors (E2F3b-E2F8). E2F1 and E2F4 appeared to exert opposite functions in cell differentiation [74]. Beyond its role as cell cycle repressor, E2F4 regulates expression of several genes involved in cell fate decisions [63,75,76]. To regulate differentiation, apoptosis, or senescence, pRb-E2F complexes can also recruit co-repressors/activators (e.g., HDAC1, HDAC2, DNMT1, HATs, BRG1) to regulate chromatin structure and transcription [76].
4.2. The p16INK4 Epigenetic Modulators and Transcription Factors in Epidermal Homeostasis
Multiple p16INK4a epigenetic regulators (described in paragraph 3) cooperatively modulate the balancing between epidermal SC maintenance and differentiation (Figure 2). Specifically, some epigenetic enzymes may preserve stemness and promote proliferation by repressing p16INK4a itself and other cell-cycle inhibitor transcription, as well as inhibiting unscheduled activation of non-lineage- or terminal differentiation-associated genes (e.g., DNMT1, HDAC1/2, PCR1, and PCR2 subunits). On the contrary, the p16INK4a transcription activators may also promote keratinocyte terminal differentiation acting on epidermal differentiation complex (EDC) genes (e.g., JMJD3, BRG1, SATB1) [28,29,38,77,78,79].
DNA methylation is relevant for tissue homeostasis as it provides functional variability to maintain the equilibrium between SC proliferation and differentiation. In skin SCs, the differentiation gene promoters are methylated and lose this repressive mark following cell fate commitment. Indeed, DNMT1 is mainly expressed in the epidermal basal layer, and decreases with keratinocyte differentiation [66,80]. Conditional ablation of DNMT1 in mouse epidermis results in marked de-repression of p16INK4a and differentiation genes. Epidermis becomes thickened, likely due to aberrant basal cell differentiation [80]. The lack of DNMT1 in human epidermal progenitor cells leads to abnormal induction of cell cycle regulator genes, such as p16INK4a, and loss of the tissue self-renewal capacity. However, depletion of DNMT1 in human keratinocytes is not sufficient to induce the loss of cell identity. Thus, DNMT1 displays a critical role in maintaining epidermal progenitor cells and tissue renewal [66]. Genes associated with cell proliferation and self-renewal are repressed by de novo methylation in differentiated cells [81]. Of note, 5-hmC is very low in SCs and differentiated keratinocytes, suggesting that TET enzymes might not play a key role in skin homeostasis [82].
The transcriptional repressor PcG proteins are essential to control SC identity and proliferation by preventing unscheduled induction of non-lineage and differentiation genes. Indeed, in keratinocyte SCs, non-epidermal and differentiation genes are H3K27me3-repressed whereas this repressive mark is lost during cell differentiation. In committed cells, stemness genes display high levels of H3K27me3 mark [29]. The role of the PRC2 complex in skin development and homeostasis has been investigated by loss-of-function studies. Knocking out both Ezh1/2 subunits in embryonic epidermal progenitors is necessary for the complete ablation of PRC2 complex activity and subsequent loss of the H3K27me3 mark. PRC-mediated repression maintains progenitor cells by repressing both suprabasal and Merkel cell lineages. Specifically, it prevents binding of AP-1 to its target genes, such as EDC genes, and represses the SOX2 gene required for Merkel cell specification. Moreover, PRC-mediated repression plays a key role in skin homeostasis: Ezh1/2 loss triggers the activation of INK4a/ARF locus, impairing proliferation and hair follicle development [68,69,83]. Loss of function of EED or Suz12 has similar skin phenotypes to Ezh1/2 loss. Thus, in the epidermis EED, Suz12 and Ezh1/2 function mainly as subunits of the PRC2 complex, although these subunits may act independently of the PRC2 complex [84]. Jarid2, a PRC2 ancillary protein, is necessary to maintain scheduled proliferation in epidermal homeostasis. Jarid2 loss leads to cell proliferation reduction and differentiation increase in progenitors, without affecting epidermal development, and hair growth delay [85].
PRC1 functions have been investigated in human keratinocytes and the most studied proteins are the PCGF4 subunit/Bmi-1 and Cbx4. Bmi-1-mediated regulation of p16INK4a expression is required for proper development, SC maintenance, and homeostasis in several tissues [86]. However, Bmi-1 is expressed in several epidermal layers, although a decrease expression gradient exists from basal to upper layers [87,88,89,90]. Of note, Bmi-1 is localized in the nucleus of proliferating keratinocytes in culture [18,90,91] and in the cytoplasm of senescent keratinocytes [18,91], suggesting that irreversible growth arrest and terminal differentiation are associated with a decrease of Bmi-1 expression and cytoplasmic distribution. Bmi-1 promotes human keratinocyte survival preventing apoptosis [90]. Bmi-1 and other PcG protein expression inversely correlate with keratinocyte clonogenic and proliferative potential even though these proteins cannot be strictly considered SC-specific proteins in the epidermis [19,69,90]. Human epidermal SC moves from a slow-cycling to an actively-proliferating status to ensure normal homeostasis. Although these states differ in their cell cycle profile, in both cases differentiation and senescence processes have to be blocked. Cbx4 uniquely possesses, among PRC1-associated Cbx subunits, both PRC1 chromodomain- and Small Ubiquitin-like Modifier (SUMO) E3 ligase-dependent activities [92]. Cbx4 ablation in mice demonstrates that it maintains epithelial identity by repressing non-epidermal lineage genes and inhibits senescence by repressing the INK4a/ARF locus in epidermal progenitor cells through its canonical PRC1 activity. On the other hand, it controls both proliferation of basal keratinocytes and inhibition of their premature differentiation by its SUMO E3 ligase activity [93]. In human epidermal SCs, Cbx4 displays an anti-senescent function by its PRC1 activity whereas inhibits SC activation and differentiation through its SUMO E3ligase activity. These findings provide evidence that differentiation and senescence are independent processes in human epidermis. Specifically, Cbx4 promotes a slow-cycling state of SCs, preventing their differentiation and senescence through repression of Bmi-1, DNMT1, and Ezh2 and, in turn, the transition to the active SC state [70]. The removal of H3K27me mark during cell differentiation is catalyzed by JMJD3. Indeed, human keratinocytes expressing JMJD3 exhibit premature differentiation whereas its loss impairs differentiation [67].
HDAC1 and HDAC2 play a critical role in the gene expression control of the epidermal progenitor cells and epidermal differentiation. They are expressed in all epidermal layers but display a higher nuclear expression level in terminally differentiated cells [71]. Conditional ablation of either HDAC1 or HDAC2 does not disrupt epidermal homeostasis [94]. However, a double knock-out HDAC1/2 mouse model displays that deficient epidermis upregulates p21 and p16INK4a, fails to differentiate and remains in single-layer, similarly to p63-null mice. Thus, HDAC1/2 mediates repressive functions of p63 in epidermal development, and suppress p53 activity [71]. Moreover, SCs of interfollicular epidermis and hair follicle exhibit hypoacethylated histone H4. When cells exit from the quiescent state to proliferate and differentiate, they display an increased acetylation level [95]. Of note, the histone deacetylase inhibitor Trichostatin A (TSA) induces premature senescence in primary human keratinocytes with concomitant Bmi-1 and Ets-1 reduction and p16INK4a increase [19], as well as terminal differentiation [96].
Altogether, these data indicate that DNMT1, PRC1, PRC2, and HDAC proteins work in concert to protect epidermal cells from senescence by repressing p16INK4a expression and preventing differentiation. Furthermore, balancing these enzymatic activities regulates the transition between epidermal SC quiescence and activation.
These epigenetic mechanisms may be regulated by specific transcription factors, such as p63, which is considered a master regulator of epidermal morphogenesis and SC maintenance [97,98]. It is regulated by the chromatin remodeler Setd8 and has an important role in transcription and higher-order chromatin remodeling through its direct targets—HBP1, LSH, SATB1, and BRG1—which also modulate p16INK4a expression. Setd8 is a Myc target and is weakly expressed in skin. Deletion of Setd8 during embryogenesis results in p63 loss and a failure to form the epidermis. Setd8 activity is mainly present in dividing basal progenitor cells and is required for normal tissue homeostasis [35]. Indeed, inducible and conditional ablation of Setd8 in the epidermal basal layer results in cell-cycle arrest, apoptosis of progenitor cells and disruption of skin homeostasis. Thus, Setd8 is required for epidermal progenitor cell survival by inducing p63 and repressing p53 levels [99]. In the epidermal basal layer, p63 directly represses HBP1 and, in turn, p16INK4a expression. HBP1 is also required for differentiation and stratification processes [100]. Moreover, the chromatin remodeler LSH is a direct target of both p63 [101] and DNMT1 in primary keratinocytes. Thus, p63 and DNMT1 may cooperate to repress p16INK4a transcription and maintain basal progenitor proliferation. The p63-target SATB1 plays a key role in epidermal morphogenesis, controlling tissue-specific chromatin remodeling and p16INK4a repression in epidermal progenitor cells [102,103]. p63 directly regulates the expression of the SWI/SNF complex BRG1 that displays a critical role in skin homeostasis. It remodels the higher-order chromatin structure of the EDC for the efficient expression of differentiation genes in epidermal progenitor cells [104]. BRG1 is expressed in all epidermal layers but its activity is restricted to suprabasal layers and BRG1 deficiency leads to defects in terminal differentiation accompanied by the formation of non-functional epidermal barrier [104,105]. Indeed, the ACTL6a subunit, which binds the SWI/SNF complex, is differentially expressed and regulates epidermal differentiation [106]. The remodeler CTFC is mainly expressed in the nuclei of suprabasal keratinocytes and its expression diminishes in the more differentiated layers of the epidermis according to its function to limit cell growth [107].
Several p16INK4a promoter transcription factor regulators (described in paragraph 3) may have a role in epidermal proliferation and differentiation. Ets1 is expressed in the epidermal basal layer, blocks keratinocyte terminal differentiation, and induces expression of matrix metalloproteases and innate immune mediators [108]. Ets1 transgene induction in basal layer of mouse epidermis results in tissue hyperplasia and impaired differentiation. Indeed, Ets1 disrupts Notch signalling in part through p63 upregulation [109]. Ets2 is highly expressed in differentiated keratinocytes. The p16INK4a repressor Id-1, Id-2, and Id-3 proteins are expressed in proliferating human keratinocytes and downregulated in differentiated cells. Of note, a cytoplasmic Id-1 expression and nuclear Id-2 and Id-3 expression has been observed in the epidermal basal layer [110]. The p16INK4a-negative regulator B-Myb plays an important role in maintaining the undifferentiated phenotype of keratinocytes in the basal epidermal layer [111]. The ubiquitously expressed Sp1 and AP-1 (c-Jun, JunB, JunD, c-fos, FosB, Fra-1, and Fra-2) transcription factors are expressed at different levels in multiple epidermal layers and regulate competing processes in the epidermis (proliferation, survival, and differentiation) to maintain physiological homeostasis [112]. Of note, Ezh2 controls the proliferative potential of epidermal progenitors by repressing the INK4A/ARF locus and the terminal differentiation by preventing the recruitment of AP-1 to promoters of genes governing barrier formation [69]. Specifically, Fra-2 is methylated on lysine residues by Ezh2 in basal keratinocytes to repress EDC genes [113]. PPARα is an important regulator of epidermal development and homeostasis as it is involved in controlling the switch from proliferating basal to differentiating suprabasal keratinocytes [114].
5. p16INK4a and Epidermal Aging
Aging is a complex process characterized by accumulation of macromolecular damage and compromised tissue renewal. Such decrease of tissue regenerative capacity is not necessarily due to a SC-number or self-renewal decline but rather to a reduced ability to produce progenitors and, in turn, differentiated effector cells [115]. Epidermal mouse SCs isolated from young and old mice do not display substantial difference in number and gene expression pattern [116,117,118]. Therefore, the epidermis is able to maintain the barrier properties for the entire organismal life-span although the tissue regenerates and heals more slowly. Of note, epidermal grafts from older individuals allow a permanent coverage of burnt skin [12,119]. Experiments in old mice show that the slow regeneration rate is likely due to changes in TA-cell kinetics following damage accumulation. This leads to a decrease in the cellular output of TA cells and a decrease in the rate of TA cell proliferation. However, the number of TA cells increases in aged epidermis likely as they progress more slowly during the cell cycle than young TA cells [117,120].
One aging hallmark is cellular senescence since the number of senescent cells increases in tissues during organismal lifespan. Cellular senescence is triggered by several different intrinsic and extrinsic stimuli that induce growth arrest and distinctive phenotypic alterations, including considerable chromatin and secretome changes. In young organisms, cellular senescence prevents the uncontrolled proliferation of damaged cells. However, in old organisms the continuous and cumulative damages and the deficient clearance of senescent cells results in their accumulation with deleterious effects on tissue homeostasis [121,122,123].
5.1. The p16INK4a/pRb Pathway in Epidermal Aging
p16INK4a has a well-established role in mediating and maintaining cellular senescence during both replicative and stress-induced senescence processes. Thus, it may be considered an effector of aging [122,123]. Indeed, the expression of p16INK4a is undetectable in unstressed and healthy tissues of young mammals [124,125]. p16INK4a accumulates during tissue aging and therefore is considered one of the most robust aging biomarkers characterized to date [124,125,126,127]. Specifically, data from progeroid and calorically restricted rodents suggest that p16INK4a may be a marker of biological rather than chronological aging [125,128]. Similarly, smoking and chemotherapy are associated with elevated p16INK4a expression in humans [129,130]. Assessing its effect on aging has been difficult since p16INK4a-null mice are tumor-prone [65]. However, in mouse models with conditional expression of p16INK4a it causes a decline of regenerative capacity of hematopoietic, neural SC and intestinal cells of young adult mice, rapidly inducing several features of premature aging. De-induction of p16INK4a revealed that these features are strikingly reversible [124,127,131]. Inducible elimination of p16INK4a-positive senescent cells in a progeroid mouse model delays the onset of premature aging phenotypes in different tissues, such as adipose tissue, skeletal muscle, and eyes; attenuates the progression of already present age-related disorders; and extends healthspan [128].
p16INK4a expression directly correlates with chronological aging of human skin in vivo. The number of p16INK4a positive cells increases with age in both epidermal and dermal compartments [87]. Furthermore, the number of p16INK4a positive cells in human skin is also a marker of biological age in middle-aged individuals with longevity propensity compared to their age-matched partners [132]. A strong positive correlation between paraclone increase and p16INK4a expression at first culture passage has been observed in human keratinocyte from elderly donors [19]. p16INK4a levels increase in the epidermal basal layer in the aged model of skin equivalent in concomitance with a proliferation marker decrease. In genetically-modified skin equivalents, p16INK4a overexpression in cultures from young donors results in a decrease of proliferation markers whereas p16INK4a silencing results in morphology improvement and proliferation markers restoration in cultures from old donors [18,133].
