Notch Signaling in Endothelial Cells: Is It the Therapeutic Target for Vascular Neointimal Hyperplasia?

1
Trainee Brigade, Third Military Medical University, Chongqing 400038, China
2
Department of Cell Biology, Third Military Medical University, Chongqing 400038, China
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2017, 18(8), 1615; https://doi.org/10.3390/ijms18081615
Submission received: 19 June 2017
/
Revised: 5 July 2017
/
Accepted: 21 July 2017
/
Published: 25 July 2017
(This article belongs to the Section Molecular Pathology, Diagnostics, and Therapeutics)
Abstract
:Blood vessels respond to injury through a healing process that includes neointimal hyperplasia. The vascular endothelium is a monolayer of cells that separates the outer vascular wall from the inner circulating blood. The disruption and exposure of endothelial cells (ECs) to subintimal components initiate the neointimal formation. ECs not only act as a highly selective barrier to prevent early pathological changes of neointimal hyperplasia, but also synthesize and release molecules to maintain vascular homeostasis. After vascular injury, ECs exhibit varied responses, including proliferation, regeneration, apoptosis, phenotypic switching, interacting with other cells by direct contact or secreted molecules and the change of barrier function. This brief review presents the functional role of the evolutionarily-conserved Notch pathway in neointimal hyperplasia, notably by regulating endothelial cell functions (proliferation, regeneration, apoptosis, differentiation, cell-cell interaction). Understanding endothelial cell biology should help us define methods to prompt cell proliferation, prevent cell apoptosis and dysfunction, block neointimal hyperplasia and vessel narrowing.
1. Introduction
Neointimal hyperplasia is an exaggerated wound-healing process that occurs in the vessel wall after injury. As a major morphological feature of many cardiovascular diseases (CVD), such as atherosclerosis and hypertension, neointimal hyperplasia is also responsible for the stenosis of vascular surgery, including bypass grafting, angioplasty and arteriovenous fistula [1,2]. The development of neointimal hyperplasia is a complex process initiated by the damage of endothelial cells (ECs) and exposure of vascular smooth muscle cells (VSMCs) to circulating blood elements. The process is further characterized by proliferative and inflammatory responses including VSMC proliferation and migration, platelet aggregation, leukocyte recruitment and extracellular matrix (ECM) deposition. Finally, EC proliferation or regeneration occurs at the lesion [3].
One of the important candidates for triggering neointimal formation is the dysfunction of endothelium. In the cardiovascular system, the endothelium is not only a barrier between the circulating blood and VSMCs, but also, it releases factors that regulate vascular tone, vessel growth, platelet function and coagulation [4]. For the underlying VSMCs, ECs could harmonize their growth and regression, through direct contact with VSMCs or secreted mediators that affect their proliferation, migration and death. In addition, ECs can regulate the thickness of intimal ECM through secreting enzymes, or inhibitors of these enzymes, which are able to degrade its components. The balance of these endothelial-derived activities regulates vessel development and vascular remodeling [5].
Recent advances in the understanding of the biology of neointimal formation indicate that ECs play a central role in the development of intimal hyperplasia during the process of vascular reconstruction. However, the mechanism of vascular neointimal hyperplasia is complicated, and a number of different intercellular signaling pathways has been implicated in this process. These pathways include the vascular endothelial growth factor (VEGF) pathway, the transforming growth factor-β (TGF-β) pathway, the Notch pathway, the Wnt pathway and many other pathways [6,7,8]. Among these pathways, the evolutionarily-conserved Notch signaling pathway controls cell fate through local cell-cell interactions. It plays a key role in the development of the cardiovascular system, as well as in the stability and remodeling of the vessel wall [9,10]. The purpose of this review is to summarize certain aspects of Notch signaling in endothelial cell biology and suggest how this knowledge might be used to reduce neointimal hyperplasia in cardiovascular disease and vascular surgical procedures.
2. The Notch Signaling Pathway
Notch signaling is significant in determining cell fate and regulating cell proliferation, apoptosis and differentiation [11,12]. It was originally identified in Drosophila, in which a mutant allele gives rise to a notched wing [13]. Mammals express four Notch transmembrane receptors (Notch-1, Notch-2, Notch-3 and Notch-4) and five typical transmembrane ligands (Delta-like 1 (Dll-1), Delta-like 3 (Dll-3) and Delta-like 4 (Dll-4), Jagged-1 and Jagged-2). Notch receptors are synthesized as single-chain precursors and cleaved into an extracellular and a transmembrane subunit by furin like convertase in the Golgi apparatus (Figure 1). These two subunits are held together on cell membrane by non-covalent bonds. Interaction of Notch receptors with their ligands leads to the transmembrane Notch receptor cleaved by a disintegrin and metalloproteinases (ADAM) proteases to remove the extracellular subunit. After that, a multisubunit membrane protease γ-secretase is responsible for the second proteolytic event that gives rise to the translocation of the Notch intracellular domain (NICD) into the nucleus. In the nucleus, NICD binds with a transcription factor, RBP-Jκ (also known as CSL for CBF1/Su(H)/Lag-1), and forms an activated transcriptional complex. Then, the activated complex upregulates the expression of target genes, such as hairy and enhancer of split (HES)-1, -5, -7 and HES-related repressor protein (HERP)-1 to -3 [14].
The fact that Notch signaling plays a crucial role in vascular biology has been clearly demonstrated. Abnormalities in vascular system caused by mutations of Notch receptors (Notch-1, -2, -4), ligands (Dll-1, -3, Jagged-1, -2) and effectors (HES-1, -5, -7, HERP-1) in mice have been reviewed in detail [15]. The disruption of Dll-4 or RBP-Jκ in mice also results in lethality due to defects in vascular remodeling or angiogenesis [16]. Human hereditary vascular disorders, such as cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) and Alagille syndrome (AGS), which manifest abnormalities in the cardiovascular system, are caused by mutations of Notch-3 and Jagged-1, respectively [17,18].
Recent studies support the emerging concept that Notch signaling is also involved in the development of neointimal hyperplasia. The increased Notch-1 signaling mediates neointimal formation in integrin β3(−/−)-induced arteriovenous graft occlusion through impairing EC regeneration [19]. Dll-4-mediated Notch activation promotes VSMC proliferation and migration in vein graft lesions and leads to vein graft failure [20,21]. Moreover, blocking the Notch pathway by using soluble Jagged-1 or by genetic deletion of the RBP-Jκ gene can inhibit neointimal formation after vessel injury [22,23].
As mentioned above, the triggering event in neointimal hyperplasia is EC damage. The ligands, receptors and other components of Notch signaling are expressed in ECs of different vascular origins [15,24]. During vascular injury, the Notch signal is definitely modulated, resulting in EC proliferation, apoptosis and differentiation. How these physiological alterations and barrier function impairments of ECs contribute to neointimal formation will be discussed later.
3. Endothelial Cell Proliferation and Regeneration
Notch signaling is an important regulator of EC proliferation and regeneration. During angiogenesis, Notch signaling suppresses EC proliferation and acts as an angiogenic “off” switch by making ECs unresponsive to VEGF [25,26]. It is estimated that only 0.01% of cells are actively proliferating in the vasculature of the adult [27,28]. Notch activation seems absent in vessels when ECs are proliferating at the early stages of angiogenesis; however, Notch is reactivated when ECs stop proliferating and vessels begin to stabilize [29,30]. Activation of Notch-1 and Notch-4 by Jagged-1 or Dll-4 reduces EC proliferation and contributes to contact inhibition of ECs [24,31]. Consistently, an extensive literature also reported that genetic or shRNA-mediated Dll-4 blockade in ECs leads to increased proliferation [32].
Compared with its role in angiogenesis, the role of Notch signaling during neointimal formation is more complicated. As shown in Figure 2, Notch activation suppresses EC proliferation, regeneration and promotes neointimal formation (Figure 2A). In integrin β3 knockout mice, the increased Notch-1 signaling inhibits circulating angiogenic cells’ (CACs) homing and differentiation, delays endothelial regeneration and promotes neointimal formation at the sites of arteriovenous grafts [19]. It is also reported that endothelial progenitor cells’ (EPCs) activity is greater in Notch-1(+/−) EPCs than in wild type (WT) EPCs, and subsequently, transplantation of Notch-1(+/−) bone marrow accelerates endothelial recovery after arterial injury in WT mice. Consistently, inhibition of Notch-1 mRNA expression in EPCs by cholesterol enhances EPCs’ activity and accelerates EC regeneration after arterial injury in atherosclerosis mice [33].
One of the characteristics of pulmonary hypertension is vascular remodeling and vascular neointimal thickening. Notch-1 and Notch-4 are detected in rat pulmonary artery ECs. Following the pulmonary hypertension induction, mRNA expression levels of Notch-1 and Notch-4 are all upregulated in rat pulmonary artery. Furthermore, an in vitro experiment also showed that the vessel wall thickness of cultured vascular strips from rats increases after hypoxia treatment, which can be decreased approximately 30% by N-(N-(3,5-difluorophenacetyl)-l-alanyl)-S-phenylglycine t-butyl ester (DAPT), a specific inhibitor of the γ-secretase [34]. These studies indicated to us that Notch signal in ECs is involved in neointimal thickness.
