Allelic Complexity in Long QT Syndrome: A Family-Case Study

 , ,
, ,
Abstract
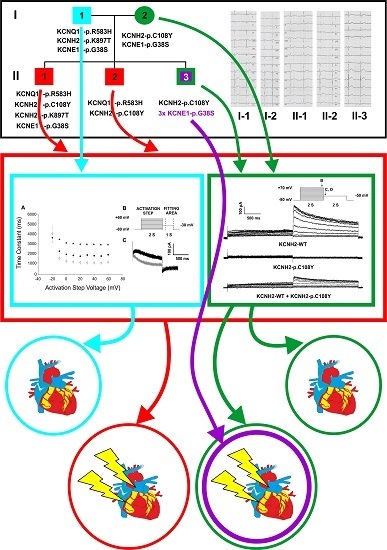
:
1. Introduction
2. Results
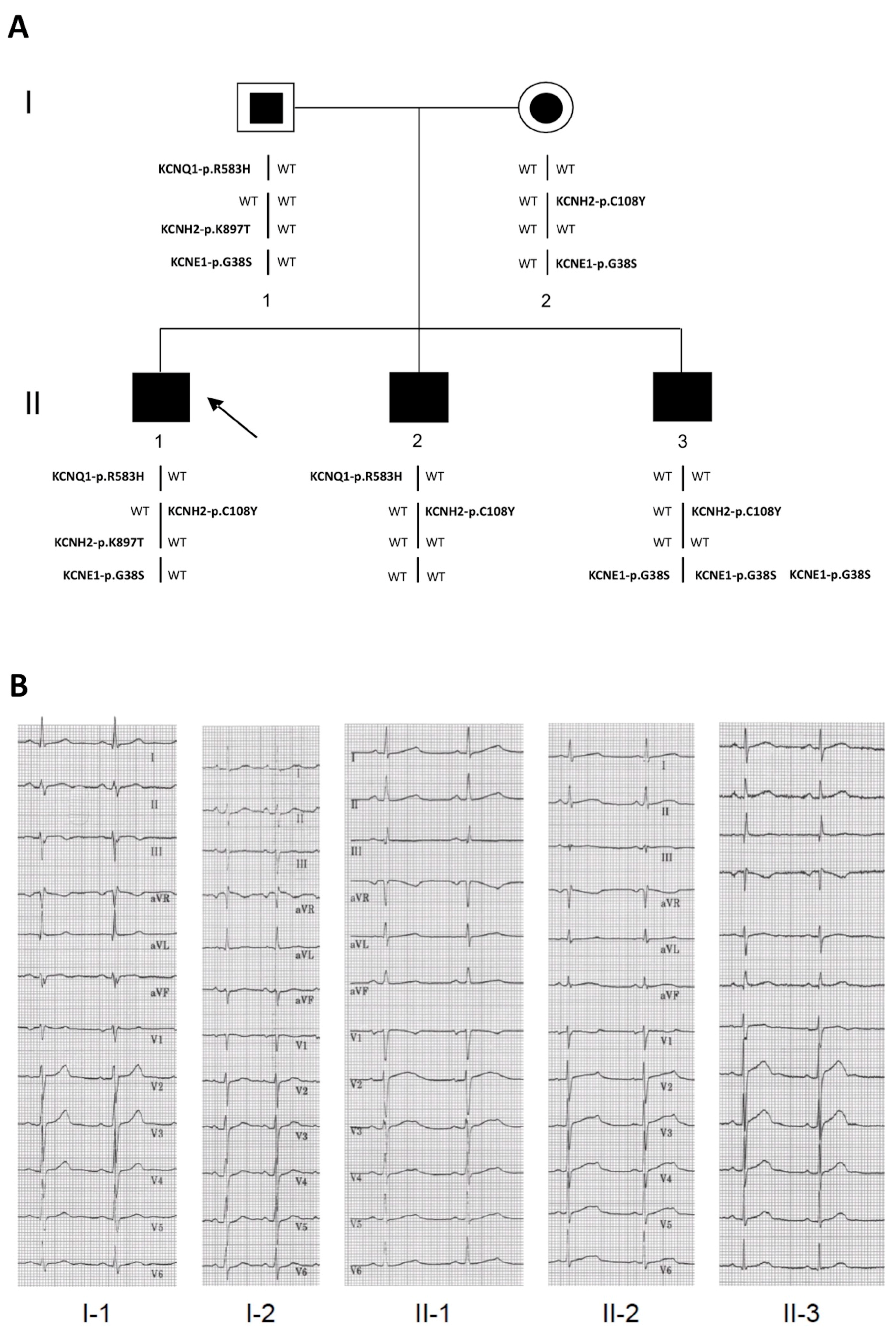