5.2. The p16INK4 Epigenetic Modulators and Transcription Factors in Epidermal Aging
An additional common age marker is genomic and epigenomic instability due to damage accumulation throughout life. Alterations in DNA methylation or histone acetylation/methylation, as well as other chromatin-associated protein modification, can induce epigenetic changes [121]. Thus, epidermal aging may depend on the impairment of epigenetic factor activity on p16INK4a promoter regulation that represses the INK4a/ARF locus. Indeed, oxidative stress induces demethylation of p16INK4a promoter and, in turn, human keratinocyte senescence [134]. The expression of PcG proteins declines in aged epidermis and their decrease is associated with keratinocyte senescence both in vivo and in cell culture models [19,87]. PRC2 expression gradually decreases with the age of basal epidermal progenitors. Ezh2 decrease leads to reduction of H3K27 trimethylation levels, disassociation of PRC1 from the chromatin and increased transcription of INK4a/ARF genes [78]. Indeed, early expression of p16INK4a in human keratinocytes from old donors strongly correlates with Bmi-1 downregulation. What is more, Bmi-1 modulates both p16INK4a levels and clonal conversion in primary human keratinocytes from elderly donors. Specifically, Bmi-1 overexpression inhibits p16INK4a-promoter activity and decreases the protein expression. These Bmi-1 overexpressing keratinocyte cultures display a delay of cell senescence and a restoration of keratinocyte clonal ability, showing a phenotype similar to that observed in age-matched healthy control subjects [19]. Of note, pRb seems necessary for Bmi-1 function in the transcription repression of p16INK4a, since the deletion of pRb from cells resulted in histone H3K27 trimethylation loss that impairs the Bmi-1 recruitment to the p16INK4a locus [135]. The enzyme MLL1 is repressed by PRC complexes during proliferation of young cells and mediates p16INK4a induction during replicative and premature senescence [36]. The chromatin remodeler Setd8 is downregulated in both oncogene-induced and replicative senescence and inhibition of Setd8 alone is sufficient to trigger senescence [35]. Deficiency of its target, p63, induces cellular senescence in human proliferating keratinocytes [136] and leads to accelerated skin aging in p63 heterozygous mice [137]. Also, the p63-targets LSH and SATB1 play a key role in senescence and aging. Indeed, LSH-deficient mice exhibit accelerated aging and enhanced cellular senescence [138]. E2F1 regulates LSH transcription, therefore its repression during senescence decreases LSH expression [139]. SATB1 expression decreases during human keratinocyte replicative senescence and its silencing is able to induce a G1 cell cycle arrest concomitant with an increase of senescence-associated markers, such as 16INK4a [103]. Of note, 16INK4a and the pRB/E2F pathway are critical for SATB1-induced cell cycle arrest. Indeed, SATB1 causes anchorage-independent growth and invasive phenotype in 16INK4a deficient cells [47]. The chromatin remodeler BRG1 may drive SAHF formation via its chromatin remodeling activity and/or by upregulating p16INK4a expression. During senescence BRG1 is overexpressed and interacts with pRB to drive SAHF formation. On the other hand, this interaction is dispensable for upregulating p16INK4a and p21 by BRG1, which depends upon its chromatin remodeling activity [42]. Interestingly, age-related metabolic pathologies are associated with PPARs activation that induces the 16INK4a expression and leads to subsequent cell cycle arrest and senescence [61].
Following acute stress stimuli, the Ras/MAPK pathway induces Ets1 and HBP1 upregulation that triggers premature senescence via 16INK4a transcription [52,140]. The age-related increase of 16INK4a expression in mouse and rat tissues has been attributed mainly to Ets-1 expression [125]. Moreover, the marked increase in 16INK4a levels observed in senescent human fibroblasts is consistent with the reciprocal reduction of Id-1 and accumulation of Ets-1 [52]. However, although Id-1 overexpression is able to extend the keratinocytes’ lifespan, it does not prevent the onset of replicative senescence [141,142]. Despite the ability of Ets-1 and Id-1 to modulate the 16INK4a promoter in primary human keratinocytes, no correlation between their expression and 16INK4a expression or SC depletion during skin chronological aging has been detected [19]. Ras/MAPK pathway also promotes senescence through the activation of JMJD3 and down regulation of Ezh2 that, in turn, cooperatively activates 16INK4a transcription [143]. Moreover, Ras pathway induces ROS increase that inhibits DNMT1 expression through E2F inhibition. Thus, reduced methylation activity may derepress the 16INK4a promoter [23]. The 16INK4a-repressor B-Myb is regulated by cyclin D1. Thus, its decrease following stress may regulates entry into senescence [144]. During replicative senescence and upon RAS overexpression, ANRIL expression is suppressed. Thus, cell-cycle inhibitors p14ARF, p15INK4b, and 16INK4a are upregulated, likely by reduced interaction with PRC complexes, and senescence is triggered [23].
6. p16INK4a and Non-Melanoma Skin Cancers
Non-melanoma skin cancers (NMSCs) are the most frequent cancers in the Caucasian population. NMSCs, mainly squamous-cell carcinomas (SCCs) and basal-cell carcinomas (BCCs), originate from keratinocytes and are the most common age-associated malignancies [145,146]. The exponential increase of tumor frequency with age may be in part due to the presence of senescent cells next to preneoplastic cells. Senescent cells accumulate in skin and secrete several enzymes, which disrupt the cellular microenvironment, and many inflammatory cytokines and growth factors that can stimulate tumor cell growth. Thus, relatively few senescent cells might compromise skin function and integrity [123,147].
Cutaneous SCCs (cSCCs), especially less well-differentiated tumors, have metastatic potential. They represent an interfollicular epidermis neoplasia and may originate from premalignant actinic keratoses (AKs) and “in situ” SCC (ISSCCs). The most important alterations in cSCC etiology involve RAS, p53 and 16INK4a/pRb pathways [148]. BCCs rarely metastasize but are locally invasive and can be disfiguring. BCCs likely arise from hair follicle SCs and their etiology is highly dependent on Hedgehog signaling pathway dysregulation [149]. However, a coupled role of 16INK4a and Hedgehog signaling pathway is observed in the BCC pathogenesis [150].
Cells with dysfunctional 16INK4a display uncontrolled proliferation bypassing G1/S transition checkpoint. The 16INK4a gene is frequently mutated in human cancers [20] and mice lacking 16INK4a are prone to spontaneous or induced tumorigenesis [151,152]. However, studies performed on NMSCs led to controversial results about 16INK4a expression. Some reports correlated higher malignancy with 16INK4a loss [153,154] and others with 16INK4a overexpression [155,156,157,158,159]. Numerous mutations of 16INK4a have been found in AKs and cSCCs, such as homozygous gene deletions, hypermethylation of its promoter, and mutations in the INK4a/ARF locus. Loss of heterozygosity in at least one locus of the 9p21 region (containing the CDKN2A gene) is observed in 20% of AKs and 46% of cSCCs, suggesting that 16INK4a inactivation may have a critical role in skin lesion progression [160]. 16INK4a gene may be mutated in up to 24% of cSCCs and these mutations may be hereditary or acquired following UV-induced damage [156]. In BCC, 16INK4a may also be inactivated apart from loss of heterozygosity, promoter hypermethylation, and/or mutations in the INK4a/ARF locus [161]. On the contrary, several immunohistochemical studies report an increased 16INK4a expression in ISSCCs compared to AKs and in poor differentiated cSCC compared to well differentiated [157,159,162,163,164]. In premalignant AKs, 16INK4a expression correlates with a functional pRb pathway, suggesting that upregulation is the result of senescence induction following stress stimuli [165]. Human papillomavirus (HPV) is well established as a risk factor for oropharynx SCCs. Neverthless, improved survival is observed in patients with HPV-positive SCCs [166,167]. HPV oncoprotein E7 directly inactivates pRb [168], thus 16INK4a overexpression may be a compensatory feedback effect of the inability to control cell proliferation and has been suggested as marker for HPV-associated SCCs [169]. However, a subgroup of aggressive cSCCs displays high 16INK4a expression in the absence of HPV [165,170]. p16INK4a upregulation might occur as a response to pRb pathway dysfunction. Indeed, 16INK4a expression correlates with the lack of pRb phosphorylation in ISSCCs and invasive cSCCs [165]. Alteration in the pRb pathway can also induce an aberrant cytoplasmic localization of p16INK4a due to CDK4 sequestration [171]. Alternatively, the protein might be present in a mutated but inactive form [161,172]. Moreover, a strong 16INK4a expression is observed in BCC [173]. In highly invasive cSCC and BCC subtypes, p16INK4a is overexpressed at the infiltrative front followed by ceased proliferation, suggesting that p16INK4a could be involved in tumor invasion independent by proliferation effects. Indeed, 16INK4a is implicated in the regulation of matrix-dependent cell migration [165,174]. However, although the expression of p16INK4a and Ki67 is associated with both BCCs and cSCCs, their expression does not seem to correlate with the degree of proliferation and malignancy. Instead, p16INK4a overexpression is significantly associated with sun-exposed areas, suggesting a possible induction of p16INK4a overexpression by UV radiation [172]. Interestingly, UV exposure induces DNMT expression in the epidermis [175], suggesting that mainly histone modification might be the cause of p16INK4a expression.
6.1. Oncomine Analysis
Further analyses concerning expression of epigenetic modulators and transcription factors (described in paragraph 3) are needed to study p16INK4a expression in-depth in NMSCs, independently from HPV infection. Here, we examined the expression of 50 genes, including p16INK4, downstream pRb pathway proteins and p16INK4 promoter modulators, as reported in human skin cancer datasets available at Oncomine (www.oncomine.org) as previously described [176,177]. Two independent datasets (Nindl skin and Riker melanoma) [178,179] from epidermal cancers (AKs, cSCCs and BCCs) were investigated as indicated in details in Table 1 and Table 2. Gene expression in tissue biopsies from 45 patients was analyzed, namely, 10 control normal tissues and 35 cancer samples. Moreover, a comparative analysis on datasets from other epithelial cancers (75 samples from head and neck SCCs (HNSCCs) and 73 samples from oral cavity SCCs (oSCCs) vs. 43 normal samples) has been performed. Four independent datasets (Ginos Head-Neck, Cromer Head-Neck, Peng Head-Neck and Toruner Head-Neck) [180,181,182,183] from HNSCCs and from oSCCs were investigated as indicated in details in Table 1 and Table 3. Gene expression was evaluated by setting “Cancer vs. Normal analysis” and choosing as Cancer Type: “Actinic (Solar) Keratosis”, “Skin Squamous Cell Carcinoma”, “Skin Basal Cell Carcinoma”, “Head and Neck Squamous Cell Carcinoma”, and “Oral Squamous Cell Carcinoma”. Expression fold change (Cancer vs. Normal samples) and p values are reported for each analysis. We mainly focus the discussion on expression values showing fold changes >2 and statistical significance (p < 0.01).
6.1.1. p16INK4 Downregulation in Epithelial Tumors
A p16INK4 decrease is observed in one dataset from HNSCCs (Cromer dataset—Table 3). Accordingly, the cell cycle regulators CDK4 and Cyclin D1 and the pRb pathway effectors E2F1 and E2F4 are significantly upregulated in these samples. Cyclin D1 upregulation may also be a sign of Ras activation [184]. Of note, the expression of some p16INK4-regulators (Ets1/2, YB1, AP-1 proteins), which are downstream targets of the Ras pathway, is significantly increased (Cromer dataset—Table 3), suggesting that dysregulation of pRb and Ras pathways might work in concert to sustain tumor progression in these samples.
6.1.2. p16INK4 Upregulation in Epithelial Tumors
A p16INK4 increase is observed in NMSCs, HNSCCs (Ginos data set) and oral cavity SCCs (oSCCs) (Table 2 and Table 3). Information regarding patient HPV-positivity is not available from these datasets. In the Nindl dataset, skin biopsies are obtained from organ-transplanted patients with multiple NMSC lesions mainly located on sun-exposed areas. Of note, immunosuppressed patients display an increased HPV prevalence in cSCC compared to immunocompetent patients [185].
The p16INK4a/pRb Pathway in Epithelial Tumors
Different from AK samples, cSCCs, and BCCs display a significant increase of CDK4 expression as HNSCCs and oSCCs samples (Table 2 and Table 3). CDK4 is overexpressed in human cSCCs and is sufficient to induce epidermal tumorigenesis concomitant with oncogenic Ras expression [184]. In AKs and cSCCs, Cyclin D1 slightly increases (Table 2) as previously described [162]. Manojlovic-Gacic and colleagues show that Cyclin D1 is more expressed in ISSCC compared to AKs and in poor-differentiated cSCC compared to well-differentiated cSCC. Thus, the increase of its distribution through pre-invasive phases in cSCC development may reflect its role in the skin-lesion progression. Of note, the Cyclin D1 expression distribution correlates with that of 16INK4a [162]. In poor differentiated HNSCC samples, Cyclin D1 expression is significantly increased. Moreover, pRb is significantly upregulated in all datasets (Table 2 and Table 3). The Rb pathway is disrupted in most tumors. Deletion and inactivating mutations of pRb are restricted to very few specific cancer types. pRb absence in mouse epidermis is characterized by moderate hyperplasia and hyperkeratosis associated with increased proliferation and altered differentiation. However, no spontaneous tumor development was observed, even after a long latency, likely for the overlapping tumor suppressor roles of p107 and p130 [76]. Here, these proteins are slightly upregulated (Table 2 and Table 3). Interestingly, high pRb or combined pRb/CyclinD1 expression in HNSCCs are strong indicators for HPV-negative status [186], suggesting that the p16INK4a upregulation observed in these datasets may be independent of HPV infection. The pRb pathway effectors, E2F1 and E2F3, significantly increase in NMSCs. E2F2 displays a similar trend (Table 2 and Table 3), indicating a deregulation of this pathway. Indeed, amplification and increased expression of specific E2F members is frequent in NMSCs. Indeed, deregulation of E2F activity may be involved in the unregulated proliferation of skin tumor cells [187]. In HNSCC samples also E2F4 displays a significant increase (Table 3).
The p16INK4 Epigenetic Modulators and Transcription Factors in Epithelial Tumors
Epigenetic modulators and transcription factors are dysregulated in cancers. However, poor data concerning modulation of epigenetic enzymes have been collected from skin tumors.
Analyzing these datasets, DNMT1 appears to be upregulated in cSCCs, BCCs, HNSCCs, and oSCCs. DNMT3A is mainly increased in BCCs whereas DNMT3B is upregulated in AKs, cSCCs, HNSCCs, and oSCCs (Table 2 and Table 3). An increase of three DNMT isoforms has been previously observed in BCCs where they contribute to hypermethylation and silencing of p16INK4a. This hypermethylation is accompanied by increased occupancy by MBD1, MeCP2, and HDACs [175]. Thus, other epigenetic and transcription factors might be responsible for p16INK4a upregulation in analyzed specimens.
Among PRC subunits, Ezh2, Bmi-1, and EED expression significantly increases in analyzed specimens (Table 2 and Table 3). Due to their role in cell cycle control and apoptosis, the PcG proteins Ezh2 and Bmi-1 are important epigenetic regulators enhancing skin cancer cell survival. Ezh2 expression increases from normal skin to AKs and further to cSCCs, suggesting a role in cSCC initiation and progression. Bmi-1 is highly expressed in both BCCs and cSCCs [88,89]. However, a strong Bmi-1 overexpression is more frequently observed in BCCs than in cSCCs and AKs [188]. In our analysis, Bmi-1 expression is significantly increased in BCCs and SCCs from one dataset (Table 2). Of note, Hedgehog pathway dysregulation upregulates Bmi-1, suggesting its critical role in BCC pathogenesis. Moreover, Bmi-1 overexpression in immortalized HaCaT cells causes malignant transformation both in vitro and in vivo [189]. Interestingly, a human cSCC cell subpopulation possesses SC-like features, has enriched expression of Ezh2, Bmi-1, and trimethylated histone H3, and display enhanced ability to drive tumor formation [190]. Specifically, Ezh2 is required for survival of these cells and tumor formation [191]. Of note, Bmi-1 overexpression restores Ezh2 levels in Epigallocatechin-3-gallate (i.e., the active agent in green tea)-treated cSCC cells, suggesting a feedback regulation to balance of PRC1 and PRC2 complex protein expression in cells [89]. Chemopreventive agents suppress proliferation by cyclin inhibition and/or induce apoptosis in cSCC cells. However, these effects are reversed by forced Bmi-1 expression [89,90]. Among the other PRC subunits, CBX4 expression is unaltered, whereas CBX7 is globally downregulated in this dataset analysis. The PRC2 ancillary protein Jarid2 significantly increases mainly in cSCCs. Of note, JMJD3 globally increases in NMSCs, whereas JDP2 is upregulated only in BCCs (Table 2 and Table 3).
MLL1 (KMT2A) expression is significantly increased in NMSCs (Table 2). Indeed, multiple mutations and copy number variations of MML1 have been found in SCCs [192].
The p63 activator Setd8 is upregulated only in SCCs. According to literature, p63 expression is significantly over-expressed in SCCs, BCCs, HNSCCs, and oSCCs (Table 2 and Table 3). Indeed, it is abundantly expressed in cSCCs and favors tumor initiation and progression [193]. For instance, p63 targets LSH to drive skin tumorigenesis [101]. Here, we find a strong and significant upregulation of LSH in NMSCs, observed also in HNSCCs and oSSCs (Table 2 and Table 3).