Notch signaling may have dual function in EC proliferation, which is depending on the relative level of p21 expression (Figure 2A). Notch-1 expression is increased in human pulmonary artery ECs after hypoxia treatment, which prompts EC proliferation via downregulation of p21 [35]. Low levels of p21 may activate cyclin D and induce cell proliferation, but p21 becomes an inhibitor at high levels [36]. Noseda and others demonstrated that when primary cultured ECs reach confluence, the activity of Notch signaling is augmented, while p21Cip1 is downregulated [24,36]. They also found that EC growth arrest is mediated by the repression of mitogen-activated protein kinase (MAPK)/PI3K (phosphatidylinositol 3 kinase) signaling and by p21Cip1, which prevents nuclear localization of cyclin D/cdk4 (cyclin-dependent kinase 4) required for Rb (retinoblastoma gene product) phosphorylation and S-phase entry [24].
Furthermore, EC behavior is at least partially dependent on reactive oxygen species (ROS) level downregulated by the Notch pathway. Blockade of Notch signaling can increase ROS in human umbilical vein endothelial cells (HUVECs), in contrast, suppression of ROS generation abolishes Notch blockade-induced HUVEC proliferation [37].
4. Endothelial Cell Apoptosis
Except for regulating cell proliferation and regeneration, Notch also affects EC apoptosis and survival, key cellular behaviors associated with vascular remodeling processes (Figure 2B). Vascular injury activates Notch signaling promptly, which further destroys the balance between EC proliferation and apoptosis, eventually influencing neointimal hyperplasia.
During the development of transplant arteriosclerosis (TA), EC apoptosis leads to neointimal hyperplasia in aortic allografts and allograft dysfunction. EC apoptosis induces the production of TGF-β1 in both apoptotic and neighboring viable cells, resulting in increased TGF-β1 in the culture media. In transgenic rat aorta transplantation models, inhibition of EC apoptosis in B-cell lymphoma (Bcl)-xL(+/+) knock-in rat aortic allografts significantly reduces TGF-β1 production in both allograft endothelia and blood plasma, which in turn decreases accumulation of SM22α+ cells from transgenic recipient ECs in neointima and alleviated TA [38].
It has been reported that the transcript levels of Notch-2, -3, and -4 are markedly downregulated in TA. In Quillard et al.’s research, TA correlates with high levels of tumor necrosis factor (TNF), TGF-β and interleukin (IL)-10. They also found that Notch-4 expression is decreased in transplants and cultured ECs. Further knockdown of Notch-4 and HES-1 by small interfering RNA (siRNA) promotes ECs apoptosis. As expected, silencing Notch-4 or HES-1 drastically inhibits repair of endothelial injury [39]. Mackenzie et al.’s studies demonstrated that Notch-4 provides endothelial protection in two ways: inhibition of the c-Jun N-terminal kinase (JNK)-dependent pro-apoptotic pathway in an RBP-Jκ-dependent manner and induction of an anti-apoptotic pathway through an RBP-Jκ-independent upregulation of Bcl-2 [40].
However, after Notch-2 ICD transduction in cultured human arterial endothelial cells (HAEC) and HUVECs or induced Notch-2 expression in HUVECs, EC apoptosis is promoted, notably through inhibiting the expression of survivin [41,42]. In addition, using a mouse model of pulmonary arterial hypertension, Li et al. showed that activation of nuclear factor kappa B (NF-κB) upregulates the expression levels of Notch-3, proapoptotic gene caspase 3 and Bax, downregulates antiapoptotic gene Bcl-2 expression in lung microvascular endothelial cells, which leads to EC apoptosis and endothelial-mesenchymal transition (EndMT) occurring in the lung [43]. Moreover, through activating Notch-1, HES-1 and caspase-3, accelerated cell apoptosis has been observed in human Eahy926 cells treated with high glucose [44].
All of this evidence supports that Notch activation is involved in neointimal formation through regulating EC apoptosis and survival. It is possible that the four different Notch receptors have different roles in EC apoptosis.
5. Endothelial-Mesenchymal Transition
EndMT is a specific form of epithelial-mesenchymal transition (EMT), which is an important biologic transdifferentiation process that participates in embryogenesis, organ development, tissue regeneration, organ fibrosis and cancer metastasis [45]. Similar to the process of EMT, when undergoing EndMT, ECs lose their endothelial specific markers, such as CD31, also known as platelet endothelial cell adhesion molecule-1 (PECAM-1) or VE-cadherin, gain mesenchymal markers, such as α-smooth muscle actin (α-SMA) or fibroblast-specific protein 1 (FSP-1, also known as S100A4), lose cell-cell junctions and acquire invasive and migratory properties [46,47].
EndMT has emerged as a player in the pathogenesis of vascular neointimal hyperplasia. Notintimal cells may arise through migration and proliferation of VSMCs; recent studies showed that the endothelium is also a source of smooth muscle-like cells [48]. It has been reported that EndMT occurs in myoendothelial cells in human atherosclerotic plaques and porcine aortic tissues. In vitro and in vivo experiments all showed that ECs exposed to disturbed flow undergo EndMT, which contributes to neointimal hyperplasia and induces atherogenic differentiation of ECs [49]. Through introducing endothelial-specific deletion of fibroblast growth factor receptor substrate 2 α (Frs2α) in atherosclerotic ApoE(−/−) mice, Chen et al. reported that these double-knockout mice exhibit extensive development of EndMT and increased neointimal formation. Furthermore, patients with coronary atherosclerosis showed that the loss of endothelial fibroblast growth factor receptor1 (FGFR1) expression leads to activation of endothelial TGF-β signaling and the development of EndMT in atherosclerotic plaques [50]. In vivo murine cell lineage-tracing models also presented that endothelial-derived cells contribute to neointimal formation through EndMT, which is dependent on the activation of the TGF-β mediated Smad2/3-Slug signaling pathway [51].
As a major regulator of cell phenotype, Notch is involved in the process of EndMT. Noseda et al. provided the first evidence that Jagged-1/Notch interactions induce endothelial-to-mesenchymal transformation. Notch activation in ECs results in morphological, phenotypic and functional changes, which is consistent with mesenchymal transformation [52]. Notch and TGF-β/smad3 signaling synergistically induce Snail expression in ECs and promote EndMT in cardiac cushion morphogenesis [53]. Blocking the Notch signaling pathway by using DAPT, EndMT in rat corneal ECs induced by TGF β1, -β2 or -β3 is prevented and the transformed ECs are reversed to a normal phenotype [54].
Neointimal hyperplasia occurs seriously in arteriovenous fistulas (AVFs) of chronic kidney disease (CKD) mice or patients. ECs of AVFs in CKD mice or patients express mesenchymal markers (FSP-1 and/or α-SMA) and exhibit increased expression and nuclear localization of the Notch intracellular domain. Uremic mice also show a decreased expression of VE-cadherin, whereas the expressions of Notch-1, -4, RBP-Jκ and Notch target genes are increased in ECs of AVFs. Blockade of the Notch pathway by DAPT or by RBP-Jκ knockout suppresses neointimal formation in mice [23]. Thus, data in the literature suggest that the Notch pathway is correlated with EndMT and contributes to the neointimal hyperplasia in vascular remodeling (Figure 2C).
6. The Contact Interaction between Endothelial Cells and Smooth Muscle Cells
Vessel wall is mainly composed of ECs and parietal cells (VSMCs and pericytes). The direct communication between ECs and VSMCs through myoendothelial gap junctions and microprojections has been widely known [55,56]. In addition, Notch ligand-receptor interaction is another direct communication means between ECs and VSMCs. Contact-mediated activation of Notch signaling plays important roles in cell and vessel maturation, survival and homeostasis. In these processes, different Notch receptors may have specific roles [57].
In vertebrates, ECs can express three Notch ligands (Dll-4, Jagged-1 and -2) and all of the known Notch receptors (Notch-1, -2, -3 and -4), while VSMCs express ligand Jagged-1 and three receptors (Notch-1, -2 and -3) [15]. We summarized the effects of ECs-VSMCs interaction mediated by different Notch receptor-ligand on neointimal formation in Table 1A. During vascular development, VSMCs recognize Notch ligand Jagged-1 on ECs and induce the expression of integrin αvβ3 in VSMCs, which facilitates VSMCs adhering to endothelial basement membrane and promotes vessel maturation [58]. In post-development vessels, the endothelial protein kinase B (PKB, also known as Akt) deletion reduces the expression of endothelial Jagged-1 and leads to the gradual loss of VSMCs due to diminished Jagged-1/Notch signaling. It sustains that contact-mediated activation of Notch signaling is critical in maintaining vascular stability and homeostasis [59]. Among the Notch signaling molecules, EC membrane ligand Jagged-1 is required for the induction of Notch-3 in VSMCs and inducing VSMC differentiation [60,61]. Neither the addition of soluble Jagged-1 nor EC-conditioned medium induces VSMC differentiation, while co-cultured ECs with VSMCs induce VSMC differentiation; furthermore, knockdown Jagged-1 expression in ECs can abrogate the co-cultured VSMC phenotype change. All of this evidence strongly supports that the direct heterocellular cell-cell contact is necessary for regulating VSMC differentiation via Jagged-1/Notch-3 signaling [62].
Except for Notch-3, Notch-2 is also activated in VSMCs co-cultured with ECs. Both Notch-2 and Notch-3 in VSMCs are mediators of EC-induced differentiated phenotype and contribute to increased contractile protein expression. However, the two receptors have separate and distinct functions. Notch-2 is specifically required for the suppression of cell proliferation, while Notch-3 is mainly responsible for VSMC secretory function [63].
VSMC proliferation, phenotype change and ECM secretion are hallmarks of neointimal formation in cardiovascular diseases and vascular surgery. Balloon injury induced Notch-1, Notch-3 and Jagged-1 expression in rat carotid arteries. However, soluble Jagged-1 inhibits neointimal formation after balloon injury or vein graft by decreasing VSMC proliferation and migration through interference with the Notch signaling pathway [64]. This evidence suggested that inhibition of neointimal formation may be due to inactivation of the Notch signaling pathway through soluble Jagged-1 competing with EC Jagged-1 to bind with the Notch receptor.
Reciprocally, VSMCs’ Jagged-1 activate Notch pathway in ECs through the Notch-1 receptor and induce EC proliferation. Notch signaling-deficient primary VSMCs have reduced proliferation and migration capacities and a diminished expression of Jagged-1 ligand. After being co-cultured with such VSMCs, ECs exhibit reduced growth rates and lower levels of activated Notch-1 receptor (Notch-1ICD) [65]. As discussed previously, EC proliferation and regeneration are critical events in neointimal formation. Through regulating the contact between ECs and VSMCs, activation of Notch could be manipulated, which may represent a unique therapeutic target to improve neointimal formation after vascular injury.
Notch signaling between cells can also be transmitted by exosomes from a distance. Dll-4 is incorporated into endothelial exosomes; the Dll-4 containing exosomes can freely travel through the 3D collagen matrix, transfer Dll-4 protein to distant tip cells and induce tip cell retraction [72]. However, whether there exists exosome-mediated Notch signaling between ECs and VSMCs in neointimal formation has not yet been reported.
Taken together, Jagged-1 seems to be the only ligand in the Notch pathway that plays an important role in both ECs and VSMCs during neointimal formation development. Notch-1 may be the only receptor in the EC lineage that recognizes the Notch ligand in VSMCs, whereas several receptors (Notch-2, -3) are involved in the VSMC lineage to recognize the Notch ligand in ECs (Figure 3).
7. Endothelial Cell Secretory Function
Although the vascular endothelium is made up from only a single layer of ECs, it works as an “endocrine organ” via an autocrine and/or paracrine process and contributes to vascular homeostasis, such as angiogenesis, inflammation, platelet aggregation and vascular remodeling [73,74]. The regulatory molecules derived from ECs include non-growth factors and growth factors, such as nitric oxide (NO), VEGF, platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF) and insulin-like growth factor-1, etc. [75,76] (Figure 3). There are many reports about crosstalk between Notch signaling and the regulatory molecules mentioned above; we refer the readers to the three main kinds of factors derived from ECs regulated by Notch signaling and their effects on neointimal hyperplasia (Table 1B).
Mainly produced by endothelial NO synthase (eNOS), NO derived from EC is an important mediator of normal and pathologic vascular remodeling [77]. NO not only confers anti-platelet and anti-inflammatory properties to the vessels, but also promotes EC proliferation [66], inhibits VSMC proliferation and migration [68], controls M1 macrophage polarization [78], regulates redox balance [79], keeps the stability and function of blood vessels and suppresses neointimal hyperplasia [80]. A recent study reported that Notch induces Activin A expression, thereby activating the PI3K/Akt pathway to phosphorylate eNOS and promoting NO production [67]. Inhibition of Notch decreases endothelial NO production by reduced eNOS expression [81]. In vein grafts of aged rats, the reduced expression of Dll-4 and Notch-4 has been found, which is associated with the decreased eNOS protein expression, reduced eNOS membrane targeting and colocalization with caveolin-1, as well as significantly thicker neointima [82].
The contribution of VEGF in neointimal formation has been widely evaluated. Recent studies demonstrated that VEGF can block neointimal formation through inducing EC growth and reducing VSMC growth after vascular injury [83]. In addition, bone marrow-derived mesenchymal stem cells treated with VEGF differentiate into endothelial-like cells and significantly attenuate neointimal thickness [84]. However, lentivirus-mediated VEGF-A inhibition can decrease the venous neointimal hyperplasia of AVFs [85]. This evidence suggested that the effect of VEGF on neointimal formation is complicated. Although there is no direct evidence to support that Notch signaling mediates neointimal hyperplasia through autocrine or paracrine of VEGF in ECs, inhibition of Notch-1 or Notch-4 can block thymosin β 4-induced VEGF expression in HUVECs [86]. In contrast, many studies uncovered that VEGF can activate Notch signaling in ECs and act as an upstream mediator of the Notch pathway [87,88].
As a smooth muscle cell growth and survival factor, PDGF also plays a prominent role in VSMCs migrating into the neointima following acute injury or in atherosclerosis. High shear stress inhibits arterial wall thickening in vivo, which may be related to enhanced activation of PDGF-R alpha in VSMCs by PDGF isoforms secreted from the endothelium. The neutralizing antibody against PDGF-AA enhances VSMC migration; in contrast, antibodies against PDGF-BB abolish VSMC migration [70]. VSMC-rich neointimal formation is accelerated in the ligated carotid artery of mice treated with erythropoietin delta, by which the expression and release of PDGF-B is induced in HUVECs [69]. Findings also showed that PDGF-BB and PDGF-DD are all VSMC phenotypic modulators. PDGF-DD expression is increased in neointimal lesions in the aortic arch region of apolipoprotein C-deficient ApoE(−/−) mice. In addition, human ECs exposed to an atherosclerosis-prone flow pattern, as in vascular regions susceptible to the development of atherosclerosis, exhibit a significant increase in PDGF-DD expression [71].
Several recent studies reported that Notch signaling is involved in regulating PDGF production from ECs. Exposure of human brain microvascular endothelial cells to DAPT or silencing of Notch-1 results in abrogation of cocaine-mediated induction of PDGF-B. The study provided the first evidence of the involvement of Notch-1 activation in PDGF-B expression [89]. β-catenin, a key signal molecule of the Wnt/β-catenin-Dll4/Notch signaling cascade in endothelia, its transcriptional activity directly regulates the endothelial expression of PDGF-B [90]. Conditional medium from matricellular protein, secreted protein acidic and rich in cysteine (SPARC) overexpressed neuroblastoma cells show suppressed expression of VEGF, PDGF, FGF and matrix metalloprotein 9 in ECs, which is mediated by the inhibition of the Notch signaling pathway [91]. Taken together, these findings suggest that PDGF secretion induced by Notch may be involved in neointimal hyperplasia.
There are also many other EC-derived molecules that have been proven or speculated to influence neointimal hyperplasia. For instance, apoptotic ECs actively release paracrine mediators’ C-terminal fragment of perlecan and epidermal growth factor, which inhibit apoptosis of mesenchymal stem cells (MSC), which are pivotal to vascular repair and neointimal formation [92]. As a secreted glycoprotein that has been implicated in regulating VSMC proliferation and migration, the downregulated expression of apolipoprotein D (APOD) is partly caused by paracrine secretion of ECs. In addition, Notch-3 on mural cells also promotes the downregulation of APOD, possibly through interaction with the Jagged-1 ligands on ECs [93].
8. Endothelial Cell Barrier Dysfunction
The development of functional blood vessel requires an integrated layer of endothelial cells. Based on the EC junction, the structural and functional integrity of the endothelium is fundamental for maintaining vascular homeostasis [94]. Destruction of the protective endothelial barrier will subsequently lead to vascular injury and neointimal formation. Changes in shear and/or hoop stress, direct drug-induced cytotoxicity, mechanical device implant-induced injury or inflammatory response in vascular surgery or cardiovascular surgery induce vascular injury and cause EC dysfunction [95]. As discussed above, vascular injury is a complex cascade of events involving endothelial denudation, the release of growth factors and cytokines which triggers platelet degranulation and aggregation, subsequent inflammatory cells or mediators invading the injured locations, smooth muscle cell proliferation and migration to form neointimal hyperplasia at the subendothelial space. Reciprocally, platelet activation and inflammation response lead to delayed re-endothelialization and endothelial dysfunction [96].
Endothelial dysfunction is characterized by EC phenotype change with impaired endothelium-dependent barrier and imbalance between re-endothelialization and apoptosis or growth inhibiting and growth-promoting substances. Notch signaling cascades are involved in and partially responsible for EC dysfunction events (Figure 4). For example, apoptotic or senescent phenotype ECs lose their barrier function. Activation of Notch increases myosin light chain phosphorylation by activating Rho kinase, which further triggers EC acquiring senescence phenotype and leads to hyperpermeability of the endothelium [97]. In atherosclerosis, Notch activation induces EC senescence and prompts the expression and secretion of pro-inflammatory cytokines such as IL-6, IL-8, IL-1α. Among these factors, the upregulated IL-6 may mediate leukocyte transendothelial migration [98]. In addition, Notch activation also causes EC to acquire VSMC phenotype, which leads to ECs losing their barrier function and induces neointimal hyperplasia [23]. Consequently, attenuation of Notch signaling in ECs might provide a treatment strategy for neointimal formation.
As mentioned above, activation of Notch-2 induces EC apoptosis while Notch-4 plays a protective role [39,43]. In arterial ECs, pro-inflammatory cytokine TNF-α elicits a switch in Notch expression, which is characterized by Notch-2 predominance over Notch-4. The events lead to a reduced Notch activity, then promoting caspase-dependent EC apoptosis and vascular dysfunction [99]. In most cases, the upregulated expressions of Jagged-1, Dll-4, Notch-1 and Notch-4 are associated with inhibition of EC proliferation and vascular dysfunction [100,101]. This evidence proved that loss of ECs could lead to increased platelet reactivity and VSMC proliferation, thereby facilitating neointimal formation.