The HAT p300 is mainly upregulated in NMSCs whereas the HDACs are globally upregulated in all analyzed pathologies (Table 2 and Table 3). The p300 increase plays an important role in the development and progression of cSCC [194]. Moreover, high p300 expression correlates with aggressive features of cSCC, suggesting that p300 may be a promising biomarker to predict clinical outcomes [195]. Overexpression of the HDACs is frequent in oral cancer. Indeed, ΔNp63α/HDAC complex is necessary for tumor maintenance in SCCs as it serves as a direct repressor of the apoptotic transcriptional program [196].
Among the other p63 targets, BRG1 is significantly upregulated in NMSC specimens, differently from HNSCCs and oSCCs (Table 2 and Table 3). BRG1 is upregulated in esophageal SCCs and is necessary for matrix metalloproteinases expression [197]. Interestingly, SATB1 is slightly downregulated in all analyzed pathologies (Oncomine results). Of note, high SATB1 expression is predictive of poor prognosis in oral, laryngeal, and esophageal SCCs [198,199,200]. HBP1 is upregulated in cSCCs and BCCs differently from HNSCCs and oSCCs (Table 2 and Table 3). The differential expression of BRG1, Satb1, HBP1, and LSH might be responsible for p16INK4 upregulation in cSCCs and BCCs.
B-myb is slightly upregulated in cSCCs and downregulated in the other specimens. CTFC expression is significantly increased in BCCs and cSCCs from one dataset (Table 2 and Table 3).
Ets transcription factors are upregulated in cSCCs, HNSCCs, and oSCCs from one dataset (Oncomine results). Ets2 is a major driver of SCCs. Ets2 binds to the super-enhancers and reprograms gene expression to induce inflammation and promote the tumor growth and development [201]. The p16INK4 repressor Id1 is significantly upregulated in AKs, cSCCs, HNSCCs, and oSCCs whereas Id2 and Id3 increase only in AKs and HNSCCs (Table 2 and Table 3). In cSCCs, Id protein expression increase is observed in the majority of malignant poorly differentiated tumors compared to these well-differentiated ones. It might contribute to the escape of the relatively undifferentiated tumor cells in BCC from immune surveillance [110,202,203]. Id1 protects keratinocytes from apoptosis via the NF-kB/survivin and phosphoinositide 3-kinase/Akt signaling pathways [203].
The transcription factor AP-1 promotes the invasive growth and metastasis of various tumors such as SCCs [204]. AP-1 is composed of a mixture of homo- and hetero-dimers formed between JUN and FOS proteins that may have both opposite and overlapping functions in cellular proliferation and cell fate. Thus, we analyzed the expression of single proteins. JunB and JunD slightly increased in HNSCCs, AKs, and cSCCs from one dataset and downregulated in BCCs and SCCs of the other one. c-JUN is significantly increased in HNSCCs, AKs and cSCCs/oSCCs from one dataset (Table 2 and Table 3). JunB and JunD are negative regulators of the cell cycle, are not induced in a majority of SCC cells and inhibit the Ras-driven tumorigenesis. JunB directly induces p16INK4a expression and epidermal senescence. However, the dominant-negative JunB mutant promotes tumorigenesis [60,205]. Recently, JunB has been considered a key molecule in promoting cell invasion, migration, and distant metastasis in head and neck SCCs [206]. On the contrary, c-Jun is a positive regulator of cell cycle, is activated in SCCs and its coexpression with oncogenic Ras is sufficient to transform primary human epidermal cells into malignancy in a regenerated human skin grafting model [60,205].
c-Fos and FosB are downregulated in BCCs, cSCCs, and oSCC whereas they are strongly upregulated in HNSCCs. FRA1 and FRA2 are mainly upregulated in AKs and cSCCs (Table 2 and Table 3). Federico and colleagues do not find differential expression of c-Fos or FosB between tumors and normal tissues. On the contrary, a strong Fra-1 and Fra-2 expression is observed in those samples [207]. c-Fos is found in the specimens of patients with lymph node metastasis and is downregulated in poorly differentiated SCCs [208,209]. Moreover, c-Fos is a regulator of EMT and cancer stem cell reprogramming in head and neck SCC cells [210]. Fra-1 is highly expressed in SCCs, promotes tumor growth through the AKT pathway and enhances cell migration through JNK/c-Jun [211].
Clinical studies define ANRIL as a potential diagnostic and prognostic biomarkers in various cancers [212]. ANRIL can be considered an oncogene since it promotes cancer progression via increasing proliferation, reprogramming cell glucose metabolism and inducing CSCs [212,213]. Inhibition of ANRIL suppressed the cancer cell proliferation, migration, and invasion [213]. ANRIL has been found dysregulated in BCC [214]. However, in our analysis, ANRIL does not significantly vary in skin tumors and increases only in oSCCs from one dataset (Table 2 and Table 3).
In these specimens, p16INK4 upregulation seems a compensatory mechanism to suppress tumor development by senescence. However, senescence of the non-epithelial component of developing skin tumours might enhance growth and invasion of the pre-malignant epidermal cells via other upregulated pathways, such as p63 and Ras. For instance, p63 is induced by oxidative stress. In the analyzed cutaneous tumors p63 targets (BRG1, Satb1, LSH) are globally upregulated. Ras is a transducer of extracellular mitogenic signals. Of note, in all analyzed tumors Ras targets (Cyclin D1, Ets1/2, HBP1, YB1, AP-1 proteins) are upregulated. Moreover, some of these proteins are also able to induce p16INK4 expression to block uncontrolled growth.
As new data sets are deposited, it will be possible to analyze larger data sets and get more in-depth information.
7. Conclusions
p16INK4a is a potent inhibitor of the G1/S-phase transition of the cell cycle and exhibits a fundamental role in epidermal homeostasis, aging, and tumorigenesis.
Keratinocytes are equipped with several enzymes that shape the epigenetic signatures of different epidermal layers, allowing a dynamic and coordinated expression change of gene sets involved in self-renewal and differentiation. In the basal layer, epigenetic modifiers and transcription factors converge their activity to repress p16INK4a expression, protecting cells from senescence and preserving epidermal homeostasis and regeneration. Several stress stimuli may activate p16INK4a expression that orchestrates cell cycle exit and senescence response. Cellular senescence is a potent tumor suppressor mechanism, acting in coordination with the immune system to clear potentially malignant cells from the tissues. Thus, it may contribute to aging by depleting functional cells required to maintain tissue homeostasis. However, aging and cancer might be considered related endpoints of accumulating damages within self-renewing compartment. Indeed, age-associated changes in chromatin remodeling and gene expression are responsible for the deterioration of multiple cellular functions, including the senescence response. Accumulation of p16INK4a positive senescent cells in the aged tissue may be due to synergic contribution of the genomic instability, senescence-associated secretory phenotype dysregulation, and decline in the immune system function, and may contribute to worsening the senescence response efficacy in tumor suppression.
Unveiling the precise mechanisms regulating p16INK4a expression in human epidermal cell types may provide new targeted approaches to protect the epidermis from regenerative capacity decline and premature aging and new protocols for regenerative medicine. However, balancing the expression of p16INK4a and its regulators has to be carefully tuned to avoid the risk of malignant transformation, given that the response to oncogenic stimuli changes at advanced ages. Indeed, better understanding of p16INK4 regulation in skin tumors may be relevant to standardizing new diagnostic or prognostic tools and in designing new cancer therapies.
Acknowledgments
This work was supported by grants from the Italian Ministry of Health (RC17-4.3 and RC17-4.5).
Author Contributions
Daniela D’Arcangelo and Elena Dellambra conceived and designed the review; Daniela D’Arcangelo, Lavinia Tinaburri, Elena Dellambra performed the bibliographic research; Daniela D’Arcangelo analyzed the Oncomine data; Elena Dellambra wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fuchs, E. Chapter nineteen—Epithelial skin biology: Three decades of developmental biology, a hundred questions answered and a thousand new ones to address. Curr. Top. Dev. Biol. 2016, 116, 357–374. [Google Scholar] [PubMed]
- Mascré, G.; Dekoninck, S.; Drogat, B.; Youssef, K.K.; Brohée, S.; Sotiropoulou, P.A.; Simons, B.D.; Blanpain, C. Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature 2012, 489, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Potten, C.S. The epidermal proliferative unit: The possible role of the central basal cell. Cell Tissue Kinet. 1974, 7, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Ghazizadeh, S.; Taichman, L.B. Multiple classes of stem cells in cutaneous epithelium: A lineage analysis of adult mouse skin. EMBO J. 2001, 20, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Clayton, E.; Doupé, D.P.; Klein, A.M.; Winton, D.J.; Simons, B.D.; Jones, P.H. A single type of progenitor cell maintains normal epidermis. Nature 2007, 446, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Doupé, D.P.; Klein, A.M.; Simons, B.D.; Jones, P.H. The ordered architecture of murine ear epidermis is maintained by progenitor cells with random fate. Dev. Cell 2010, 18, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M.; Haase, I.; Nestle, F.O. Mechanisms regulating skin immunity and inflammation. Nat. Rev. Immunol. 2014, 14, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Kolodka, T.M.; Garlick, J.A.; Taichman, L.B. Evidence for keratinocyte stem cells in vitro: Long term engraftment and persistence of transgene expression from retrovirus-transduced keratinocytes. Proc. Natl. Acad. Sci. USA 1998, 95, 4356–4461. [Google Scholar] [CrossRef] [PubMed]
- Ghazizadeh, S.; Taichman, L.B. Organization of stem cells and their progeny in human epidermis. J. Investig. Dermatol. 2005, 124, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Rheinwald, J.G.; Green, H. Serial cultivation of strains of human epidermal keratinocytes: The formation of keratinizing colonies from single cells. Cell 1975, 6, 331–343. [Google Scholar] [CrossRef]
- Barrandon, Y.; Grasset, N.; Zaffalon, A.; Gorostidi, F.; Claudinot, S.; Droz-Georget, S.L.; Nanba, D.; Rochat, A. Capturing epidermal stemness for regenerative medicine. Semin. Cell Dev. Biol. 2012, 23, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, G.; Ranno, R.; Stracuzzi, G.; Bondanza, S.; Guerra, L.; Zambruno, G.; Micali, G.; de Luca, M. The control of epidermal stem cells (holoclones) in the treatment of massive full-thickness burns with autologous keratinocytes cultured on fibrin. Transplantation 1999, 68, 868–879. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, G.; Golisano, O.; Paterna, P.; Lambiase, A.; Bonini, S.; Rama, P.; de Luca, M. Location and clonal analysis of stem cells and their differentiated progeny in the human ocular surface. J. Cell Biol. 1999, 145, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Rama, P.; Matuska, S.; Paganoni, G.; Spinelli, A.; de Luca, M.; Pellegrini, G. Limbal stem-cell therapy and long-term corneal regeneration. N. Engl. J. Med. 2010, 363, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Rama, P.; Bonini, S.; Lambiase, A.; Golisano, O.; Paterna, P.; de Luca, M.; Pellegrini, G. Autologous fibrin-cultured limbal stem cells permanently restore the corneal surface of patients with total limbal stem cell deficiency. Transplantation 2001, 72, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, L.; de Luca, M. Cell biology: Dormant and restless skin stem cells. Nature 2012, 489, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Dellambra, E.; Golisano, O.; Bondanza, S.; Siviero, E.; Lacal, P.; Molinari, M.; D’Atri, S.; de Luca, M. Downregulation of 14-3-3σ prevents clonal evolution and leads to immortalization of primary human keratinocytes. J. Cell Biol. 2000, 149, 1117–1130. [Google Scholar] [CrossRef] [PubMed]
- Maurelli, R.; Zambruno, G.; Guerra, L.; Abbruzzese, C.; Dimri, G.; Gellini, M.; Bondanza, S.; Dellambra, E. Inactivation of p16INK4a (inhibitor of cyclin-dependent kinase 4A) immortalizes primary human keratinocytes by maintaining cells in the stem cell compartment. FASEB J. 2006, 20, 1516–1518. [Google Scholar] [CrossRef] [PubMed]
- Cordisco, S.; Maurelli, R.; Bondanza, S.; Stefanini, M.; Zambruno, G.; Guerra, L.; Dellambra, E. Bmi-1 reduction plays a key role in physiological and premature aging of primary human keratinocytes. J. Investig. Dermatol. 2010, 130, 1048–1062. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Sharpless, N.E. The regulation of INK4/ARF in cancer and aging. Cell 2006, 127, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E. INK4a/ARF: A multifunctional tumor suppressor locus. Mutat. Res. 2005, 576, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Narita, M. Cellular senescence and chromatin organisation. Br. J. Cancer 2007, 96, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Poi, M.J.; Tsai, M. Regulatory mechanisms of tumor suppressor P16INK4A and their relevance to cancer. Biochemistry 2011, 50, 5566–5582. [Google Scholar] [CrossRef] [PubMed]
- LaPak, K.M.; Burd, C.E. The molecular balancing act of p16INK4a in cancer and aging. Mol. Cancer Res. 2014, 12, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Adam, R.C.; Fuchs, E. The Yin and Yang of chromatin dynamics in stem cell fate selection. Trends Genet. 2016, 32, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Perdigoto, C.N.; Valdes, V.J.; Bardot, E.S.; Ezhkova, E. Epigenetic regulation of epidermal differentiation. Cold Spring Harb. Perspect. Med. 2014, 4, a015263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Bardot, E.; Ezhkova, E. Epigenetic regulation of skin: Focus on the polycomb complex. Cell. Mol. Life Sci. 2012, 69, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Bracken, A.P.; Kleine-Kohlbrecher, D.; Dietrich, N.; Pasini, D.; Gargiulo, G.; Beekman, C.; Theilgaard-Mönch, K.; Minucci, S.; Porse, B.T.; Marine, J.C.; et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007, 21, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; O’Loghlen, A. PRC1 complex diversity: Where is it taking us? Trends Cell Biol. 2014, 24, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Nakagawa, T.; Kitagawa, K.; Suzuki, S.; Liu, N.; Kitagawa, M.; Xiong, Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15INK4B tumor suppressor gene. Oncogene 2011, 30, 1956–1962. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Takebayashi, S.I.; Sakamoto, A.; Igata, T.; Nakatsu, Y.; Saitoh, N.; Hino, S.; Nakao, M. The SETD8/PR-Set7 methyltransferase functions as a barrier to prevent senescence-associated metabolic remodeling. Cell Rep. 2017, 18, 2148–2161. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Zeng, Y.; Xiong, Y. DDB1-CUL4 and MLL1 mediate oncogene-induced p16INK4a activation. Cancer Res. 2009, 69, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Pan, K.; Chen, Y.; Huang, C.; Zhang, X. The acetylation of transcription factor HBP1 by p300/CBP enhances p16INK4A expression. Nucleic Acids Res. 2012, 40, 981–995. [Google Scholar] [CrossRef] [PubMed]
- Botchkarev, V.A.; Gdula, M.R.; Mardaryev, A.N.; Sharov, A.A.; Fessing, M.Y. Epigenetic regulation of gene expression in keratinocytes. J. Investig. Dermatol. 2012, 132, 2505–2521. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Feng, Y.; Xu, L.; Chen, Y.; Zhang, Y.; Su, D.; Ren, G.; Lu, J.; Huang, B. YY1 restrained cell senescence through repressing the transcription of p16. Biochim. Biophys. Acta 2008, 1783, 1876–1883. [Google Scholar] [CrossRef] [PubMed]
- Clapier, C.R.; Cairns, B.R. The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 2009, 78, 273–304. [Google Scholar] [CrossRef] [PubMed]
- Kia, S.K.; Gorski, M.M.; Giannakopoulos, S.; Verrijzer, C.P. SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol. Cell. Biol. 2008, 28, 3457–3464. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Zhuang, X.; Yao, Y.G.; Zhang, R. BRG1 is required for formation of senescence-associated heterochromatin foci induced by oncogenic RAS or BRCA1 loss. Mol. Cell. Biol. 2013, 33, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- Muegge, K. Lsh, a guardian of heterochromatin at repeat elements. Biochem. Cell Biol. 2005, 83, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Xi, S.; Zhu, H.; Xu, H.; Schmidtmann, A.; Geiman, T.M.; Muegge, K. Lsh controls Hox gene silencing during development. Proc. Natl. Acad. Sci. USA 2007, 104, 14366–14371. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Han, L.; Li, G.; Tong, T. Senescence delay and repression of p16INK4a by Lsh via recruitment of histone deacetylases in human diploid fibroblasts. Nucleic Acids Res. 2009, 37, 5183–5196. [Google Scholar] [CrossRef] [PubMed]
- Han, H.J.; Russo, J.; Kohwi, Y.; Kohwi-Shigematsu, T. SATB1 reprogrammes gene expression to promote breast tumour growth and metastasis. Nature 2008, 452, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Agrelo, R.; Kishimoto, H.; Novatchkova, M.; Peraza, V.; Paolino, M.; Souabni, A.; Wutz, A. SATB1 collaborates with loss of p16 in cellular transformation. Oncogene 2013, 32, 5492–5500. [Google Scholar] [CrossRef] [PubMed]
- Witcher, M.; Emerson, B.M. Epigenetic silencing of the p16INK4a tumor suppressor is associated with loss of CTCF binding and a chromatin boundary. Mol. Cell 2009, 34, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Raisner, R.M.; Madhani, H.D. Patterning chromatin: Form and function for H2A.Z variant nucleosomes. Curr. Opin. Genet. Dev. 2006, 16, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Filippova, G.N. Genetics and epigenetics of the multifunctional protein CTCF. Curr. Top. Dev. Biol. 2008, 80, 337–360. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.A.; Felsenfeld, G. We gather together: Insulators and genome organization. Curr. Opin. Genet. Dev. 2007, 17, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, N.; Zebedee, Z.; Huot, T. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature 2001, 409, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Ansieau, S.; Bastid, J.; Doreau, A.; Morel, A.P.; Bouchet, B.P.; Thomas, C.; Fauvet, F.; Puisieux, I.; Doglioni, C.; Piccinin, S.; et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 2008, 14, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Gil, J.; Peters, G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: All for one or one for all. Nat. Rev. Mol. Cell Biol. 2006, 7, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Ozawa, Y.; Harada, M.; Kitagawa, K.; Niida, H.; Morita, Y.; Tanaka, K.; Suda, T.; Kitagawa, M. YB1 binds to and represses the p16 tumor suppressor gene. Genes Cells 2013, 18, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.A.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef] [PubMed]
- Cakouros, D.; Isenmann, S.; Cooper, L.; Zannettino, A.; Anderson, P.; Glackin, C.; Gronthos, S. Twist-1 induces Ezh2 recruitment regulating histone methylation along the Ink4A/Arf locus in mesenchymal stem cells. Mol. Cell. Biol. 2012, 32, 1433–1441. [Google Scholar] [CrossRef] [PubMed]
- Pan, K.; Chen, Y.; Roth, M.; Wang, W.; Wang, S.; Yee, A.S.; Zhang, X. HBP1-mediated transcriptional regulation of DNA methyltransferase 1 and its impact on cell senescence. Mol. Cell. Biol. 2013, 33, 887–903. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wu, J.; Li, R.; Wang, P.; Han, L.; Zhang, Z.; Tong, T. B-MYB delays cell aging by repressing p16INK4α transcription. Cell. Mol. Life Sci. 2011, 68, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Mechta-Grigoriou, F.; Gerald, D.; Yaniv, M. The mammalian Jun proteins: Redundancy and specificity. Oncogene 2001, 20, 2378–2389. [Google Scholar] [CrossRef] [PubMed]
- Gan, Q.; Huang, J.; Zhou, R.; Niu, J.; Zhu, X.; Wang, J.; Zhang, Z.; Tong, T. PPARγ accelerates cellular senescence by inducing p16INK4α expression in human diploid fibroblasts. J. Cell Sci. 2008, 121, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- Woods, M.; Pant, R.; Mallya, S.M. Cyclin D1 and cyclin D-dependent kinases enhance oral keratinocyte proliferation but do not block keratinocyte differentiation. Int. J. Oncol. 2010, 37, 1471–1475. [Google Scholar] [PubMed]
- Ivanova, I.A.; D’Souza, S.J.; Dagnino, L. Signalling in the epidermis: The E2F cell cycle regulatory pathway in epidermal morphogenesis, regeneration and transformation. Int. J. Biol. Sci. 2005, 1, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Magenta, A.; Dellambra, E.; Ciarapica, R.; Capogrossi, M.C. Oxidative stress, microRNAs and cytosolic calcium homeostasis. Cell Calcium 2016, 60, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.; DePinho, R. Telomeres, stem cells, senescence, and cancer. J. Clin. Investig. 2004, 113, 160. [Google Scholar] [CrossRef] [PubMed]
- Sen, G.L.; Reuter, J.A.; Webster, D.E.; Zhu, L.; Khavari, P.A. DNMT1 maintains progenitor function in self-renewing somatic tissue. Nature 2010, 463, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Sen, G.L.; Webster, D.E.; Barragan, D.I.; Chang, H.Y.; Khavari, P.A. Control of differentiation in a self-renewing mammalian tissue by the histone demethylase JMJD3. Genes Dev. 2008, 22, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Ezhkova, E.; Lien, W.H.; Stokes, N.; Pasolli, H.A.; Silva, J.M.; Fuchs, E. EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes Dev. 2011, 25, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Ezhkova, E.; Pasolli, H.A.; Parker, J.S.; Stokes, N.; Su, I.H.; Hannon, G.; Tarakhovsky, A.; Fuchs, E. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 2009, 136, 1122–1135. [Google Scholar] [CrossRef] [PubMed]
- Luis, N.M.; Morey, L.; Mejetta, S.; Pascual, G.; Janich, P.; Kuebler, B.; Cozutto, L.; Roma, G.; Nascimento, E.; Frye, M.; et al. Regulation of human epidermal stem cell proliferation and senescence requires polycomb-dependent and -independent functions of Cbx4. Cell Stem Cell 2011, 9, 233–246. [Google Scholar] [CrossRef] [PubMed]
- LeBoeuf, M.; Terrell, A.; Trivedi, S.; Sinha, S.; Epstein, J.A.; Olson, E.N.; Morrisey, E.E.; Millar, S.E. Hdac1 and Hdac2 act redundantly to control p63 and p53 functions in epidermal progenitor cells. Dev. Cell 2010, 19, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Bartolome, R.; Callejas-Valera, J.L.; Gutkind, J.S. Control of the epithelial stem cell epigenome: The shaping of epithelial stem cell identity. Curr. Opin. Cell Biol. 2013, 25, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, S. Unique and overlapping functions of pRb and p107 in the control of proliferation and differentiation in epidermis. Development 2004, 131, 2737–2748. [Google Scholar] [CrossRef] [PubMed]
- Paramio, J.M.; Segrelles, C.; Casanova, M.L.; Jorcano, J.L. Opposite functions for E2F1 and E2F4 in human epidermal keratinocyte differentiation. J. Biol. Chem. 2000, 275, 41219–41226. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Santos, M.; Martinez-Fernandez, M.; Lorz, C.; Lazaro, S.; Paramio, J.M. Deregulation of the pRb-E2F4 axis alters epidermal homeostasis and favors tumor development. Oncotarget 2016, 7, 75712–75728. [Google Scholar] [CrossRef] [PubMed]
- Costa, C.; Paramio, J.M.; Santos, M. Skin tumors Rb(eing) uncovered. Front. Oncol. 2013, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.L.; Adhikary, G.; Rorke, E.A.; Chew, Y.C.; Balasubramanian, S. Polycomb group proteins are key regulators of keratinocyte function. J. Investig. Dermatol. 2011, 131, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Benitah, S.A. Chromatin regulators in mammalian epidermis. Semin. Cell Dev. Biol. 2012, 23, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Avgustinova, A.; Benitah, S.A. Epigenetic control of adult stem cell function. Nat. Rev. Mol. Cell Biol. 2016, 17, 643–658. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jiang, T.X.; Hughes, M.W.; Wu, P.; Yu, J.; Widelitz, R.B.; Fan, G.; Chuong, C.M. Progressive alopecia reveals decreasing stem cell activation probability during aging of mice with epidermal deletion of DNA methyltransferase 1. J. Investig. Dermatol. 2012, 132, 2681–2690. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Beerman, I.; Lien, W.H.; Smith, Z.D.; Gu, H.; Boyle, P.; Gnirke, A.; Fuchs, E.; Rossi, D.J.; Meissner, A. DNA methylation dynamics during in vivo differentiation of blood and skin stem cells. Mol. Cell 2012, 47, 633–647. [Google Scholar] [CrossRef] [PubMed]
- Mulder, K.W.; Wang, X.; Escriu, C.; Ito, Y.; Schwarz, R.F.; Gillis, J.; Sirokmány, G.; Donati, G.; Uribe-Lewis, S.; Pavlidis, P.; et al. Diverse epigenetic strategies interact to control epidermal differentiation. Nat. Cell Biol. 2012, 14, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Bardot, E.S.; Valdes, V.J.; Zhang, J.; Perdigoto, C.N.; Nicolis, S.; Hearn, S.A.; Silva, J.M.; Ezhkova, E. Polycomb subunits Ezh1 and Ezh2 regulate the Merkel cell differentiation program in skin stem cells. EMBO J. 2013, 32, 1990–2000. [Google Scholar] [CrossRef] [PubMed]
- Dauber, K.L.; Perdigoto, C.N.; Valdes, V.J.; Santoriello, F.J.; Cohen, I.; Ezhkova, E. Dissecting the roles of polycomb repressive complex 2 subunits in the control of skin development. J. Investig. Dermatol. 2016, 136, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Mejetta, S.; Morey, L.; Pascual, G.; Kuebler, B.; Mysliwiec, M.R.; Lee, Y.; Shiekhattar, R.; di Croce, L.; Benitah, S.A. Jarid2 regulates mouse epidermal stem cell activation and differentiation. EMBO J. 2011, 30, 3635–3646. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, R.; Mustafi, S.B.; Street, M.; Dey, A.; Dwivedi, S.K.D. Bmi-1: At the crossroads of physiological and pathological biology. Genes Dis. 2015, 2, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Ressler, S.; Bartkova, J.; Niederegger, H.; Bartek, J.; Scharffetter-Kochanek, K.; Jansen-Dürr, P.; Wlaschek, M. p16INK4A is a robust in vivo biomarker of cellular aging in human skin. Aging Cell 2006, 5, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Reinisch, C.M.; Uthman, A.; Erovic, B.M.; Pammer, J. Expression of BMI-1 in normal skin and inflammatory and neoplastic skin lesions. J. Cutan. Pathol. 2007, 34, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Lee, K.; Adhikary, G.; Gopalakrishnan, R.; Rorke, E.A.; Eckert, R.L. The Bmi-1 polycomb group gene in skin cancer: Regulation of function by (−)-epigallocatechin-3-gallate. Nutr. Rev. 2008, 66 (Suppl. S1), S65–S68. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Adhikary, G.; Balasubramanian, S.; Gopalakrishnan, R.; McCormick, T.; Dimri, G.P.; Eckert, R.L.; Rorke, E. A expression of Bmi-1 in epidermis enhances cell survival by altering cell cycle regulatory protein expression and inhibiting apoptosis. J. Investig. Dermatol. 2008, 128, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.; García, J.M.; Peña, C.; García, V.; Domínguez, G.; Suárez, D.; Camacho, F.I.; Espinosa, R.; Provencio, M.; España, P. Implication of polycomb members Bmi-1, Mel-18, and Hpc-2 in the regulation of p16INK4a, p14ARF, h-TERT, and c-Myc expression in primary breast carcinomas. Clin. Cancer Res. 2006, 12, 6929–6936. [Google Scholar] [CrossRef] [PubMed]
- Roscic, A.; Möller, A.; Calzado, M.A.; Renner, F.; Wimmer, V.C.; Gresko, E.; Lüdi, K.S.; Schmitz, M.L. Phosphorylation-dependent control of Pc2 SUMO E3 ligase activity by its substrate protein HIPK2. Mol. Cell 2006, 24, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Mardaryev, A.N.; Liu, B.; Rapisarda, V.; Poterlowicz, K.; Malashchuk, I.; Rudolf, J.; Sharov, A.A.; Jahoda, C.A.; Fessing, M.Y.; Benitah, S.A.; et al. Cbx4 maintains the epithelial lineage identity and cell proliferation in the developing stratified epithelium. J. Cell Biol. 2016, 212, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Winter, M.; Moser, M.A.; Meunier, D.; Fischer, C.; Machat, G.; Mattes, K.; Lichtenberger, B.M.; Brunmeir, R.; Weissmann, S.; Murko, C.; et al. Divergent roles of HDAC1 and HDAC2 in the regulation of epidermal development and tumorigenesis. EMBO J. 2013, 32, 3176–3191. [Google Scholar] [CrossRef] [PubMed]
- Frye, M.; Fisher, A.G.; Watt, F.M. Epidermal stem cells are defined by global histone modifications that are altered by Myc-induced differentiation. PLoS ONE 2007, 2, e763. [Google Scholar] [CrossRef] [PubMed]
- Markova, N.G.; Karaman-Jurukovska, N.; Pinkas-Sarafova, A.; Marekov, L.N.; Simon, M. Inhibition of histone deacetylation promotes abnormal epidermal differentiation and specifically suppresses the expression of the late differentiation marker profilaggrin. J. Investig. Dermatol. 2007, 127, 1126–1139. [Google Scholar] [CrossRef] [PubMed]
- Mills, A.A.; Zheng, B.; Wang, X.J.; Vogel, H.; Roop, D.R.; Bradley, A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999, 398, 708–713. [Google Scholar] [CrossRef] [PubMed]
- McKeon, F.; Yang, A.; Schweitzer, R.; Sun, D.; Kaghad, M.; Walker, N.; Bronson, R.T.; Tabin, C.; Sharpe, A.; Caput, D.; et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999, 398, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Driskell, I.; Oda, H.; Blanco, S.; Nascimento, E.; Humphreys, P.; Frye, M. The histone methyltransferase Setd8 acts in concert with c-Myc and is required to maintain skin. EMBO J. 2012, 31, 616–629. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, S.; Candi, E.; Hu, B.; Dolfini, D.; Ravo, M.; Grober, O.M.V.; Grober, O.M.V.; Weisz, A.; Dotto, G.P.; Melino, G.; et al. The p63 target HBP1 is required for skin differentiation and stratification. Cell Death Differ. 2010, 17, 1896–1907. [Google Scholar] [CrossRef] [PubMed]