EC dysfunction may also be related to the change of the secretory function of ECs. Among the substances released from ECs, NO is involved in the impairment of endothelium-dependent vasodilatation. Endovascular interventions are associated with diminished bioavailability of NO and increased local inflammatory response, which triggers EC apoptosis [102,103,104]. Reducing eNOS expression or promoting inducible nitric oxide synthase (iNOS) expression induces EC apoptosis and dysfunction, which can be mediated through the Jagged-1/Notch pathway [105]. To conclude, enhanced degradation of NO and decreased eNOS expression and/or activity leads to EC dysfunction and contributes to neointimal formation. As shown in Figure 4, we summarized the relationship between Notch signaling and EC dysfunction during neointimal formation development.
9. Limits and Perspectives
We have reviewed here the accumulated evidence that the Notch pathway is involved in multiple aspects of EC key functions (proliferation, regeneration, apoptosis, differentiation, cell-cell interaction) and contributes to neointimal formation. However, our review also found a number of apparent inconsistencies and problems in these studies. Although the relative level of p21 expression may decide EC growth after Notch activation, no data show the exact expression level or range of p21. The patterns of Notch-1 to -4 expression can play different roles in EC apoptosis, but which components function at what apoptosis steps in ECs? Notch signaling seems to induce endothelial mesenchymal phenotype switch, so does there exist a specific relationship among ligands, receptors, and target genes in the process of transdifferentiation? How does Notch play a role in EC regeneration and differentiation? Except NO, how does the Notch pathway play any role in the regulation of other cytokines secreted from ECs to cause EC dysfunction? How about the crosstalk between Notch and other signaling pathways? Given that the Notch has a significant role in vascular development, further understanding of the Notch signaling pathway in the context of vascular biology will likely provide novel insights into the mechanisms of neointimal formation and new opportunities for rational therapeutic intervention.
Acknowledgments
This work was supported by grants from National Nature Science Foundation of China 81470963, the Natural Science Foundation Project of Chongqing cstc2014jcyjA10098 and the Student’s Platform for Innovation and Entrepreneurship Training Program 201690031045.
Author Contributions
Yun Wang conceptualized this review. Ding-Yuan Tian wrote the manuscript and prepared the table. Xu-Rui Jin drew the figures. Xi Zeng edited the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ADAM | a disintegrin and metalloproteinases |
| AGS | alagille syndrome |
| APOD | apolipoprotein D |
| AVF | arteriovenous fistula |
| AVG | arteriovenous graft |
| bFGF | basic fibroblast growth factor |
| CACs | circulating angiogenic cells |
| CADASIL | cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy |
| CKD | chronic kidney disease |
| CVD | cardiovascular diseases |
| DAPT | N-[N-(3,5-Difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester |
| Dll | delta-like |
| EC | endothelial cell |
| ECM | extracellular matrix |
| EMT | Epithelial-mesenchymal transition |
| EndMT | Endothelial-mesenchymal transition |
| eNOS | endothelial NO synthase |
| EPC | endothelial progenitor cell |
| FGFR1 | Fibroblast growth factor receptor1 |
| Frs2α | Fibroblast growth factor receptor substrate 2 α |
| FSP-1 | fibroblast-specific protein 1 |
| HAEC | human arterial endothelial cell |
| HERP | HES-related repressor protein |
| HES | hairy and enhancer of split |
| HUVEC | human umbilical vein endothelial cell |
| IFNγ | Interferon γ |
| IL | interleukin |
| iNOS | inducible nitric oxide synthase |
| MAML | Mastermind-like |
| MAPK | mitogen-activated protein kinase |
| MSC | mesenchymal stem cell |
| NF-κB | nuclear factor κB |
| NICD | Notch intracellular domain |
| NO | Nitric oxide |
| PDGF | platelet-derived growth factor |
| PECAM-1 | platelet endothelial cell adhesion molecule-1 |
| Rb | retinoblastoma |
| ROS | reactive oxygen species |
| α-SMA | α-smooth muscle actin |
| SPARC | secreted protein acidic and rich in cysteine |
| TA | transplant arteriosclerosis |
| TGF-β | transforming growth factor-β |
| TNF | tumor necrosis factor |
| VEGF | vascular endothelial growth factor |
| VSMC | vascular smooth muscle cell |
| WT | wild type |
References
- Brahmbhatt, A.; Remuzzi, A.; Franzoni, M.; Misra, S. The molecular mechanisms of hemodialysis vascular access failure. Kidney Int. 2016, 89, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Zhao, X.J.; Xiang, X.Y. Gene therapy for vein graft failure. J. Card. Surg. 2013, 28, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Ahanchi, S.S.; Tsihlis, N.D.; Kibbe, M.R. The role of nitric oxide in the pathophysiology of intimal hyperplasia. J. Vasc. Surg. 2007, 45, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Butt, M.; Dwivedi, G.; Blann, A.; Khair, O.; Lip, G.Y. Endothelial dysfunction: Methods of assessment & implications for cardiovascular diseases. Curr. Pharm. Des. 2010, 16, 3442–3454. [Google Scholar] [PubMed]
- Allaire, E.; Clowes, A.W. Endothelial cell injury in cardiovascular surgery: The intimal hyperplastic response. Ann. Thorac. Surg. 1997, 63, 582–591. [Google Scholar] [PubMed]
- Bhardwaj, S.; Roy, H.; Heikura, T.; Yla-Herttuala, S. VEGF-A, VEGF-D and VEGF-D (DeltaNDeltaC) induced intimal hyperplasia in carotid arteries. Eur. J. Clin. Investig. 2005, 35, 669–676. [Google Scholar] [CrossRef] [PubMed]
- DiRenzo, D.M.; Chaudhary, M.A.; Shi, X.; Franco, S.R.; Zent, J.; Wang, K.; Guo, L.W.; Kent, K.C. A crosstalk between TGF-β/Smad3 and Wnt/β-catenin pathways promotes vascular smooth muscle cell proliferation. Cell Signal. 2016, 28, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, J.; Xu, C.; Yang, J.; Guo, Q.; Hu, Q.; Jiang, H. Resveratrol inhibits phenotypic switching of neointimal vascular smooth muscle cells after balloon injury through blockade of Notch pathway. J. Cardiovasc. Pharmacol. 2014, 63, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Benedito, R.; Hellstrom, M. Notch as a hub for signaling in angiogenesis. Exp. Cell. Res. 2013, 319, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Krebs, L.T.; Xue, Y.; Norton, C.R.; Shutter, J.R.; Maguire, M.; Sundberg, J.P.; Gallahan, D.; Closson, V.; Kitajewski, J.; Callahan, R.; et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000, 14, 1343–1352. [Google Scholar] [PubMed]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Gama-Norton, L.; Ferrando, E.; Ruiz-Herguido, C.; Liu, Z.; Guiu, J.; Islam, A.B.; Lee, S.U.; Yan, M.; Guidos, C.J.; Lopez-Bigas, N.; et al. Notch signal strength controls cell fate in the haemogenic endothelium. Nat. Commun. 2015, 6, 8510. [Google Scholar] [CrossRef] [PubMed]
- De Celis, J.F.; Garcia-Bellido, A. Roles of the Notch gene in Drosophila wing morphogenesis. Mech. Dev. 1994, 46, 109–122. [Google Scholar] [CrossRef]
- Hofmann, J.J.; Iruela-Arispe, M.L. Notch signaling in blood vessels: Who is talking to whom about what? Circ. Res. 2007, 100, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Iso, T.; Hamamori, Y.; Kedes, L. Notch signaling in vascular development. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Krebs, L.T.; Shutter, J.R.; Tanigaki, K.; Honjo, T.; Stark, K.L.; Gridley, T. Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev. 2004, 18, 2469–2473. [Google Scholar] [CrossRef] [PubMed]
- Louvi, A.; Arboleda-Velasquez, J.F.; Artavanis-Tsakonas, S. CADASIL: A critical look at a Notch disease. Dev. Neurosci. 2006, 28, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Gridley, T. Notch signaling and inherited disease syndromes. Hum. Mol. Genet. 2003, 12, 9–13. [Google Scholar] [CrossRef]