- Keyes, W.M.; Pecoraro, M.; Aranda, V.; Vernersson-Lindahl, E.; Li, W.; Vogel, H.; Guo, X.; Garcia, E.L.; Michurina, T.V.; Enikolopov, G.; et al. A ΔNp63α is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell 2011, 8, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Fessing, M.Y.; Mardaryev, A.N.; Gdula, M.R.; Sharov, A.A.; Sharova, T.Y.; Rapisarda, V.; Gordon, K.B.; Smorodchenko, A.D.; Poterlowicz, K.; Ferone, G.; et al. p63 regulates Satb1 to control tissue-specific chromatin remodeling during development of the epidermis. J. Cell Biol. 2011, 194, 825–839. [Google Scholar] [CrossRef] [PubMed]
- Lena, A.M.; Mancini, M.; Rivetti di Val Cervo, P.; Saintigny, G.; Mahé, C.; Melino, G.; Candi, E. Micro RNA-191 triggers keratinocytes senescence by SATB1 and CDK6 downregulation. Biochem. Biophys. Res. Commun. 2012, 423, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Mardaryev, A.N.; Gdula, M.R.; Yarker, J.L.; Emelianov, V.U.; Emelianov, V.N.; Poterlowicz, K.; Sharov, A.A.; Sharova, T.Y.; Scarpa, J.A.; Joffe, B.; et al. p63 and Brg1 control developmentally regulated higher-order chromatin remodelling at the epidermal differentiation complex locus in epidermal progenitor cells. Development 2014, 141, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Indra, A.K.; Dupé, V.; Bornert, J.M.; Messaddeq, N.; Yaniv, M.; Mark, M.; Chambon, P.; Metzger, D. Temporally controlled targeted somatic mutagenesis in embryonic surface ectoderm and fetal epidermal keratinocytes unveils two distinct developmental functions of BRG1 in limb morphogenesis and skin barrier formation. Development 2005, 132, 4533–4544. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Tang, J.; Lopez-Pajares, V.; Tao, S.; Qu, K.; Crabtree, G.R.; Khavari, P.A. ACTL6a enforces the epidermal progenitor state by suppressing SWI/SNF-dependent induction of KLF4. Cell Stem Cell 2013, 12, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Rosa-Garrido, M.; Ceballos, L.; Alonso-Lecue, P.; Abraira, C.; Delgado, M.D.; Gandarillas, A. A cell cycle role for the epigenetic factor CTCF-L/BORIS. PLoS ONE 2012, 7, e39371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagarajan, P.; Chin, S.S.; Wang, D.; Liu, S.; Sinha, S.; Garrett-Sinha, L.A. Ets1 blocks terminal differentiation of keratinocytes and induces expression of matrix metalloproteases and innate immune mediators. J. Cell Sci. 2010, 123, 3566–3575. [Google Scholar] [CrossRef] [PubMed]
- Chin, S.S.; Romano, R.A.; Nagarajan, P.; Sinha, S.; Garrett-Sinha, L.A. Aberrant epidermal differentiation and disrupted ΔNp63/Notch regulatory axis in Ets1 transgenic mice. Biol. Open 2013, 2, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Langlands, K.; Down, G.A.; Kealey, T. Id proteins are dynamically expressed in normal epidermis and dysregulated in squamous cell carcinoma. Cancer Res. 2000, 60, 5929–5933. [Google Scholar] [PubMed]
- Maruyama, H.; Ishitsuka, Y.; Fujisawa, Y.; Furuta, J.; Sekido, M.; Kawachi, Y. B-Myb enhances proliferation and suppresses differentiation of keratinocytes in three-dimensional cell culture. Arch. Dermatol. Res. 2014, 306, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Eckert, R.L.; Adhikary, G.; Young, C.A.; Jans, R.; Crish, J.F.; Xu, W.; Rorke, E.A. AP1 transcription factors in epidermal differentiation and skin cancer. J. Skin Cancer 2013, 2013, 537028. [Google Scholar] [CrossRef] [PubMed]
- Wurm, S.; Zhang, J.; Guinea-Viniegra, J.; Garcia, F.; Munoz, J.; Bakiri, L.; Ezhkova, E.; Wagner, E.F. Terminal epidermal differentiation is regulated by the interaction of Fra-2/AP-1 with Ezh2 and ERK1/2. Genes Dev. 2015, 29, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Yamada, A.; Kimura, S.; Peters, J.M.; Gonzalez, F.J. Alterations in skin and stratified epithelia by constitutively activated PPARalpha. J. Investig. Dermatol. 2006, 126, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; dePinho, R.A. How stem cells age and why this makes us grow old. Nat. Rev. Mol. Cell Biol. 2007, 8, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Giangreco, A.; Qin, M.; Pintar, J.E.; Watt, F.M. Epidermal stem cells are retained in vivo throughout skin aging. Aging Cell 2008, 7, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Winter, M.C.; Bickenbach, J.R. Aging epidermis is maintained by changes in transit-amplifying cell kinetics, not stem cell kinetics. J. Investig. Dermatol. 2009, 129, 2541–2543. [Google Scholar] [CrossRef] [PubMed]
- Stern, M.M.; Bickenbach, J.R. Epidermal stem cells are resistant to cellular aging. Aging Cell 2007, 6, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Gallico, G.G.; O’Connor, N.E.; Compton, C.C.; Kehinde, O.; Green, H. Permanent coverage of large burn wounds with autologous cultured human epithelium. N. Engl. J. Med. 1984, 311, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Charruyer, A.; Barland, C.O.; Yue, L.; Wessendorf, H.B.; Lu, Y.; Jeffrey Lawrence, H.; Mancianti, M.L.; Ghadially, R. Transit-amplifying cell frequency and cell cycle kinetics are altered in aged epidermis. J. Investig. Dermatol. 2009, 129, 2574–2583. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, J.; Ramsey, M.R.; Ligon, K.L.; Torrice, C.; Koh, A.; Bonner-Weir, S.; Sharpless, N.E. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 2006, 443, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 2004, 114, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.V.; Slutsky, S.G.; Joseph, N.M.; He, S.; Pardal, R.; Krishnamurthy, J.; Sharpless, N.E.; Morrison, S.J. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 2006, 443, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Janzen, V.; Forkert, R.; Fleming, H.E.; Saito, Y.; Waring, M.T.; Dombkowski, D.M.; Cheng, T.; dePinho, R.A.; Sharpless, N.E.; Scadden, D.T. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16INK4a. Nature 2006, 443, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16INK4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sanoff, H.K.; Cho, H.; Burd, C.E.; Torrice, C.; Ibrahim, J.G.; Thomas, N.E.; Sharpless, N.E. Expression of p16INK4a in peripheral blood T-cells is a biomarker of human aging. Aging Cell 2009, 8, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Muss, H.B.J.K.; Alston, S.M.; Lacy, A.C.; Jolly, T.A.; Williams, G.; Carey, L.A.; Dees, E.C.; Anders, C.K.; Irvin, W.J.S.N. Chemotherapy in older women with breast cancer. N. Engl. J. Med. 2009, 361, 1023–1024. [Google Scholar] [CrossRef]
- Boquoi, A.; Arora, S.; Chen, T.; Litwin, S.; Koh, J.; Enders, G.H. Reversible cell cycle inhibition and premature aging features imposed by conditional expression of p16INK4a. Aging Cell 2015, 14, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Waaijer, M.E.C.; Parish, W.E.; Strongitharm, B.H.; van Heemst, D.; Slagboom, P.E.; de Craen, A.J.M.; Sedivy, J.M.; Westendorp, R.G.J.; Gunn, D.A.; Maier, A.B. The number of p16INK4a positive cells in human skin reflects biological age. Aging Cell 2012, 11, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Adamus, J.; Aho, S.; Meldrum, H.; Bosko, C.; Lee, J.M. p16INK4a influences the aging phenotype in the living skin equivalent. J. Investig. Dermatol. 2014, 134, 1131–1133. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Kajiya, H.; Ozeki, S.; Okabe, K.; Ikebe, T. Reactive oxygen species promotes cellular senescence in normal human epidermal keratinocytes through epigenetic regulation of p16INK4a. Biochem. Biophys. Res. Commun. 2014, 452, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Kotake, Y.; Cao, R.; Viatour, P.; Sage, J.; Zhang, Y.; Xiong, Y. pRB family proteins are required for H3K27 trimethylation and polycomb repression complexes binding to and silencing p16INK4a tumor suppressor gene. Genes Dev. 2007, 21, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Rivetti di val Cervo, P.; Lena, A.M.; Nicoloso, M.; Rossi, S.; Mancini, M.; Zhou, H.; Saintigny, G.; Dellambra, E.; Odorisio, T.; Mahé, C.; et al. p63-microRNA feedback in keratinocyte senescence. Proc. Natl. Acad. Sci. USA 2012, 109, 1133–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keyes, W.M.; Wu, Y.; Vogel, H.; Guo, X.; Lowe, S.W.; Mills, A.A. p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Genes Dev. 2005, 19, 1986–1999. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.Q. Growth retardation and premature aging phenotypes in mice with disruption of the SNF2-like gene, PASG. Genes Dev. 2004, 18, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Chen, T.; Han, L.; Wang, P.; Li, N.; Tong, T. Transcriptional activation of the senescence regulator Lsh by E2F1. Mech. Ageing Dev. 2011, 132, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, W.; Liu, X.; Paulson, K.E.; Yee, A.S.; Zhang, X. Transcriptional factor HBP1 targets p16INK4a, upregulating its expression and consequently is involved in Ras-induced premature senescence. Oncogene 2010, 29, 5083–5094. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, B.J.; Chaturvedi, V.; Bacon, P.; Qin, J.Z.; Denning, M.F.; Diaz, M.O. Id-1 delays senescence but does not immortalize keratinocytes. J. Biol. Chem. 2000, 275, 27501–27504. [Google Scholar] [CrossRef] [PubMed]
- Alani, R.M.; Hasskarl, J.; Grace, M.; Hernandez, M.C.; Israel, M.A.; Münger, K. Immortalization of primary human keratinocytes by the helix-loop-helix protein, Id-1. Proc. Natl. Acad. Sci. USA 1999, 96, 9637–9641. [Google Scholar] [CrossRef] [PubMed]
- Barradas, M.; Anderton, E.; Acosta, J.C.; Li, S.; Banito, A.; Rodriguez-Niedenführ, M.; Maertens, G.; Banck, M.; Zhou, M.M.; Walsh, M.J.; et al. Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev. 2009, 23, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Mowla, S.N.; Lam, E.W.F.; Jat, P.S. Cellular senescence and aging: The role of B-MYB. Aging Cell 2014, 13, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Lear, J.T.; Szeimies, R. Non-melanoma skin cancer. Lancet 2010, 375, 673–685. [Google Scholar] [CrossRef]
- Diffey, B.L.; Langtry, J.A.A. Skin cancer incidence and the ageing population. Br. J. Dermatol. 2005, 153, 679–680. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; Robert, L. Cell senescence: role in aging and age-related diseases. Interdiscip. Top. Gerontol. 2014, 39, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Ruini, C.; Witkowski, A.M.; Cesinaro, A.; Teixeira de Carvalho, N.; Pellacani, G. From actinic keratosis to squamous cell carcinoma: Evidence of morphologic and biologic progression. J. Am. Acad. Dermatol. 2015, 72, S8–S10. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Chi, S.; Xie, J. Hedgehog signaling in skin cancers. Cell Signal. 2011, 23, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Cretnik, M.; Poje, G.; Musani, V.; Kruslin, B.; Ozretic, P.; Tomas, D.; Situm, M.; Levanat, S. Involvement of p16 and PTCH in pathogenesis of melanoma and basal cell carcinoma. Int. J. Oncol. 2009, 34, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Krimpenfort, P.; Quon, K.C.; Mooi, W.J.; Loonstra, A.; Berns, A. Loss of p16INK4a confers susceptibility to metastatic melanoma in mice. Nature 2001, 413, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Bardeesy, N.; Lee, K.H.; Carrasco, D.; Castrillon, D.H.; Aguirre, A.J.; Wu, E.A.; Horner, J.W.; dePinho, R.A. Loss of p16INK4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001, 413, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Mortier, L.; Marchetti, P.; Delaporte, E.; Martin de Lassalle, E.; Thomas, P.; Piette, F.; Formstecher, P.; Polakowska, R.; Danzé, P.M. Progression of actinic keratosis to squamous cell carcinoma of the skin correlates with deletion of the 9p21 region encoding the p16INK4a tumor suppressor. Cancer Lett. 2002, 176, 205–214. [Google Scholar] [CrossRef]
- Chang, T.G.; Wang, J.; Chen, L.W.; Hsu, C.Y.; Chang, H.W.; Chen, J.S.; Cho, C.L. Loss of expression of the p16 gene is frequent in malignant skin tumors. Biochem. Biophys. Res. Commun. 1997, 230, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Kalof, A.N.; Evans, M.F.; Simmons-Arnold, L.; Beatty, B.G.; Cooper, K. p16INK4a immunoexpression and HPV in situ hybridization signal patterns: Potential markers of high-grade cervical intraepithelial neoplasia. Am. J. Surg. Pathol. 2005, 29, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Saridaki, Z.; Liloglou, T.; Zafiropoulos, A.; Koumantaki, E.; Zoras, O.; Spandidos, D.A. Mutational analysis of CDKN2A genes in patients with squamous cell carcinoma of the skin. Br. J. Dermatol. 2003, 148, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Hodges, A.; Smoller, B.R. Immunohistochemical comparison of p16 expression in actinic keratoses and squamous cell carcinomas of the skin. Mod. Pathol. 2002, 15, 1121–1125. [Google Scholar] [CrossRef] [PubMed]
- Salama, M.E.; Mahmood, M.N.; Qureshi, H.S.; Ma, C.; Zarbo, R.J.; Ormsby, A.H. p16INK4a expression in actinic keratosis and Bowen’s disease. Br. J. Dermatol. 2003, 149, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Blokx, W.A.M.; de Jong, E.M.G.J.; de Wilde, P.C.M.; Bulten, J.; Link, M.M.G.M.; Ruiter, D.J.; van de Kerkhof, P.C.M. p16 and p53 expression in (pre)malignant epidermal tumors of renal transplant recipients and immunocompetent individuals. Mod. Pathol. 2003, 16, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Cabral, L.S.; Festa Neto, C.; Sanches, J.A.; Ruiz, I.R.G. Genomic instability in human actinic keratosis and squamous cell carcinoma. Clinics (Sao Paulo) 2011, 66, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Kanellou, P.; Zaravinos, A.; Zioga, M.; Spandidos, D.A. Deregulation of the tumour suppressor genes p14(ARF), p15(INK4b), p16(INK4a) and p53 in basal cell carcinoma. Br. J. Dermatol. 2009, 160, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Brasanac, D.; Stojkovic-Filipovic, J.; Bosic, M.; Tomanovic, N.; Manojlovic-Gacic, E. Expression of G1/S-cyclins and cyclin-dependent kinase inhibitors in actinic keratosis and squamous cell carcinoma. J. Cutan. Pathol. 2016, 43, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Bagazgoitia, L.; Cuevas, J.; Juarranz, A. Expression of p53 and p16 in actinic keratosis, bowenoid actinic keratosis and Bowen’s disease. J. Eur. Acad. Dermatol. Venereol. 2010, 24, 228–230. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, K.; Svensson, S.; Landberg, G. Retinoblastoma protein function and p16INK4a expression in actinic keratosis, squamous cell carcinoma in situ and invasive squamous cell carcinoma of the skin and links between p16INK4a expression and infiltrative behavior. Mod. Pathol. 2004, 17, 1464–1474. [Google Scholar] [CrossRef] [PubMed]
- Grønhøj Larsen, C.; Gyldenløve, M.; Jensen, D.H.; Therkildsen, M.H.; Kiss, K.; Norrild, B.; Konge, L.; von Buchwald, C. Correlation between human papillomavirus and p16 overexpression in oropharyngeal tumours: A systematic review. Br. J. Cancer 2014, 110, 1587–1594. [Google Scholar] [CrossRef] [PubMed]