- Liang, M.; Wang, Y.; Liang, A.; Dong, J.F.; Du, J.; Cheng, J. Impaired integrin β3 delays endothelial cell regeneration and contributes to arteriovenous graft failure in mice. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Koga, J.; Nakano, T.; Dahlman, J.E.; Figueiredo, J.L.; Zhang, H.; Decano, J.; Khan, O.F.; Niida, T.; Iwata, H.; Aster, J.C.; et al. Macrophage notch ligand delta-like 4 promotes vein graft lesion development: Implications for the treatment of vein graft failure. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2343–2353. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Wang, Y.; Liang, A.; Mitch, W.E.; Roy-Chaudhury, P.; Han, G.; Cheng, J. Migration of smooth muscle cells from the arterial anastomosis of arteriovenous fistulas requires Notch activation to form neointima. Kidney Int. 2015, 88, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Caolo, V.; Schulten, H.M.; Zhuang, Z.W.; Murakami, M.; Wagenaar, A.; Verbruggen, S.; Molin, D.G.; Post, M.J. Soluble Jagged-1 inhibits neointima formation by attenuating Notch-Herp2 signaling. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liang, A.; Luo, J.; Liang, M.; Han, G.; Mitch, W.E.; Cheng, J. Blocking notch in endothelial cells prevents arteriovenous fistula failure despite CKD. J. Am. Soc. Nephrol. 2014, 25, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Noseda, M.; Chang, L.; McLean, G.; Grim, J.E.; Clurman, B.E.; Smith, L.L.; Karsan, A. Notch activation induces endothelial cell cycle arrest and participates in contact inhibition: Role of p21Cip1 repression. Mol. Cell Biol. 2004, 24, 8813–8822. [Google Scholar] [CrossRef] [PubMed]
- Leslie, J.D.; Ariza-McNaughton, L.; Bermange, A.L.; McAdow, R.; Johnson, S.L.; Lewis, J. Endothelial signalling by the Notch ligand Delta-like 4 restricts angiogenesis. Development 2007, 134, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Sainson, R.C.; Aoto, J.; Nakatsu, M.N.; Holderfield, M.; Conn, E.; Koller, E.; Hughes, C.C. Cell-autonomous notch signaling regulates endothelial cell branching and proliferation during vascular tubulogenesis. FASEB J. 2005, 19, 1027–1029. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Hobson, B.; Denekamp, J. Endothelial proliferation in tumours and normal tissues: Continuous labelling studies. Br. J. Cancer 1984, 49, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Henderson, A.M.; Wang, S.J.; Taylor, A.C.; Aitkenhead, M.; Hughes, C.C. The basic helix-loop-helix transcription factor HESR1 regulates endothelial cell tube formation. J. Biol. Chem. 2001, 276, 6169–6176. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.L.; Henderson, A.M.; Hughes, C.C. Notch activation during endothelial cell network formation in vitro targets the basic HLH transcription factor HESR-1 and downregulates VEGFR-2/KDR expression. Microvasc. Res. 2002, 64, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Xiao, M.; Balint, K.; Soma, A.; Pinnix, C.C.; Capobianco, A.J.; Velazquez, O.C.; Herlyn, M. Inhibition of endothelial cell proliferation by Notch1 signaling is mediated by repressing MAPK and PI3K/Akt pathways and requires MAML1. FASEB J. 2006, 20, 1009–1011. [Google Scholar] [CrossRef] [PubMed]
- Benedito, R.; Trindade, A.; Hirashima, M.; Henrique, D.; Da Costa, L.L.; Rossant, J.; Gill, P.S.; Duarte, A. Loss of Notch signalling induced by Dll4 causes arterial calibre reduction by increasing endothelial cell response to angiogenic stimuli. BMC Dev. Biol. 2008, 8, 117. [Google Scholar] [CrossRef] [PubMed]
- Ii, M.; Takeshita, K.; Ibusuki, K.; Luedemann, C.; Wecker, A.; Eaton, E.; Thorne, T.; Asahara, T.; Liao, J.K.; Losordo, D.W. Notch signaling regulates endothelial progenitor cell activity during recovery from arterial injury in hypercholesterolemic mice. Circulation 2010, 121, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Xie, L.; Shi, K.; Zhou, T.; Hua, Y.; Liu, H. Notch signaling change in pulmonary vascular remodeling in rats with pulmonary hypertension and its implication for therapeutic intervention. PLoS ONE 2012, 7, e51514. [Google Scholar] [CrossRef] [PubMed]
- Dabral, S.; Tian, X.; Kojonazarov, B.; Savai, R.; Ghofrani, H.A.; Weissmann, N.; Florio, M.; Sun, J.; Jonigk, D.; Maegel, L.; et al. Notch1 signalling regulates endothelial proliferation and apoptosis in pulmonary arterial hypertension. Eur. Respir. J. 2016, 48, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Emuss, V.; Lagos, D.; Pizzey, A.; Gratrix, F.; Henderson, S.R.; Boshoff, C. KSHV manipulates Notch signaling by DLL4 and JAG1 to alter cell cycle genes in lymphatic endothelia. PLoS Pathog. 2009, 5, e1000616. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.X.; Liang, L.; Wang, L.; Han, J.T.; Zhu, X.X.; Han, H.; Hu, D.H.; Zhang, P. Inhibition of Notch signaling leads to increased intracellular ROS by up-regulating Nox4 expression in primary HUVECs. Cell. Immunol. 2014, 287, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xiong, J.; Yang, B.; Zhou, Q.; Wu, Y.; Luo, H.; Zhou, H.; Liu, N.; Li, Y.; Song, Z.; et al. Endothelial cell apoptosis induces TGF-β signaling-dependent host endothelial-mesenchymal transition to promote transplant arteriosclerosis. Am. J. Transplant. 2015, 15, 3095–3111. [Google Scholar] [CrossRef] [PubMed]
- Quillard, T.; Coupel, S.; Coulon, F.; Fitau, J.; Chatelais, M.; Cuturi, M.C.; Chiffoleau, E.; Charreau, B. Impaired Notch4 activity elicits endothelial cell activation and apoptosis: Implication for transplant arteriosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2258–2265. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, F.; Duriez, P.; Wong, F.; Noseda, M.; Karsan, A. Notch4 inhibits endothelial apoptosis via RBP-Jkappa-dependent and -independent pathways. J. Biol. Chem. 2004, 279, 11657–11663. [Google Scholar] [CrossRef] [PubMed]
- Kiriakidis, S.; Henze, A.T.; Kruszynska-Ziaja, I.; Skobridis, K.; Theodorou, V.; Paleolog, E.M.; Mazzone, M. Factor-inhibiting HIF-1 (FIH-1) is required for human vascular endothelial cell survival. FASEB J. 2015, 29, 2814–2827. [Google Scholar] [CrossRef] [PubMed]
- Quillard, T.; Devalliere, J.; Chatelais, M.; Coulon, F.; Seveno, C.; Romagnoli, M.; Barille Nion, S.; Charreau, B. Notch2 signaling sensitizes endothelial cells to apoptosis by negatively regulating the key protective molecule survivin. PLoS ONE 2009, 4, e8244. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wei, C.; Kim, I.K.; Janssen-Heininger, Y.; Gupta, S. Inhibition of nuclear factor-kappaB in the lungs prevents monocrotaline-induced pulmonary hypertension in mice. Hypertension 2014, 63, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Du, B.; Xie, F.; Cai, W.; Liu, Y.; Li, Y.; Feng, L.; Qiu, L. Vaccarin attenuates high glucose-induced human EA*hy926 endothelial cell injury through inhibition of Notch signaling. Mol. Med. Rep. 2016, 13, 2143–2150. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Kovacic, J.C.; Mercader, N.; Torres, M.; Boehm, M.; Fuster, V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: From cardiovascular development to disease. Circulation 2012, 125, 1795–1808. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Wang, N.; Zhang, T.C. The role of endothelial-mesenchymal transition in development and pathological process. IUBMB Life 2012, 64, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Hong, M.S.; Fu, C.; Schmit, B.M.; Su, Y.; Berceli, S.A.; Jiang, Z. Preexisting smooth muscle cells contribute to neointimal cell repopulation at an incidence varying widely among individual lesions. Surgery 2016, 159, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Moonen, J.R.; Lee, E.S.; Schmidt, M.; Maleszewska, M.; Koerts, J.A.; Brouwer, L.A.; Van Kooten, T.G.; Van Luyn, M.J.; Zeebregts, C.J.; Krenning, G.; et al. Endothelial-to-mesenchymal transition contributes to fibro-proliferative vascular disease and is modulated by fluid shear stress. Cardiovasc. Res. 2015, 108, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.Y.; Qin, L.; Baeyens, N.; Li, G.; Afolabi, T.; Budatha, M.; Tellides, G.; Schwartz, M.A.; Simons, M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J. Clin. Investig. 2015, 125, 4514–4528. [Google Scholar] [CrossRef] [PubMed]
- Cooley, B.C.; Nevado, J.; Mellad, J.; Yang, D.; St Hilaire, C.; Negro, A.; Fang, F.; Chen, G.; San, H.; Walts, A.D.; et al. TGF-β signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci. Transl. Med. 2014, 6, 227ra34. [Google Scholar] [CrossRef] [PubMed]
- Noseda, M.; McLean, G.; Niessen, K.; Chang, L.; Pollet, I.; Montpetit, R.; Shahidi, R.; Dorovini-Zis, K.; Li, L.; Beckstead, B.; et al. Notch activation results in phenotypic and functional changes consistent with endothelial-to-mesenchymal transformation. Circ. Res. 2004, 94, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chang, A.; Chang, L.; Niessen, K.; Eapen, S.; Setiadi, A.; Karsan, A. Differential regulation of transforming growth factor β signaling pathways by Notch in human endothelial cells. J. Biol. Chem. 2009, 284, 19452–19462. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Dong, F.; Jia, Y.; Du, H.; Dong, N.; Xu, Y.; Wang, S.; Wu, H.; Liu, Z.; Li, W. Notch signal regulates corneal endothelial-to-mesenchymal transition. Am. J. Pathol. 2013, 183, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, S.; Kapela, A.; Tran, C.H.; Welsh, D.G.; Tsoukias, N.M. Role of microprojections in myoendothelial feedback—A theoretical study. J. Physiol. 2013, 591, 2795–2812. [Google Scholar] [CrossRef] [PubMed]
- Sandow, S.L.; Haddock, R.E.; Hill, C.E.; Chadha, P.S.; Kerr, P.M.; Welsh, D.G.; Plane, F. What’s where and why at a vascular myoendothelial microdomain signalling complex. Clin. Exp. Pharmacol. Physiol. 2009, 36, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Rostama, B.; Peterson, S.M.; Vary, C.P.; Liaw, L. Notch signal integration in the vasculature during remodeling. Vascul. Pharmacol. 2014, 63, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Scheppke, L.; Murphy, E.A.; Zarpellon, A.; Hofmann, J.J.; Merkulova, A.; Shields, D.J.; Weis, S.M.; Byzova, T.V.; Ruggeri, Z.M.; Iruela-Arispe, M.L.; et al. Notch promotes vascular maturation by inducing integrin-mediated smooth muscle cell adhesion to the endothelial basement membrane. Blood 2012, 119, 2149–2158. [Google Scholar] [CrossRef] [PubMed]
- Kerr, B.A.; West, X.Z.; Kim, Y.W.; Zhao, Y.; Tischenko, M.; Cull, R.M.; Phares, T.W.; Peng, X.D.; Bernier-Latmani, J.; Petrova, T.V.; et al. Stability and function of adult vasculature is sustained by Akt/Jagged1 signalling axis in endothelium. Nat. Commun. 2016, 7, 10960. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Lin, S.; Sandig, M.; Mequanint, K. Regulation of vascular smooth muscle cell phenotype in three-dimensional coculture system by Jagged1-selective Notch3 signaling. Tissue Eng. Part A 2014, 20, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Kennard, S.; Lilly, B. NOTCH3 expression is induced in mural cells through an autoregulatory loop that requires endothelial-expressed JAGGED1. Circ. Res. 2009, 104, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Bhattacharyya, A.; Roszell, E.E.; Sandig, M.; Mequanint, K. The role of endothelial cell-bound Jagged1 in Notch3-induced human coronary artery smooth muscle cell differentiation. Biomaterials 2012, 33, 2462–2472. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Lilly, B. Notch signaling governs phenotypic modulation of smooth muscle cells. Vascul. Pharmacol. 2014, 63, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xiao, Y.; Mao, Z.; Huang, J.; Geng, Q.; Wang, W.; Dong, P. Soluble Jagged-1 inhibits restenosis of vein graft by attenuating Notch signaling. Microvasc. Res. 2015, 100, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Proweller, A. Vascular smooth muscle Notch signals regulate endothelial cell sensitivity to angiogenic stimulation. J. Biol. Chem. 2011, 286, 13741–13753. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Huang, L.; Fang, Y.; Guo, R.; Yin, Y.; Zhao, X. Transplantation of endothelial progenitor cells overexpressing endothelial nitric oxide synthase enhances inhibition of neointimal hyperplasia and restores endothelium-dependent vasodilatation. Microvasc. Res. 2011, 81, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.C.; Fu, Y.; Garside, V.C.; Niessen, K.; Chang, L.; Fuller, M.; Setiadi, A.; Smrz, J.; Kyle, A.; Minchinton, A.; et al. Notch initiates the endothelial-to-mesenchymal transition in the atrioventricular canal through autocrine activation of soluble guanylyl cyclase. Dev. Cell 2011, 21, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Tsihlis, N.D.; Oustwani, C.S.; Vavra, A.K.; Jiang, Q.; Keefer, L.K.; Kibbe, M.R. Nitric oxide inhibits vascular smooth muscle cell proliferation and neointimal hyperplasia by increasing the ubiquitination and degradation of UbcH10. Cell Biochem. Biophys. 2011, 60, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Janmaat, M.L.; Heerkens, J.L.; De Bruin, A.M.; Klous, A.; De Waard, V.; De Vries, C.J. Erythropoietin accelerates smooth muscle cell-rich vascular lesion formation in mice through endothelial cell activation involving enhanced PDGF-BB release. Blood 2010, 115, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, R.; Gaetano, C.; Antonini, A.; Pompilio, G.; Bracco, E.; Ronnstrand, L.; Heldin, C.H.; Capogrossi, M.C. Different effects of high and low shear stress on platelet-derived growth factor isoform release by endothelial cells: Consequences for smooth muscle cell migration. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.A.; Deaton, R.A.; Hastings, N.E.; Shang, Y.; Moehle, C.W.; Eriksson, U.; Topouzis, S.; Wamhoff, B.R.; Blackman, B.R.; Owens, G.K. PDGF-DD, a novel mediator of smooth muscle cell phenotypic modulation, is upregulated in endothelial cells exposed to atherosclerosis-prone flow patterns. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Sharghi-Namini, S.; Tan, E.; Ong, L.L.; Ge, R.; Asada, H.H. Dll4-containing exosomes induce capillary sprout retraction in a 3D microenvironment. Sci. Rep. 2014, 4, 4031. [Google Scholar] [CrossRef] [PubMed]
- Feletou, M. The endothelium: Part 1: Multiple functions of the endothelial cells-focus on endothelium-derived vasoactive mediators. In Integrated Systems Physiology: From Molecule to Function to Disease; Granger, D.N., Granger, J.P., Eds.; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2011. [Google Scholar]
- Triggle, C.R.; Samuel, S.M.; Ravishankar, S.; Marei, I.; Arunachalam, G.; Ding, H. The endothelium: Influencing vascular smooth muscle in many ways. Can. J. Physiol. Pharmacol. 2012, 90, 713–738. [Google Scholar] [CrossRef] [PubMed]
- Peiro, C.; Redondo, J.; Rodriguez-Martinez, M.A.; Angulo, J.; Marin, J.; Sanchez-Ferrer, C.F. Influence of endothelium on cultured vascular smooth muscle cell proliferation. Hypertension 1995, 25, 748–751. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Nico, B.; Crivellato, E. The role of pericytes in angiogenesis. Int. J. Dev. Biol. 2011, 55, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Miller, T.W.; Isenberg, J.S.; Roberts, D.D. Molecular regulation of tumor angiogenesis and perfusion via redox signaling. Chem. Rev. 2009, 109, 3099–3124. [Google Scholar] [CrossRef] [PubMed]
- Lavin, B.; Gomez, M.; Pello, O.M.; Castejon, B.; Piedras, M.J.; Saura, M.; Zaragoza, C. Nitric oxide prevents aortic neointimal hyperplasia by controlling macrophage polarization. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1739–1746. [Google Scholar] [CrossRef] [PubMed]
- Bahnson, E.S.; Koo, N.; Cantu-Medellin, N.; Tsui, A.Y.; Havelka, G.E.; Vercammen, J.M.; Jiang, Q.; Kelley, E.E.; Kibbe, M.R. Nitric oxide inhibits neointimal hyperplasia following vascular injury via differential, cell-specific modulation of SOD-1 in the arterial wall. Nitric Oxide 2015, 44, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Pearce, C.G.; Najjar, S.F.; Kapadia, M.R.; Murar, J.; Eng, J.; Lyle, B.; Aalami, O.O.; Jiang, Q.; Hrabie, J.A.; Saavedra, J.E.; et al. Beneficial effect of a short-acting NO donor for the prevention of neointimal hyperplasia. Free Radic. Biol. Med. 2008, 44, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Patenaude, A.; Fuller, M.; Chang, L.; Wong, F.; Paliouras, G.; Shaw, R.; Kyle, A.H.; Umlandt, P.; Baker, J.H.; Diaz, E.; et al. Endothelial-specific Notch blockade inhibits vascular function and tumor growth through an eNOS-dependent mechanism. Cancer Res. 2014, 74, 2402–2411. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Muto, A.; Kudo, F.A.; Model, L.; Eghbalieh, S.; Chowdhary, P.; Dardik, A. Age-related Notch-4 quiescence is associated with altered wall remodeling during vein graft adaptation. J. Surg. Res. 2011, 171, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; McRobb, L.S.; Khachigian, L.M. Inhibition of intimal thickening after vascular injury with a cocktail of vascular endothelial growth factor and cyclic Arg-Gly-Asp peptide. Int. J. Cardiol. 2016, 220, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wu, D.; Yang, Y.; Ji, K.; Gao, P. Endotheliallike cells differentiated from mesenchymal stem cells attenuate neointimal hyperplasia after vascular injury. Mol. Med. Rep. 2016, 14, 4830–4836. [Google Scholar] [PubMed]