- Beadle, B.M.; William, W.N.; McLemore, M.S.; Sturgis, E.M.; Williams, M.D. p16 Expression in cutaneous squamous carcinomas with neck metastases: A potential pitfall in identifying unknown primaries of the head and neck. Head Neck 2013, 35, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.S.; Patrick, D.R.; Edwards, G.; Goodhart, P.J.; Huber, H.E.; Miles, L.; Garsky, V.M.; Oliff, A.; Heimbrook, D.C. Protein domains governing interactions between E2F, the retinoblastoma gene product, and human papillomavirus type 16 E7 protein. Mol. Cell. Biol. 1993, 13, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Romagosa, C.; Simonetti, S.; López-Vicente, L.; Mazo, A.; Lleonart, M.E.; Castellvi, J.; Ramon y Cajal, S. p16(Ink4a) overexpression in cancer: A tumor suppressor gene associated with senescence and high-grade tumors. Oncogene 2011, 30, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Küsters-Vandevelde, H.V.N.; de Koning, M.N.C.; Melchers, W.J.G.; Quint, W.G.V.; de Wilde, P.C.M.; de Jong, E.M.G.J.; van de Kerkhof, P.C.M.; Blokx, W.A.M. Expression of p14ARF, p16INK4a and p53 in relation to HPV in (pre-)malignant squamous skin tumours. J. Cell. Mol. Med. 2009, 13, 2148–2157. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Mao, X.; Talbot, I.C. Aberrant cytological localization of p16 and CDK4 in colorectal epithelia in the normal adenoma carcinoma sequence. World J. Gastroenterol. 2006, 12, 6391–6396. [Google Scholar] [CrossRef] [PubMed]
- Conscience, I.; Jovenin, N.; Coissard, C.; Lorenzato, M.; Durlach, A.; Grange, F.; Birembaut, P.; Clavel, C.; Bernard, P. p16 is overexpressed in cutaneous carcinomas located on sun-exposed areas. Eur. J. Dermatology 2006, 16, 518–522. [Google Scholar] [CrossRef]
- Eshkoor, S.A.; Ismail, P.; Rahman, S.A.; Oshkour, S.A. p16 gene expression in basal cell carcinoma. Arch. Med. Res. 2008, 39, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Svensson, S.; Nilsson, K.; Ringberg, A.; Landberg, G. Invade or proliferate? Two contrasting events in malignant behavior governed by p16(INK4a) and an intact Rb pathway illustrated by a model system of basal cell carcinoma. Cancer Res. 2003, 63, 1737–1742. [Google Scholar] [PubMed]
- Nandakumar, V.; Vaid, M.; Tollefsbol, T.O.; Katiyar, S.K. Aberrant DNA hypermethylation patterns lead to transcriptional silencing of tumor suppressor genes in UVB-exposed skin and UVB-induced skin tumors of mice. Carcinogenesis 2011, 32, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Biasiotta, A.; D’Arcangelo, D.; Passarelli, F.; Nicodemi, E.M.; Facchiano, A. Ion channels expression and function are strongly modified in solid tumors and vascular malformations. J. Transl. Med. 2016, 14, 285. [Google Scholar] [CrossRef] [PubMed]
- D’Arcangelo, D.; Facchiano, F.; Nassa, G.; Stancato, A.; Antonini, A.; Rossi, S.; Senatore, C.; Cordella, M.; Tabolacci, C.; Salvati, A.; et al. PDGFR-alpha inhibits melanoma growth via CXCL10/IP-10: A multi-omics approach. Oncotarget 2016, 7, 77257–77275. [Google Scholar] [CrossRef] [PubMed]
- Nindl, I.; Dang, C.; Forschner, T.; Kuban, R.J.; Meyer, T.; Sterry, W.; Stockfleth, E. Identification of differentially expressed genes in cutaneous squamous cell carcinoma by microarray expression profiling. Mol. Cancer 2006, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Riker, A.I.; Enkemann, S.A.; Fodstad, O.; Liu, S.; Ren, S.; Morris, C.; Xi, Y.; Howell, P.; Metge, B.; Samant, R.S.; et al. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med. Genomics 2008, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Ginos, M.A.; Page, G.P.; Michalowicz, B.S.; Patel, K.J.; Volker, S.E.; Pambuccian, S.E.; Ondrey, F.G.; Adams, G.L.; Gaffney, P.M. Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer Res. 2004, 64, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Cromer, A.; Carles, A.; Millon, R.; Ganguli, G.; Chalmel, F.; Lemaire, F.; Young, J.; Dembélé, D.; Thibault, C.; Muller, D.; et al. Identification of genes associated with tumorigenesis and metastatic potential of hypopharyngeal cancer by microarray analysis. Oncogene 2004, 23, 2484–2498. [Google Scholar] [CrossRef] [PubMed]
- Toruner, G.A.; Ulger, C.; Alkan, M.; Galante, A.T.; Rinaggio, J.; Wilk, R.; Tian, B.; Soteropoulos, P.; Hameed, M.R.; Schwalb, M.N.; et al. Association between gene expression profile and tumor invasion in oral squamous cell carcinoma. Cancer Genet. Cytogenet. 2004, 154, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.H.; Liao, C.T.; Peng, S.C.; Chen, Y.J.; Cheng, A.J.; Juang, J.L.; Tsai, C.Y.; Chen, T.C.; Chuang, Y.J.; Tang, C.Y.; et al. A novel molecular signature identified by systems genetics approach predicts prognosis in oral squamous cell carcinoma. PLoS ONE 2011, 6, e23452. [Google Scholar] [CrossRef] [PubMed]
- Lazarov, M.; Kubo, Y.; Cai, T.; Dajee, M.; Tarutani, M.; Lin, Q.; Fang, M.; Tao, S.; Green, C.L.; Khavari, P. A CDK4 coexpression with Ras generates malignant human epidermal tumorigenesis. Nat. Med. 2002, 8, 1105–1114. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Aldabagh, B.; Yu, J.; Arron, S.T. Role of human papillomavirus in cutaneous squamous cell carcinoma: A meta-analysis. J. Am. Acad. Dermatol. 2014, 70, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, J.H.; Hauck, F.; Barros, M.H.M.; Niedobitek, G. pRb and CyclinD1 Complement p16 as Immunohistochemical Surrogate Markers of HPV Infection in Head and Neck Cancer. Appl. Immunohistochem. Mol. Morphol. 2017, 25, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.J.; Dicker, A.J.; Dahler, A.L.; Saunders, N.A. E2F as a Regulator of Keratinocyte Proliferation: Implications for Skin Tumor Development. J. Investig. Dermatol. 1997, 109, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.C.; Sohng, S.H.; Shin, D.H.; Choi, J.S.; Bae, Y.K. Immunohistochemical expression of cytokeratin 15, cytokeratin 19, follistatin, and Bmi-1 in basal cell carcinoma. Int. J. Dermatol. 2016, 55, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, W.L.; You, P.; Su, J.; Zhu, M.H.; Xie, D.F.; Zhu, H.Y.; He, Z.Y.; Li, J.X.; Ding, X.Y.; et al. Oncoprotein BMI-1 induces the malignant transformation of HaCaT cells. J. Cell. Biochem. 2009, 106, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, G.; Grun, D.; Kerr, C.; Balasubramanian, S.; Rorke, E.A.; Vemuri, M.; Boucher, S.; Bickenbach, J.R.; Hornyak, T.; Xu, W.; et al. Identification of a population of epidermal squamous cell carcinoma cells with enhanced potential for tumor formation. PLoS ONE 2013, 8, e84324. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, G.; Grun, D.; Balasubramanian, S.; Kerr, C.; Huang, J.M.; Eckert, R.L. Survival of skin cancer stem cells requires the Ezh2 polycomb group protein. Carcinogenesis 2015, 36, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Abba, M.C.; Molinolo, A.A.; Vitale-Cross, L.; Wang, Z.; Zaida, M.; Delic, N.C.; Samuels, Y.; Lyons, G.J.; Gutkind, J.S. The head and neck cancer cell oncogenome: A platform for the development of precision molecular therapies. Oncotarget 2014, 5, 8906–8923. [Google Scholar] [CrossRef] [PubMed]
- Missero, C.; Antonini, D. Crosstalk among p53 family members in cutaneous carcinoma. Exp. Dermatol. 2014, 23, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Bosic, M.M.; Brasanac, D.C.; Stojkovic-Filipovic, J.M.; Zaletel, I.V.; Gardner, J.M.; Cirovic, S.L. Expression of p300 and p300/CBP associated factor (PCAF) in actinic keratosis and squamous cell carcinoma of the skin. Exp. Mol. Pathol. 2016, 100, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.K.; Cai, M.Y.; Luo, R.Z.; Tian, X.; Liao, Q.M.; Zhang, X.Y.; Han, J.D. Overexpression of p300 correlates with poor prognosis in patients with cutaneous squamous cell carcinoma. Br. J. Dermatol. 2015, 172, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, M.R.; He, L.; Forster, N.; Ory, B.; Ellisen, L.W. Physical association of HDAC1 and HDAC2 with p63 mediates transcriptional repression and tumor maintenance in squamous cell carcinoma. Cancer Res. 2011, 71, 4373–4379. [Google Scholar] [CrossRef] [PubMed]
- Torkamandi, S.; Moghbeli, M.; Farshchian, M.; Rad, A.; Abbaszadegan, M.R. Role of Brg1 in progression of esophageal squamous cell carcinoma. Iran. J. Basic Med. Sci. 2014, 17, 912–917. [Google Scholar] [PubMed]
- Li, Y.C.; Bu, L.L.; Mao, L.; Ma, S.R.; Liu, J.F.; Yu, G.T.; Deng, W.W.; Zhang, W.F.; Sun, Z.J. SATB1 promotes tumor metastasis and invasiveness in oral squamous cell carcinoma. Oral Dis. 2017, 23, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Cong, Q.X.; Zhang, H.; Sun, S.X.; Li, H.F.; Wang, Y.; Jian, S. Pilot study special AT-rich sequence-binding protein 1 investigating as a potential biomarker for esophageal squamous cell carcinoma. Dis. Esophagus 2016, 29, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.D.; Ji, W.; Zhang, W.; He, L.X.; Yang, J.; Liang, H.J.; Wang, L.L. Overexpression of SATB1 in laryngeal squamous cell carcinoma. ORL J. Otorhinolaryngol. Relat. Spec. 2010, 72, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Schramek, D.; Adam, R.C.; Keyes, B.E.; Wang, P.; Zheng, D.; Fuchs, E. ETS family transcriptional regulators drive chromatin dynamics and malignancy in squamous cell carcinomas. Elife 2015, 4, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, V.; Cesnjaj, M.; Bacon, P.; Panella, J.; Choubey, D.; Diaz, M.O.; Nickoloff, B.J. Role of INK4a/Arf locus-encoded senescent checkpoints activated in normal and psoriatic keratinocytes. Am. J. Pathol. 2003, 162, 161–170. [Google Scholar] [CrossRef]
- Lin, J.; Guan, Z.; Wang, C.; Feng, L.; Zheng, Y.; Caicedo, E.; Bearth, E.; Peng, J.R.; Gaffney, P.; Ondrey, F.G. Inhibitor of differentiation 1 contributes to head and neck squamous cell carcinoma survival via the NF-kappaB/survivin and phosphoinositide 3-kinase/Akt signaling pathways. Clin. Cancer Res. 2010, 16, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Pan, H.; Li, J.; Zhong, Q.; Chen, X.; Dry, S.M.; Wang, C.Y. Epigenetic Activation of AP1 Promotes Squamous Cell Carcinoma Metastasis. Sci. Signal. 2013, 6, ra28. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.Y.; Ke, H.; Hall, R.P.; Zhang, J. Y. c-Jun Promotes whereas JunB Inhibits Epidermal Neoplasia. J. Investig. Dermatol. 2011, 131, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Hyakusoku, H.; Sano, D.; Takahashi, H.; Hatano, T.; Isono, Y.; Shimada, S.; Ito, Y.; Myers, J.N.; Oridate, N. JunB promotes cell invasion, migration and distant metastasis of head and neck squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2016, 35, 6. [Google Scholar] [CrossRef] [PubMed]
- Mangone, F.R.R.; Brentani, M.M.; Nonogaki, S.; Begnami, M.D.F.S.; Campos, A.H.J.F.M.; Walder, F.; Carvalho, M.B.; Soares, F.A.; Torloni, H.; Kowalski, L.P.; et al. Overexpression of Fos-related antigen-1 in head and neck squamous cell carcinoma. Int. J. Exp. Pathol. 2005, 86, 205–212. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, S.O.M.; Mesquita, R.A.; Pinto, D.S.; Gutkind, S. Immunolocalization of c-Fos and c-Jun in human oral mucosa and in oral squamous cell carcinoma. J. Oral Pathol. Med. 2002, 31, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Ye, D.X.; Zhang, W.B.; Pan, H.Y.; Zhang, Z.Y.; Zhang, L. Overexpression of c-fos promotes cell invasion and migration via CD44 pathway in oral squamous cell carcinoma. J. Oral Pathol. Med. 2015, 44, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, N.; Bhattacharya, S.; Steele, R.; Phillips, N.; Ray, R.B. Involvement of c-Fos in the Promotion of Cancer Stem-like Cell Properties in Head and Neck Squamous Cell Carcinoma. Clin. Cancer Res. 2017, 23, 3120–3128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, J.; Luo, S.; Lechler, T.; Zhang, J.Y. FRA1 promotes squamous cell carcinoma growth and metastasis through distinct AKT and c-Jun dependent mechanisms. Oncotarget 2016, 7, 34371–34383. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.T.; Zhu, P.Q.; Luo, H.L.; Zhang, Y.; Hao, T.F.; Xia, G.F.; Zhu, Z.M.; Qiu, C. Long noncoding RNA ANRIL: A potential novel prognostic marker in cancer: A meta-analysis. Minerva Med. 2016, 107, 77–83. [Google Scholar] [PubMed]
- Li, Z.; Yu, X.; Shen, J. ANRIL: A pivotal tumor suppressor long non-coding RNA in human cancers. Tumour Biol. 2016, 37, 5657–5661. [Google Scholar] [CrossRef] [PubMed]
- Sand, M.; Bechara, F.G.; Sand, D.; Gambichler, T.; Hahn, S.A.; Bromba, M.; Stockfleth, E.; Hessam, S. Long-noncoding RNAs in basal cell carcinoma. Tumor Biol. 2016, 37, 10595–10608. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Epigenetic and transcriptional regulation of p16INK4a. (A) Epigenetic regulation of p16INK4a—The genomic INK/ARF locus is depicted as a bold line, with exons indicated by colored vertical lines (not drawn to scale). The coding regions of INK4b (CDKN2B) are shown in red, those of ARF in orange and those of INK4a (CDKN2A) in blue. Epigenetic repressors (red) and activators (green) have the opposite function in INK/ARF locus regulation; (B) Transcriptional activators and repressors of p16INK4a—The p16INK4a promoter is depicted as bold line with binding sites indicated by white rectangles (not drawn to scale). Expression of p16INK4a requires the action of transcription factors (green) that recruit and/or facilitate RNA polymerase association with the promoter. Transcriptional repressors (red) have an opposite function.
Figure 1.
Epigenetic and transcriptional regulation of p16INK4a. (A) Epigenetic regulation of p16INK4a—The genomic INK/ARF locus is depicted as a bold line, with exons indicated by colored vertical lines (not drawn to scale). The coding regions of INK4b (CDKN2B) are shown in red, those of ARF in orange and those of INK4a (CDKN2A) in blue. Epigenetic repressors (red) and activators (green) have the opposite function in INK/ARF locus regulation; (B) Transcriptional activators and repressors of p16INK4a—The p16INK4a promoter is depicted as bold line with binding sites indicated by white rectangles (not drawn to scale). Expression of p16INK4a requires the action of transcription factors (green) that recruit and/or facilitate RNA polymerase association with the promoter. Transcriptional repressors (red) have an opposite function.


Figure 2.
Epigenetic regulation of p16INK4a in epidermal homeostasis. The interfollicular epidermis is a stratified epithelium in which cell proliferation and differentiation are compartmentalized and tightly regulated. Cell proliferation occurs in the basal layer. When keratinocytes withdraw from cell cycle, they generate post-mitotic cells, which migrate upwards and form suprabasal layers executing their terminal differentiation program. Epigenetic modifiers (orange circles) that regulates p16INK4a (blue circle) expression and/or keratinocyte differentiation in epidermal homeostasis are indicated. Blue arrows and red lines indicate positive or negative actions, respectively.
Figure 2.
Epigenetic regulation of p16INK4a in epidermal homeostasis. The interfollicular epidermis is a stratified epithelium in which cell proliferation and differentiation are compartmentalized and tightly regulated. Cell proliferation occurs in the basal layer. When keratinocytes withdraw from cell cycle, they generate post-mitotic cells, which migrate upwards and form suprabasal layers executing their terminal differentiation program. Epigenetic modifiers (orange circles) that regulates p16INK4a (blue circle) expression and/or keratinocyte differentiation in epidermal homeostasis are indicated. Blue arrows and red lines indicate positive or negative actions, respectively.


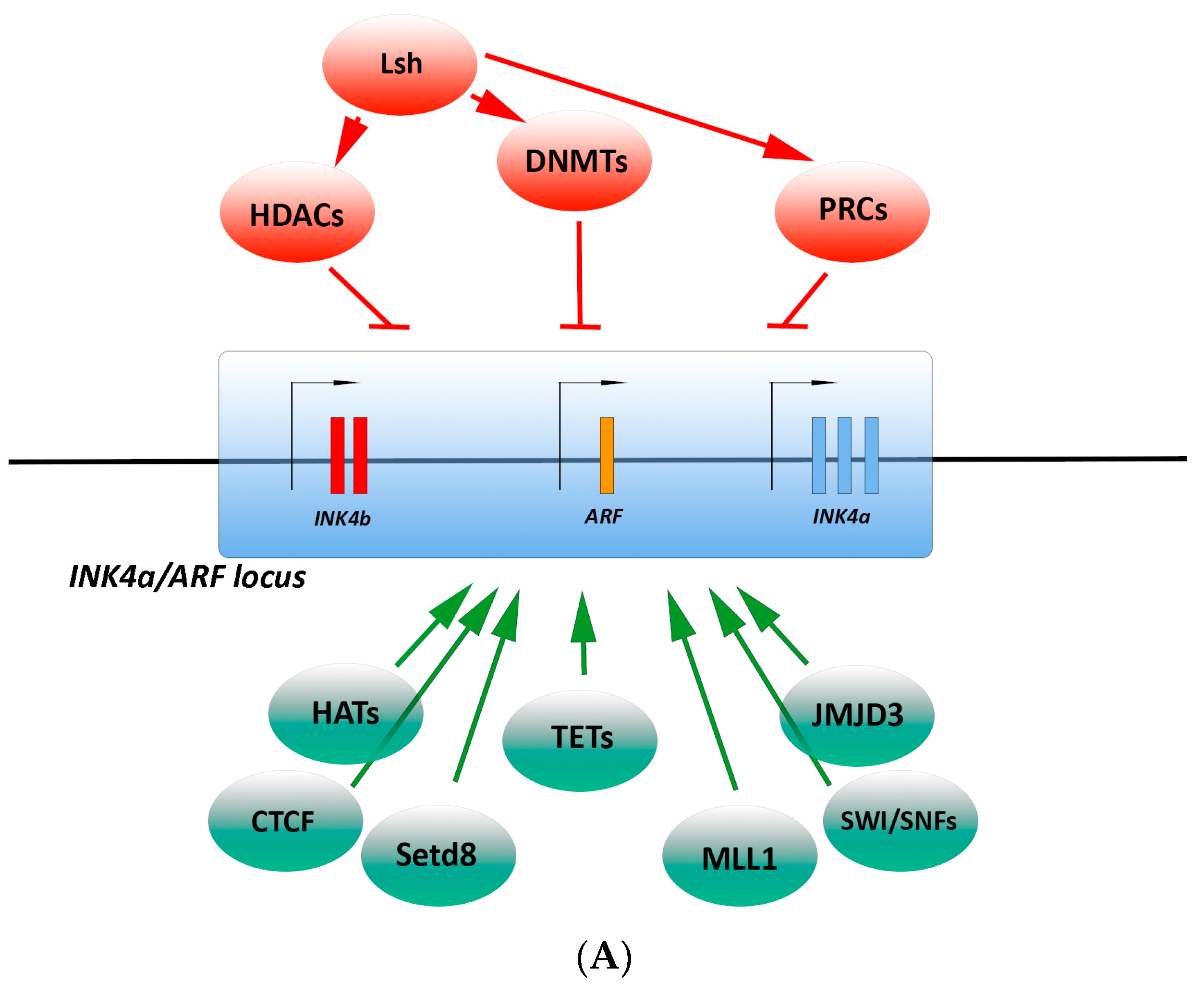{kind=link}
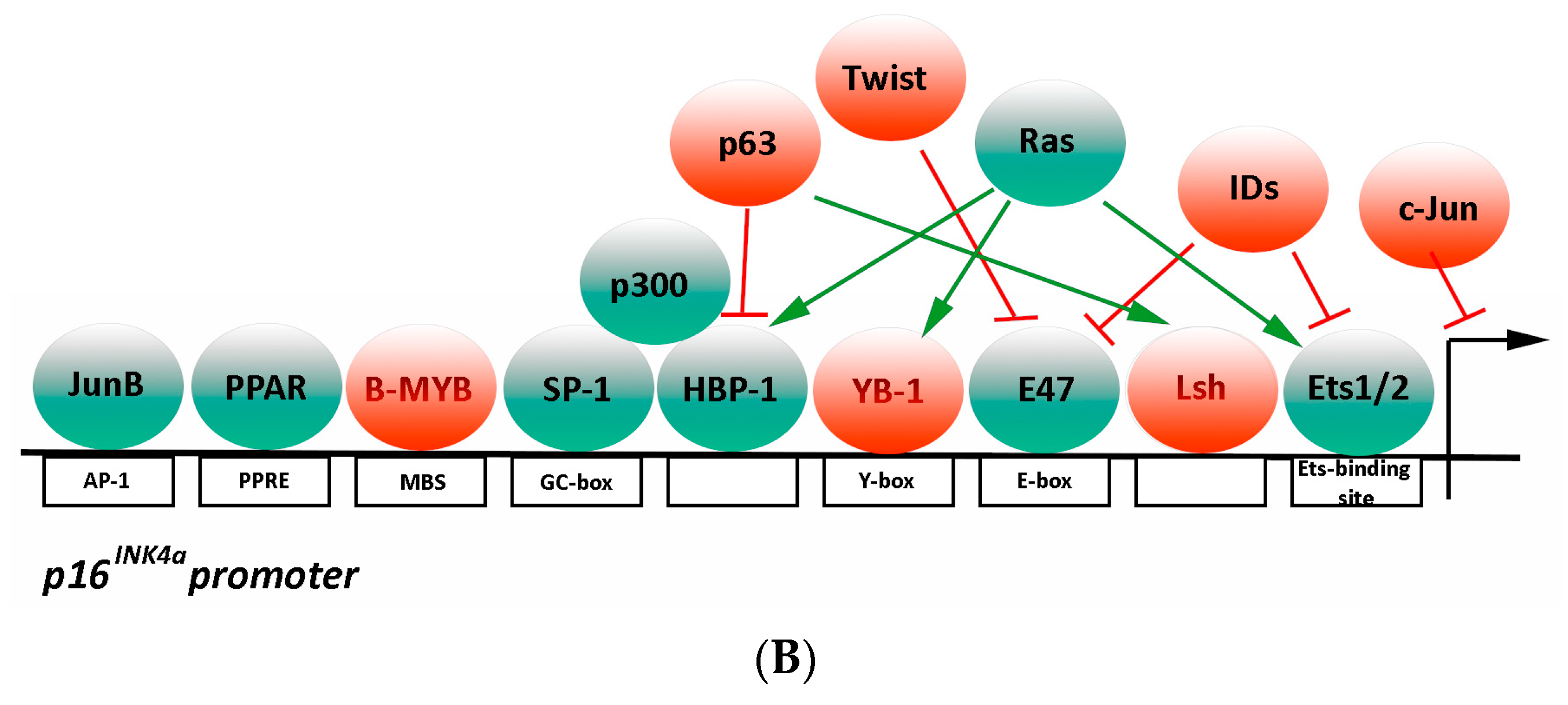{kind=link}
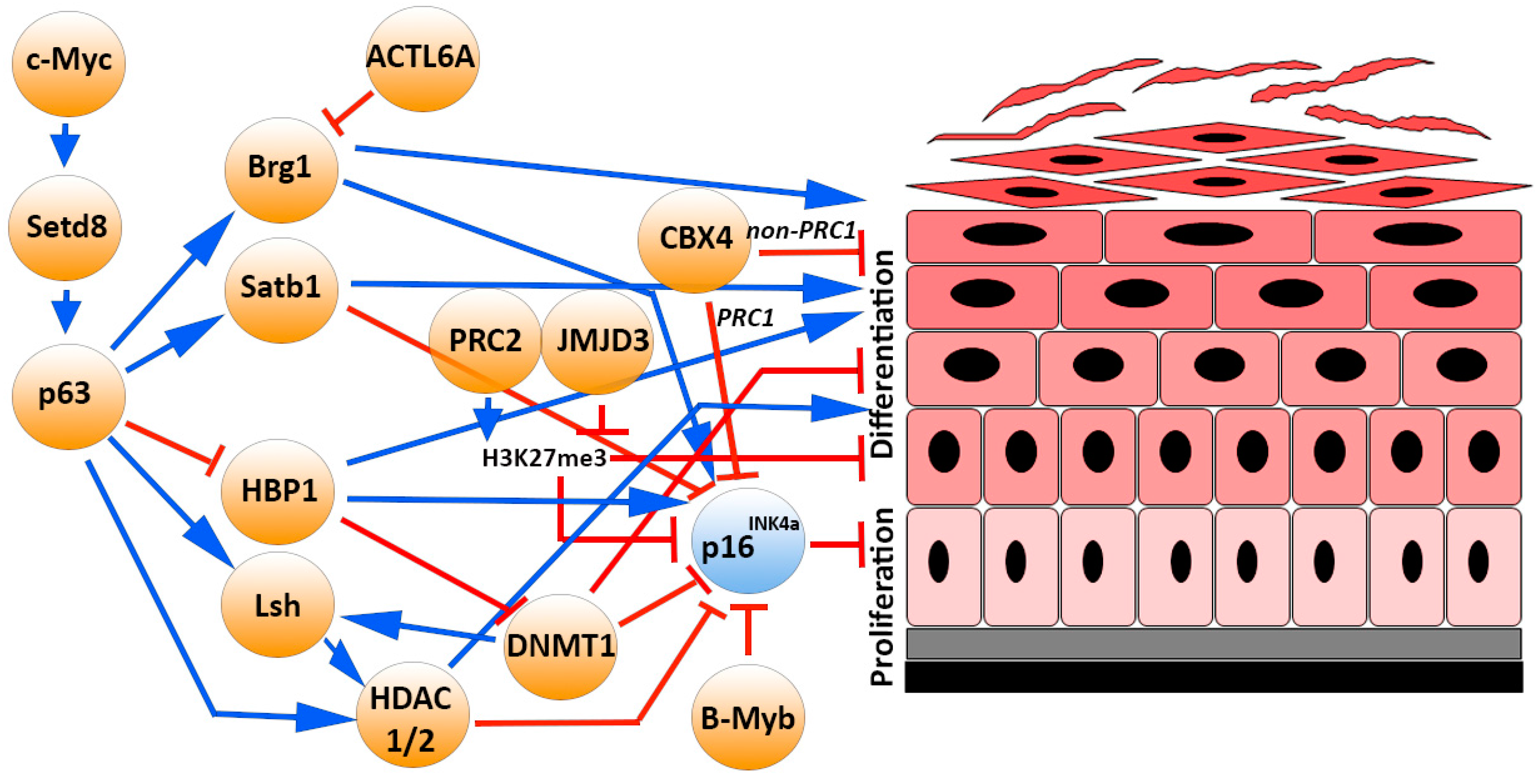{kind=link}
Table 1.
Patient numerosity and Reference of each dataset investigated in the present study from Oncomine database (www.oncomine.org)
Table 1.
Patient numerosity and Reference of each dataset investigated in the present study from Oncomine database (www.oncomine.org)
| Tumor Type | Nindl Dataset [178] | Riker Dataset [179] | Ginos Dataset [180] | Cromer Dataset [181] | Peng Dataset [183] | Toruner Dataset [182] | TOT |
|---|---|---|---|---|---|---|---|
| Normal Tissue | 6 | 4 | 13 | 4 | 22 | 4 | 53 |
| Actinic (Solar) Keratosis | 4 | 0 | 0 | 0 | 0 | 0 | 4 |
| Skin Squamous Cell Carcinoma | 5 | 11 | 0 | 0 | 0 | 0 | 16 |
| Skin Basal Cell Carcinoma | 0 | 15 | 0 | 0 | 0 | 0 | 15 |
| Head and Neck Squamous Cell Carcinoma | 0 | 0 | 41 | 34 | 0 | 0 | 75 |
| Oral Cavity Squamous Cell Carcinoma | 0 | 0 | 0 | 0 | 57 | 16 | 73 |
The table shows the tumors types, the datasets names, the patient numerosity, and the Reference of each dataset investigated in the present study, from Oncomine database (www.oncomine.org).
Table 2.
Expression fold change of the genes investigated in the present study from Actinic (solar) Keratosis, Skin Squamous Cell Carcinoma, and Skin Basal Cell Carcinoma databases.
Table 2.
Expression fold change of the genes investigated in the present study from Actinic (solar) Keratosis, Skin Squamous Cell Carcinoma, and Skin Basal Cell Carcinoma databases.