- Janardhanan, R.; Yang, B.; Kilari, S.; Leof, E.B.; Mukhopadhyay, D.; Misra, S. The Role of Repeat Administration of Adventitial Delivery of Lentivirus-shRNA-Vegf-A in Arteriovenous Fistula to Prevent Venous Stenosis Formation. J. Vasc. Interv. Radiol. 2016, 27, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Lv, S.; Cheng, G.; Zhou, Y.; Xu, G. Thymosin β4 induces angiogenesis through Notch signaling in endothelial cells. Mol. Cell. Biochem. 2013, 381, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Chiu, A.P.; Wan, A.; Lal, N.; Zhang, D.; Wang, F.; Vlodavsky, I.; Hussein, B.; Rodrigues, B. Cardiomyocyte VEGF Regulates Endothelial Cell GPIHBP1 to relocate lipoprotein lipase to the coronary lumen during diabetes mellitus. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.C.; Mouillesseaux, K.P.; Bautch, V.L. Flt-1 (vascular endothelial growth factor receptor-1) is essential for the vascular endothelial growth factor-Notch feedback loop during angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1952–1959. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Duan, M.; Hu, G.; Buch, S. Platelet-derived growth factor B chain is a novel target gene of cocaine-mediated Notch1 signaling: Implications for HIV-associated neurological disorders. J. Neurosci. 2011, 31, 12449–12454. [Google Scholar] [CrossRef] [PubMed]
- Reis, M.; Czupalla, C.J.; Ziegler, N.; Devraj, K.; Zinke, J.; Seidel, S.; Heck, R.; Thom, S.; Macas, J.; Bockamp, E.; et al. Endothelial Wnt/β-catenin signaling inhibits glioma angiogenesis and normalizes tumor blood vessels by inducing PDGF-B expression. J. Exp. Med. 2012, 209, 1611–1627. [Google Scholar] [CrossRef] [PubMed]
- Gorantla, B.; Bhoopathi, P.; Chetty, C.; Gogineni, V.R.; Sailaja, G.S.; Gondi, C.S.; Rao, J.S. Notch signaling regulates tumor-induced angiogenesis in SPARC-overexpressed neuroblastoma. Angiogenesis 2013, 16, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Soulez, M.; Sirois, I.; Brassard, N.; Raymond, M.A.; Nicodeme, F.; Noiseux, N.; Durocher, Y.; Pshezhetsky, A.V.; Hebert, M.J. Epidermal growth factor and perlecan fragments produced by apoptotic endothelial cells co-ordinately activate ERK1/2-dependent antiapoptotic pathways in mesenchymal stem cells. Stem Cells 2010, 28, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Pajaniappan, M.; Glober, N.K.; Kennard, S.; Liu, H.; Zhao, N.; Lilly, B. Endothelial cells downregulate apolipoprotein D expression in mural cells through paracrine secretion and Notch signaling. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Park, W.J. Endothelial Dysfunction: Clinical Implications in Cardiovascular Disease and Therapeutic Approaches. J. Korean Med. Sci. 2015, 30, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Remuzzi, A.; Ene-Iordache, B. Novel paradigms for dialysis vascular access: Upstream hemodynamics and vascular remodeling in dialysis access stenosis. Clin. J. Am. Soc. Nephrol. 2013, 8, 2186–2193. [Google Scholar] [CrossRef] [PubMed]
- Tesfamariam, B. Endothelial Repair and Regeneration Following Intimal Injury. J. Cardiovasc. Transl. Res. 2016, 9, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, D.; Fredette, N.; Rostama, B.; Tang, Y.; Vary, C.P.; Liaw, L.; Urs, S. RhoA-mediated signaling in Notch-induced senescence-like growth arrest and endothelial barrier dysfunction. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 876–882. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Tan, Y.; Beecham, G.W.; Seo, D.M.; Tian, R.; Li, Y.; Vazquez-Padron, R.I.; Pericak-Vance, M.; Vance, J.M.; Goldschmidt-Clermont, P.J.; et al. Notch activation induces endothelial cell senescence and pro-inflammatory response: Implication of Notch signaling in atherosclerosis. Atherosclerosis 2012, 225, 296–303. [Google Scholar] [CrossRef] [PubMed]
- Quillard, T.; Devalliere, J.; Coupel, S.; Charreau, B. Inflammation dysregulates Notch signaling in endothelial cells: Implication of Notch2 and Notch4 to endothelial dysfunction. Biochem. Pharmacol. 2010, 80, 2032–2041. [Google Scholar] [CrossRef] [PubMed]
- Rooney, P.; Connolly, M.; Gao, W.; McCormick, J.; Biniecka, M.; Sullivan, O.; Kirby, B.; Sweeney, C.; Molloy, E.; Markham, T.; et al. Notch-1 mediates endothelial cell activation and invasion in psoriasis. Exp. Dermatol. 2014, 23, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Chen, S.; Li, K.; Li, L.; Xu, C.; Xiang, B. Signaling pathways in the development of infantile hemangioma. J. Hematol. Oncol. 2014, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Blum, A.; Schneider, D.J.; Sobel, B.E.; Dauerman, H.L. Endothelial dysfunction and inflammation after percutaneous coronary intervention. Am. J. Cardiol. 2004, 94, 1420–1423. [Google Scholar] [CrossRef] [PubMed]
- Hofma, S.H.; Van der Giessen, W.J.; Van Dalen, B.M.; Lemos, P.A.; McFadden, E.P.; Sianos, G.; Ligthart, J.M.; Van Essen, D.; De Feyter, P.J.; Serruys, P.W. Indication of long-term endothelial dysfunction after sirolimus-eluting stent implantation. Eur. Heart J. 2006, 27, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Togni, M.; Windecker, S.; Cocchia, R.; Wenaweser, P.; Cook, S.; Billinger, M.; Meier, B.; Hess, O.M. Sirolimus-eluting stents associated with paradoxic coronary vasoconstriction. J. Am. Coll. Cardiol. 2005, 46, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Huang, F.; Yi, Y.; Yin, L.; Peng, D. EGCG attenuates atherosclerosis through the Jagged-1/Notch pathway. Int. J. Mol. Med. 2016, 37, 398–406. [Google Scholar] [CrossRef] [PubMed]
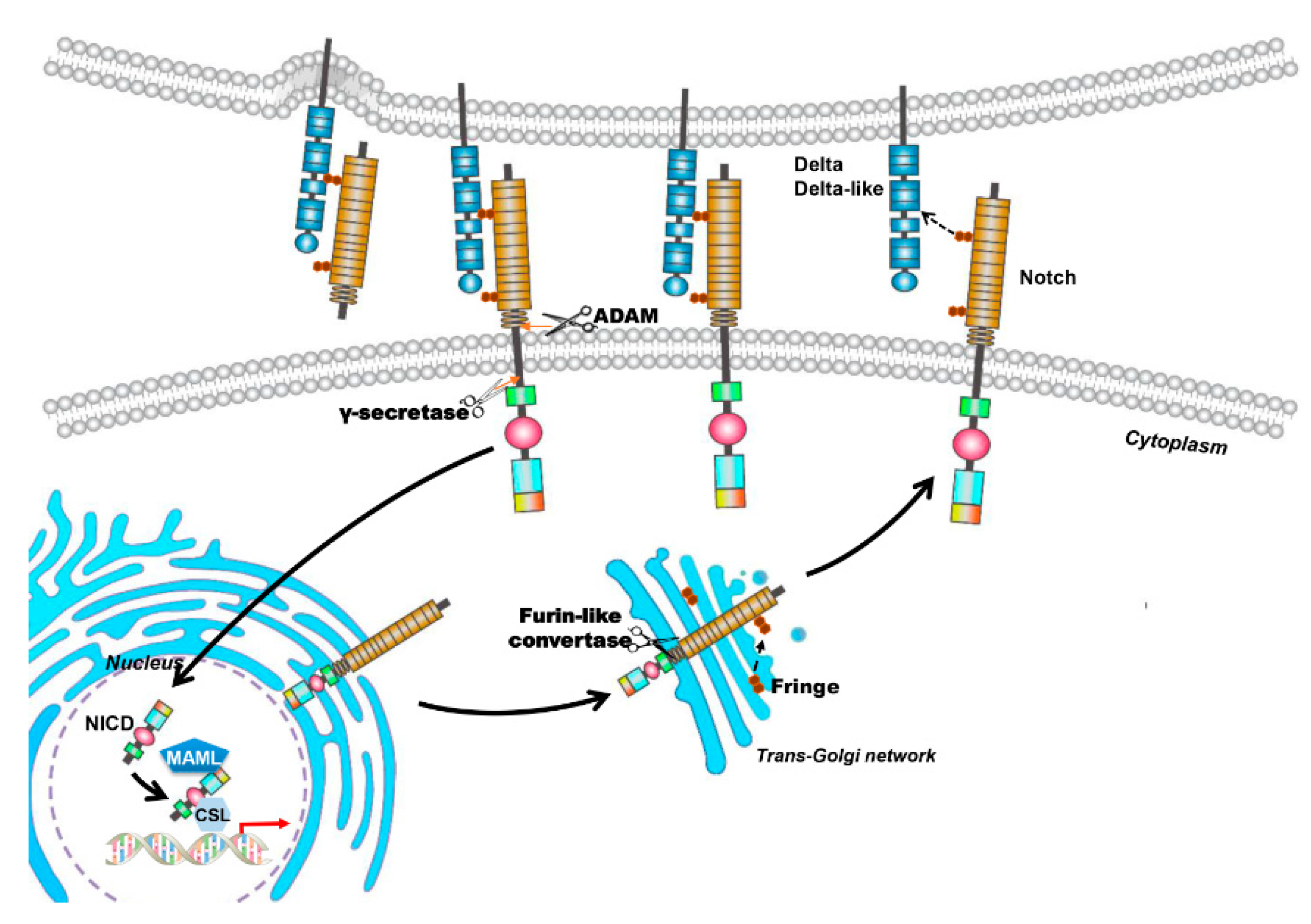
Figure 1.
The canonical Notch signaling pathway. Mammal Notch family members are composed of four Notch transmembrane receptors (Notch-1, Notch-2, Notch-3 and Notch-4) and five typical transmembrane ligands (Delta-like 1, Delta-like 3 and Delta-like 4, Jagged-1 and Jagged-2). Notch receptors are synthesized as single-chain precursors and transported to the Golgi apparatus (Black arrow points to the Golgi apparatus). In the Golgi apparatus, the precursors are cleaved into an extracellular and a transmembrane subunit by furin and modified by glycosyltransferases Fringe. Then the matured proteins are transported and inserted into cell membrane (Black arrow points to the cell membrane). Interaction between Notch receptors and their ligands triggers the canonical Notch signaling pathway. The transmembrane Notch receptor is cleaved by a disintegrin and metalloproteinases (ADAM) to remove the extracellular subunit, and then, a multisubunit membrane protease γ-secretase catalyzes the second proteolytic cleavage that gives rise to the translocation of the Notch intracellular domain (NICD) into the nucleus (Black arrow points to the nucleus). In the nucleus, NICD binds with a transcription factor (Black arrow in the nucleus), RBP-Jκ (also known as CSL for CBF1/Su(H)/Lag-1), coactivator Mastermind-like (MAML) proteins, and forms an activated transcriptional complex. Then, the activated complex upregulates the expression of target genes (red arrow), such as hairy and enhancer of split (HES)-1, -5, -7 and HES-related repressor protein (HERP)-1 to -3. The deep blue pentagon represents coactivator MAML protein and the light blue hexagon represents transcriptional factor CSL. The orange arrows indicate cleavage sites; the arrow with dotted line in Golgi aparatus indicates protein glycosylation by Fringe and the arrow with dotted line between Notch ligand and receptor indicates the interaction of the two proteins.
Figure 1.
The canonical Notch signaling pathway. Mammal Notch family members are composed of four Notch transmembrane receptors (Notch-1, Notch-2, Notch-3 and Notch-4) and five typical transmembrane ligands (Delta-like 1, Delta-like 3 and Delta-like 4, Jagged-1 and Jagged-2). Notch receptors are synthesized as single-chain precursors and transported to the Golgi apparatus (Black arrow points to the Golgi apparatus). In the Golgi apparatus, the precursors are cleaved into an extracellular and a transmembrane subunit by furin and modified by glycosyltransferases Fringe. Then the matured proteins are transported and inserted into cell membrane (Black arrow points to the cell membrane). Interaction between Notch receptors and their ligands triggers the canonical Notch signaling pathway. The transmembrane Notch receptor is cleaved by a disintegrin and metalloproteinases (ADAM) to remove the extracellular subunit, and then, a multisubunit membrane protease γ-secretase catalyzes the second proteolytic cleavage that gives rise to the translocation of the Notch intracellular domain (NICD) into the nucleus (Black arrow points to the nucleus). In the nucleus, NICD binds with a transcription factor (Black arrow in the nucleus), RBP-Jκ (also known as CSL for CBF1/Su(H)/Lag-1), coactivator Mastermind-like (MAML) proteins, and forms an activated transcriptional complex. Then, the activated complex upregulates the expression of target genes (red arrow), such as hairy and enhancer of split (HES)-1, -5, -7 and HES-related repressor protein (HERP)-1 to -3. The deep blue pentagon represents coactivator MAML protein and the light blue hexagon represents transcriptional factor CSL. The orange arrows indicate cleavage sites; the arrow with dotted line in Golgi aparatus indicates protein glycosylation by Fringe and the arrow with dotted line between Notch ligand and receptor indicates the interaction of the two proteins.

Figure 2.
Notch signaling modulates endothelial cells’ (EC) fate associated with neointimal hyperplasia. (A) EC proliferation reduces neointimal hyperplasia. Notch signaling may have dual function in EC proliferation, which is dependent on the relative level of p21 expression. If the expression of p21 is above the level, Notch activation inhibits EC proliferation, however, ECs grow and proliferate when the expression of p21 is below the level. In addition, activation of Notch blocks EC regeneration and induces neointimal hyperplasia; (B) EC apoptosis contributes to neointimal hyperplasia. Upregulation of Notch-1, Notch-2, Notch-3 and downregulation of Notch-4, promote EC apoptosis, which further promotes neointimal hyperplasia; (C) Endothelial-mesenchymal transition (EndMT) contributes to neointimal hyperplasia. Notch activation can trigger EndMT and promote neointimal hyperplasia. The canonical EndMT inducer TGF-β (transforming growth factor-β) also can activate the Notch signaling pathway. FSP-1, fibroblast-specific protein 1; CD31, also known as platelet endothelial cell adhesion molecule-1 (PECAM-1).
Figure 2.
Notch signaling modulates endothelial cells’ (EC) fate associated with neointimal hyperplasia. (A) EC proliferation reduces neointimal hyperplasia. Notch signaling may have dual function in EC proliferation, which is dependent on the relative level of p21 expression. If the expression of p21 is above the level, Notch activation inhibits EC proliferation, however, ECs grow and proliferate when the expression of p21 is below the level. In addition, activation of Notch blocks EC regeneration and induces neointimal hyperplasia; (B) EC apoptosis contributes to neointimal hyperplasia. Upregulation of Notch-1, Notch-2, Notch-3 and downregulation of Notch-4, promote EC apoptosis, which further promotes neointimal hyperplasia; (C) Endothelial-mesenchymal transition (EndMT) contributes to neointimal hyperplasia. Notch activation can trigger EndMT and promote neointimal hyperplasia. The canonical EndMT inducer TGF-β (transforming growth factor-β) also can activate the Notch signaling pathway. FSP-1, fibroblast-specific protein 1; CD31, also known as platelet endothelial cell adhesion molecule-1 (PECAM-1).

Figure 3.
Notch signaling in cell-cell communication between endothelial cells (ECs) and vascular smooth muscle cells (VSMCs). In the process of direct cell-cell interaction between ECs and VSMCs, EC membrane ligand Jagged-1 is recognized by Notch-2, -3 in VSMCs; reciprocally, VSMCs Jagged-1 binds with EC Notch-1 receptor and activates the Notch downstream pathway in ECs (also see Table 1A). In addition, activation of Notch signaling induces nitric oxide (NO), platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF) and other factors’ production from ECs via autocrine and paracrine process; these factors can regulate EC and VSMC proliferation, migration, differentiation and play different roles in neointimal hyperplasia (see Table 1B).
Figure 3.
Notch signaling in cell-cell communication between endothelial cells (ECs) and vascular smooth muscle cells (VSMCs). In the process of direct cell-cell interaction between ECs and VSMCs, EC membrane ligand Jagged-1 is recognized by Notch-2, -3 in VSMCs; reciprocally, VSMCs Jagged-1 binds with EC Notch-1 receptor and activates the Notch downstream pathway in ECs (also see Table 1A). In addition, activation of Notch signaling induces nitric oxide (NO), platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF) and other factors’ production from ECs via autocrine and paracrine process; these factors can regulate EC and VSMC proliferation, migration, differentiation and play different roles in neointimal hyperplasia (see Table 1B).

Figure 4.
Endothelial cell (EC) dysfunction caused by Notch signaling leads to neointimal hyperplasia. When vascular injury occurs, Notch signaling is activated, which further triggers EC phenotype change and causes EC dysfunction. In these processes, ECs acquire the senescent phenotype, the apoptotic phenotype or the mesenchymal phenotype and lose their barrier function, which leads to endothelial hyperpermeability, leakage and inflammatory responses. Furthermore, vascular smooth muscle cell -like cells can transmigrate into the media and proliferate, resulting in neointimal hyperplasia.
Figure 4.
Endothelial cell (EC) dysfunction caused by Notch signaling leads to neointimal hyperplasia. When vascular injury occurs, Notch signaling is activated, which further triggers EC phenotype change and causes EC dysfunction. In these processes, ECs acquire the senescent phenotype, the apoptotic phenotype or the mesenchymal phenotype and lose their barrier function, which leads to endothelial hyperpermeability, leakage and inflammatory responses. Furthermore, vascular smooth muscle cell -like cells can transmigrate into the media and proliferate, resulting in neointimal hyperplasia.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The Notch pathway-mediated interaction between endothelial cells (ECs) and vascular smooth muscle cells (VSMCs).
Table 1.
The Notch pathway-mediated interaction between endothelial cells (ECs) and vascular smooth muscle cells (VSMCs).
| A. The Direct Contact between ECs and VSMCs Mediated by Notch | ||||
| Ways of Interaction | Ligand | Receptor | Function | Possible Effect on Neointimal Hyperplasia |
| Direct Contact | ND | VSMC Notch-1 | VSMC migration [64]↑ | Promotion |
| EC Jagged-1 | VSMC Notch-2 | VSMC differentiation [63]↑ | ND | |
| VSMC proliferation [63]↓ | ||||
| EC Jagged-1 | VSMC Notch-3 | VSMC differentiation [60,61,62,63]↑ | Promotion | |
| VSMC secretion [63]↑ | ||||
| VSMC migration [64]↑ | ||||
| VSMC Jagged-1 | EC Notch-1 | EC proliferation [65]↑ | Inhibition | |
| ND | EC Notch-2, -3, -4 [15] | ND | ND | |
| B. The Indirect Communication between ECs and VSMCs Mediated by Notch Activation | ||||
| Ways of Interaction | Notch Signaling | Molecules Secreted from EC | Function | Possible Effect on Neointimal Hyperplasia |
| Indirect Communication | Activation | NO | EC proliferation [66,67]↑ | Inhibition |
| VSMC proliferation [68]↓ | ||||
| VSMC migration [68]↓ | ||||
| VEGF | EC proliferation↑ | Inhibition | ||
| EC regeneration↑ | ||||
| VSMC proliferation↓ | ||||
| PDGF | VSMC proliferation [69]↑ | Promotion | ||
| VSMC migration [70]↑ | ||||
| VSMC differentiation [71]↑ | ||||
ND indicates not described; NO: nitric oxide; PDGF: platelet-derived growth factor; VEGF: vascular endothelial growth factor; ↑ indicates promotion; ↓ indicates inhibition.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tian, D.-Y.; Jin, X.-R.; Zeng, X.; Wang, Y. Notch Signaling in Endothelial Cells: Is It the Therapeutic Target for Vascular Neointimal Hyperplasia? Int. J. Mol. Sci. 2017, 18, 1615. https://doi.org/10.3390/ijms18081615
AMA Style
Tian D-Y, Jin X-R, Zeng X, Wang Y. Notch Signaling in Endothelial Cells: Is It the Therapeutic Target for Vascular Neointimal Hyperplasia? International Journal of Molecular Sciences. 2017; 18(8):1615. https://doi.org/10.3390/ijms18081615
Chicago/Turabian StyleTian, Ding-Yuan, Xu-Rui Jin, Xi Zeng, and Yun Wang. 2017. "Notch Signaling in Endothelial Cells: Is It the Therapeutic Target for Vascular Neointimal Hyperplasia?" International Journal of Molecular Sciences 18, no. 8: 1615. https://doi.org/10.3390/ijms18081615
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.