| Actinic (solar) Keratosis | Skin Squamous Cell Carcinoma | Skin Basal Cell Carcinoma | Skin Squamous Cell Carcinoma | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Nindl Dataset | Riker Dataset | |||||||||
| Cancer vs. Normal | Cancer vs. Normal | Cancer vs. Normal | Cancer vs. Normal | |||||||
| n | Protein | Gene | Fold Change | p Value | Fold Change | p Value | Fold Change | p Value | Fold Change | p Value |
| 1 | p16INK4a | CDKN2A | 2.218 | 0.004 | 3.553 | 0.003 | 2.297 | 0.009 | 4.404 | 4.09 × 10−4 |
| p16INK4a Downstream Genes | ||||||||||
| 2 | CDK4 | CDK4 | −1.033 | 0.602 | 1.125 | 0.163 | 1.976 | 5.34 × 10−5 | 1.713 | 8.07 × 10−4 |
| 3 | Cyclin D1 | CCND1 | 2.393 | 0.053 | 1.513 | 0.226 | 1.006 | 0.492 | 1.002 | 0.496 |
| 4 | pRB | RB1 | 1.974 | 0.05 | 1.349 | 0.17 | 1.251 | 0.017 | 1.579 | 5.48 × 10−4 |
| 5 | p107 | RBL1 | n.a. | n.a. | n.a. | n.a. | 1.307 | 0.284 | 1.9 | 0.11 |
| 6 | p130 | RBL2 | 1.632 | 0.152 | 1.676 | 0.163 | 1.367 | 0.141 | 1.41 | 0.046 |
| 7 | E2F1 | E2F1 | 1.885 | 0.0037 | 2.071 | 0.028 | 1.604 | 0.011 | 1.749 | 0.004 |
| 8 | E2F2 | E2F2 | 1.918 | 0.065 | 2.655 | 0.052 | 1.277 | 0.16 | 1.769 | 0.028 |
| 9 | E2F3 | E2F3 | 1.759 | 0.006 | 1.78 | 0.006 | 3.28 | 2.41 × 10−5 | 2.617 | 3.08 × 10−4 |
| 10 | E2F4 | E2F4 | −1.072 | 0.638 | 1.114 | 0.089 | −1.139 | 0.702 | 1.03 | 0.452 |
| Epigenetic Regulator Genes | ||||||||||
| 11 | DNMT1 | DNMT1 | 1.473 | 0.068 | 1.81 | 0.05 | 1.945 | 0.001 | 2.221 | 1.27 × 10−4 |
| 12 | DNMT3A | DNMT3A | −2.54 | 0.894 | −3.426 | 0.969 | 3.818 | 1.55 × 10−8 | 1.703 | 3.19 × 10−4 |
| 13 | DNMT3B | DNMT3B | 1.256 | 0.008 | 2.032 | 0.008 | 1.079 | 0.258 | 1.48 | 0.011 |
| 14 | Bmi-1 | BMI1 | −1.347 | 0.874 | −1.536 | 0.939 | 1.713 | 0.001 | 1.324 | 0.24 |
| 15 | Ezh1 | EZH1 | 1.397 | 0.259 | 1.619 | 0.272 | 1.611 | 0.251 | 1.379 | 0.331 |
| 16 | Ezh2 | EZH2 | 1.671 | 0.059 | 2.31 | 0.017 | 2.016 | 2.24 × 10−4 | 2.112 | 8.79 × 10−5 |
| 17 | EED | EED | −1.293 | 0.659 | −1.043 | 0.567 | 1.231 | 0.074 | 1.411 | 0.016 |
| 18 | CBX4 | CBX4 | −1.269 | 0.64 | 1.072 | 0.456 | −1.082 | 0.762 | 1.006 | 0.486 |
| 19 | CBX7 | CBX7 | −1.315 | 0.855 | −1.993 | 0.997 | −2.078 | 0.992 | −2.923 | 0.999 |
| 20 | Jarid2 | JARID2 | 1.167 | 0.223 | 1.409 | 0.021 | 1.091 | 0.195 | 1.382 | 0.006 |
| 21 | JMJD3 | JMJD3 | 1.568 | 0.018 | 1.562 | 0.017 | 2.822 | 0.075 | 1.853 | 0.171 |
| 22 | JDP2 | JDP2 | n.a. | n.a. | n.a. | n.a. | 1.264 | 0.052 | −1.147 | 0.821 |
| 23 | SETD8 | KMT5A | −1.282 | 0.629 | 1.411 | 0.102 | 1.06 | 0.346 | 1.505 | 0.004 |
| 24 | KMT2A | MLL | 1.657 | 0.004 | 1.479 | 0.043 | 1.413 | 0.01 | 1.184 | 0.076 |
| 25 | HAT p300 | EP300 | 1.692 | 0.041 | 1.855 | 0.025 | 1.112 | 0.039 | 1.143 | 0.03 |
| 26 | HDAC1 | HDAC1 | 1.066 | 0.414 | 1.632 | 0.025 | 1.636 | 1.39 × 10−5 | 2.309 | 2.89 × 10−7 |
| 27 | HDAC2 | HDAC2 | −1.461 | 0.909 | 1.007 | 0.486 | 2.223 | 1.19 × 10−4 | 1.682 | 0.001 |
| 28 | p63 | TP63 | 1.452 | 0.234 | 2.905 | 0.008 | 1.273 | 0.022 | 1.512 | 0.002 |
| 29 | BRG1 | SMARCA4 | 2.03 | 0.003 | 1.826 | 2.27 × 10−4 | 1.666 | 2.08 × 10−6 | 1.767 | 0.002 |
| 30 | LSH | HELLS | 4.504 | 0.039 | 8.371 | 0.016 | 5.292 | 3.02 × 10−4 | 1.948 | 0.003 |
| 31 | Satb1 | SATB1 | −1.125 | 0.853 | −1.17 | 0.835 | −1.342 | 0.865 | 1.25 | 0.813 |
| 32 | CTCF | CTCF | −1.083 | 0.756 | 1.063 | 0.35 | 1.54 | 1.72 × 10−4 | 1.277 | 0.006 |
| Transcriptional Regulator Genes | ||||||||||
| 33 | Ets1 | ETS1 | 1.31 | 0.759 | 1.446 | 0.13 | 1.139 | 0.329 | 2.421 | 0.011 |
| 34 | Ets2 | ETS2 | 1.247 | 0.102 | 1.696 | 0.007 | −1.463 | 0.823 | 1.106 | 0.393 |
| 35 | ID1 | ID1 | 1.23 | 0.243 | 1.48 | 0.103 | −1.824 | 0.995 | 1.434 | 0.041 |
| 36 | ID2 | ID2 | 1.508 | 0.042 | −1.117 | 0.698 | 1.281 | 0.163 | 1.165 | 0.262 |
| 37 | ID3 | ID3 | 1.224 | 0.184 | −1.512 | 0.92 | −2.316 | 0.999 | −1.137 | 0.687 |
| 38 | YB1 | YXB1 | 1.126 | 0.164 | 1.117 | 0.271 | 1.314 | 0.005 | 1.48 | 0.001 |
| 39 | SP1 | SP1 | -1.699 | 0.769 | 1.847 | 0.082 | 1.048 | 0.223 | 1.112 | 0.317 |
| 40 | HBP1 | PBRM1 | 1.167 | 0.156 | 1.146 | 0.192 | 1.513 | 5.96 × 10−6 | 1.984 | 0.031 |
| 41 | b-myb | MYB | −1.66 | 0.898 | 1.348 | 0.925 | −1.14 | 0.593 | 1.592 | 0.204 |
| 42 | JUNB | JUNB | 1.107 | 0.292 | 1.375 | 0.078 | −1.989 | 0.927 | −1.09 | 0.586 |
| 43 | c-Jun | C-JUN | 1.635 | 0.016 | 1.517 | 0.014 | 1.055 | 0.455 | 1.04 | 0.468 |
| 44 | JunD | JUND | 1.16 | 0.121 | 1.058 | 0.401 | −1.428 | 0.742 | −1.558 | 0.8 |
| 45 | c-Fos | C-FOS | −2.149 | 0.834 | −3.068 | 0.937 | −6.298 | 0.972 | −1.95 | 0.814 |
| 46 | FosB | FOSB | 1.265 | 0.401 | −6.929 | 0.96 | −11.992 | 0.923 | −8.365 | 0.896 |
| 47 | FRA1 | FOSL1 | 2.66 | 0.048 | 2.937 | 0.021 | 1.12 | 0.366 | 1.867 | 0.058 |
| 48 | FRA2 | FOSL2 | 1.883 | 0.015 | 2.38 | 0.005 | 1.339 | 0.065 | 1.658 | 0.013 |
| 49 | PPARalpha | PPARA | −1.556 | 0.877 | 1.132 | 0.352 | 1.229 | 0.378 | 1.153 | 0.198 |
| 50 | ANRIL | CDKN2BAS | n.a. | n.a. | n.a. | n.a. | −1.109 | 0.697 | −1.152 | 0.744 |
The table shows the tumors types, dataset names, and complete list of genes investigated in the present study, from Oncomine database (www.oncomine.org). Statistically significant expression fold changes (p ≤ 0.05) are labeled: Fold change ≥ 2 (green); fold change < 2 (light green); n.a.: Not available data.
Table 3.
Expression fold change of the genes investigated in the present study, from Head and Neck Squamous Cell Carcinoma and Oral Cavity Squamous Cell Carcinoma databases.
Table 3.
Expression fold change of the genes investigated in the present study, from Head and Neck Squamous Cell Carcinoma and Oral Cavity Squamous Cell Carcinoma databases.
| Head and Neck Squamous Cell Carcinoma | Oral Cavity SquamousCell Carcinoma | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ginos Dataset | Cromer Dataset | Peng Dataset | Toruner dataset | |||||||
| Cancer vs. Normal | Cancer vs. Normal | Cancer vs. Normal | Cancer vs. Normal | |||||||
| n | Protein | Gene | Fold Change | p Value | Fold Change | p Value | Fold Change | p Value | Fold Change | p Value |
| 1 | p16INK4a | CDKN2A | 2.297 | 6.27 × 10−5 | −1.415 | 0.712 | 1.684 | 1.21 × 10−7 | 1.372 | 0.047 |
| p16INK4a Downstream Genes | ||||||||||
| 2 | CDK4 | CDK4 | 1.842 | 8.84 × 10−12 | 2.63 | 8.00 × 10−6 | 1.498 | 3.76E−08 | 2.13 | 6.31 × 10−4 |
| 3 | Cyclin D1 | CCND1 | 1.465 | 0.023 | 2.031 | 5.97 × 10−5 | −1.112 | 0.907 | 1.201 | 0.164 |
| 4 | pRB | RB1 | 1.372 | 8.92 × 10−4 | 1.165 | 0.429 | 1.2 | 0.018 | 1.579 | 0.005 |
| 5 | p107 | RBL1 | n.a. | n.a. | −2.032 | 0.97 | 1.334 | 4.47 × 10−4 | n.a. | n.a. |
| 6 | p130 | RBL2 | 1.081 | 0.172 | −1.449 | 0.774 | −1.228 | 0.997 | 1.141 | 0.046 |
| 7 | E2F1 | E2F1 | 1.347 | 0.046 | 1.754 | 1.00 × 10−6 | 1.032 | 0.313 | 1.083 | 0.05 |
| 8 | E2F2 | E2F2 | −1.329 | 0.856 | −1.147 | 0.813 | −1.424 | 1 | 1.044 | 0.172 |
| 9 | E2F3 | E2F3 | 2.945 | 7.20 × 10−8 | 1.948 | 0.159 | 1.099 | 4.19 × 10−15 | 1.231 | 0.284 |
| 10 | E2F4 | E2F4 | 1.131 | 0.025 | 1.462 | 0.014 | 1.415 | 1.28 × 10−11 | −1.178 | 1.00 |
| Epigenetic Regulator Genes | ||||||||||
| 11 | DNMT1 | DNMT1 | 1.706 | 2.00 × 10−7 | 1.581 | 0.124 | 1.499 | 1.87 × 10−15 | 1.457 | 2.00 × 10−3 |
| 12 | DNMT3A | DNMT3A | −1.046 | 0.584 | n.a. | n.a. | 1.01 | 0.37 | −1.038 | 0.644 |
| 13 | DNMT3B | DNMT3B | 1.37 | 5.19 × 10−4 | n.a. | n.a. | 1.512 | 3.82 × 10−8 | 1.296 | 0.009 |
| 14 | Bmi-1 | BMI1 | 1.818 | 3.23 × 10−7 | 1.627 | 5.16 × 10−5 | −1.046 | 0.689 | 1.708 | 2.78 × 10−7 |
| 15 | Ezh1 | EZH1 | 1.238 | 0.011 | 1.383 | 0.083 | 1.131 | 6.00 × 10−2 | 1.259 | 0.03 |
| 16 | Ezh2 | EZH2 | −1.325 | 0.88 | −1.151 | 0.833 | −1.401 | 1.000 | 1.021 | 0.4 |
| 17 | EED | EED | 1.495 | 3.89 × 10−4 | 1.118 | 0.286 | 1.349 | 3.12 × 10−7 | 1.112 | 0.013 |
| 18 | CBX4 | CBX4 | −1.133 | 0.750 | −1.132 | 0.613 | −1.183 | 0.999 | −1.04 | 0.817 |
| 19 | CBX7 | CBX7 | −1.429 | 0.992 | n.a. | n.a. | −1.311 | 1 | −1.24 | 1 |
| 20 | Jarid2 | JARID2 | 1.123 | 0.045 | 1.17 | 0.039 | 1.2 | 0.002 | 1.365 | 0. 948 |
| 21 | JMJD3 | JMJD3 | 1.039 | 0.292 | 1.127 | 0.116 | 1.088 | 0.174 | −1.229 | 0.924 |
| 22 | JDP2 | JDP2 | n.a. | n.a. | n.a. | n.a. | −1.214 | 1 | n.a. | n.a. |
| 23 | SETD8 | KMT5A | −1.375 | 0.999 | n.a. | n.a. | 1.058 | 0.214 | 1.097 | 2.19 × 10−4 |
| 24 | KMT2A | MLL | −1.185 | 0.974 | −1.252 | 0.901 | 1.179 | 0.005 | 1.11 | 0.215 |
| 25 | HAT p300 | EP300 | 1.054 | 0.327 | −1.196 | 0.737 | 1.141 | 0.006 | −1.146 | 0.904 |
| 26 | HDAC | HDAC1 | −1.075 | 0.83 | 1.361 | 0.054 | 1.174 | 0.016 | 1.883 | 1.42 × 10−4 |
| 27 | HDAC | HDAC2 | 1.613 | 1.41 × 10−4 | 1.181 | 0.241 | 1.066 | 0.151 | 1.265 | 0.029 |
| 28 | p63 | TP63 | 1.491 | 0.001 | 1.717 | 0.019 | 1.238 | 2.68 × 10−4 | 1.12 | 0.021 |
| 29 | BRG1 | SMARCA4 | 1.047 | 0.309 | −1.543 | 0.960 | 1.001 | 0.416 | 1.175 | 9.28 × 10−4 |
| 30 | LSH | HELLS | 2.463 | 0.013 | n.a. | n.a. | 1.617 | 6.24 × 10−6 | 1.445 | 0.004 |
| 31 | Satb1 | SATB1 | 1.025 | 0.396 | −2.039 | 0.92 | −1.496 | 0.997 | −1.685 | 1 |
| 32 | CTCF | CTCF | 1.003 | 0.478 | 1.292 | 0.018 | −1.114 | 0.998 | 1.29 | 0.097 |
| Transcriptional Regulator Genes | ||||||||||
| 33 | Ets1 | ETS1 | 1.694 | 0.001 | 2.076 | 0.016 | 2.111 | 2.97 × 10−10 | −1.114 | 1 |
| 34 | Ets2 | ETS2 | 1.78 | 1.76 × 10−6 | 1.761 | 0.012 | −1.102 | 0.908 | −1.218 | 0.618 |
| 35 | ID1 | ID1 | 1.356 | 0.015 | 1.375 | 0.278 | 1.21 | 0.025 | 1.242 | 0.317 |
| 36 | ID2 | ID2 | 1.765 | 1.79 × 10−4 | 2.022 | 0.258 | 1.066 | 0.193 | 1.038 | 0.362 |
| 37 | ID3 | ID3 | 3.023 | 2.14 × 10−4 | n.a. | n.a. | 1.072 | 0.208 | 1.043 | 0.447 |
| 38 | YB1 | YXB1 | 1.421 | 0.001 | 1.644 | 6.26 × 10−4 | −1.362 | 0.998 | 1.637 | 0.001 |
| 39 | SP1 | SP1 | 1.292 | 0.95 | −1.382 | 0.717 | −1.232 | 1 | 1.008 | 0.372 |
| 40 | HBP1 | PBRM1 | −1.131 | 0.716 | n.a. | n.a. | −1.303 | 1.00 | 1.039 | 0.137 |
| 41 | b-myb | MYB | 1.003 | 0.495 | 1.414 | 0.13 | −1.842 | 1 | 1.134 | 0.229 |
| 42 | JUNB | JUNB | 1.38 | 0.006 | 1.252 | 0.093 | 1.003 | 0.485 | −1.029 | 0.552 |
| 43 | c-Jun | C-JUN | 1.549 | 1.13 × 10−5 | 1.18 | 0.083 | 1.318 | 0.008 | −1.093 | 0.508 |
| 44 | JunD | JUND | −1.048 | 0.549 | 1.176 | 0.05 | −1.227 | 0.999 | 1.045 | 0.086 |
| 45 | c-Fos | C-FOS | 13.503 | 2.88 × 10−5 | 1.481 | 0.212 | −1.415 | 0.956 | 1.225 | 0.365 |
| 46 | FosB | FOSB | 3.016 | 0.005 | 1.385 | 0.021 | −1.042 | 0.527 | −1.881 | 1 |
| 47 | FRA1 | FOSL1 | 1.663 | 0.076 | −1.194 | 0.922 | 1.2 | 0.045 | −3.719 | 0.946 |
| 48 | FRA2 | FOSL2 | −1.355 | 0.997 | 1.13 | 0.205 | −2.032 | 1 | −1.149 | 0.909 |
| 49 | PPARalpha | PPARA | −1.043 | 0.406 | −1.134 | 0.585 | −1.079 | 0.984 | 1.021 | 0.104 |
| 50 | ANRIL | CDKN2BAS | n.a. | n.a. | n.a. | n.a. | 1.802 | 8.85 × 10−12 | n.a. | n.a. |
The table shows the tumors types, dataset names, and complete list of genes investigated in the present study, from Oncomine database (www.oncomine.org). Statistically significant expression fold changes (p ≤ 0.05) are labeled: fold change ≥ 2 (green); fold change < 2 (light green); n.a.: not available data.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
D’Arcangelo, D.; Tinaburri, L.; Dellambra, E. The Role of p16INK4a Pathway in Human Epidermal Stem Cell Self-Renewal, Aging and Cancer. Int. J. Mol. Sci. 2017, 18, 1591. https://doi.org/10.3390/ijms18071591
AMA Style
D’Arcangelo D, Tinaburri L, Dellambra E. The Role of p16INK4a Pathway in Human Epidermal Stem Cell Self-Renewal, Aging and Cancer. International Journal of Molecular Sciences. 2017; 18(7):1591. https://doi.org/10.3390/ijms18071591
Chicago/Turabian StyleD’Arcangelo, Daniela, Lavinia Tinaburri, and Elena Dellambra. 2017. "The Role of p16INK4a Pathway in Human Epidermal Stem Cell Self-Renewal, Aging and Cancer" International Journal of Molecular Sciences 18, no. 7: 1591. https://doi.org/10.3390/ijms18071591
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.