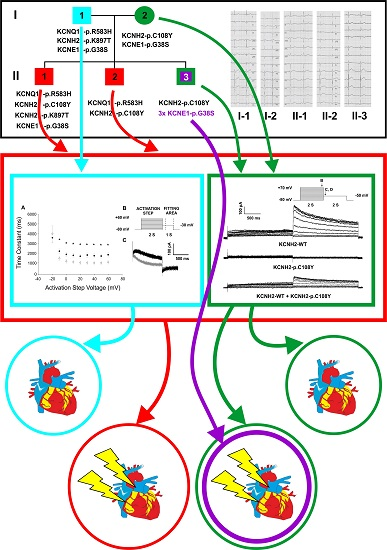
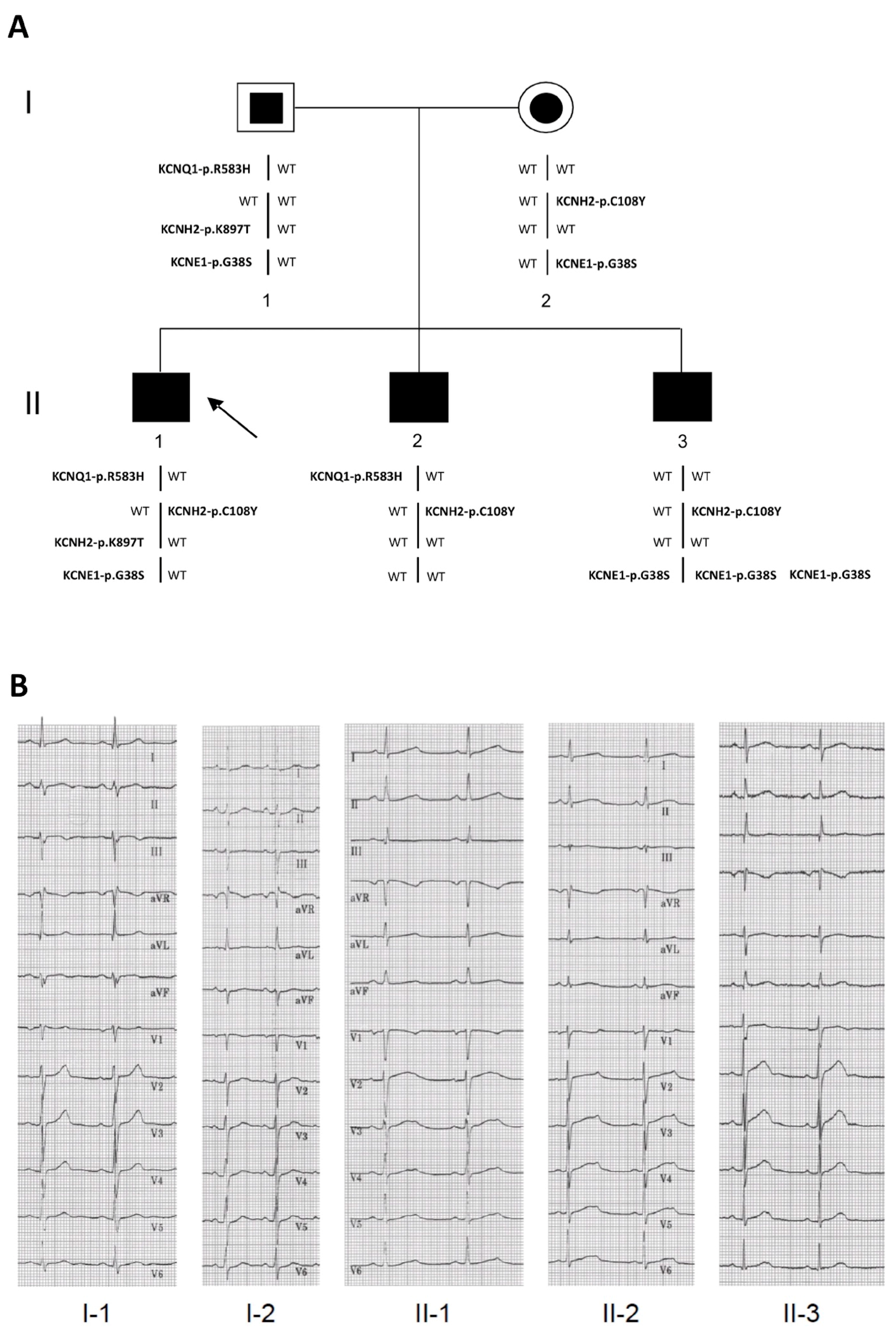
2.1. Clinical Phenotypes
2.2. Identification of the KCNQ1, KCNH2 and KCNE1 Variants
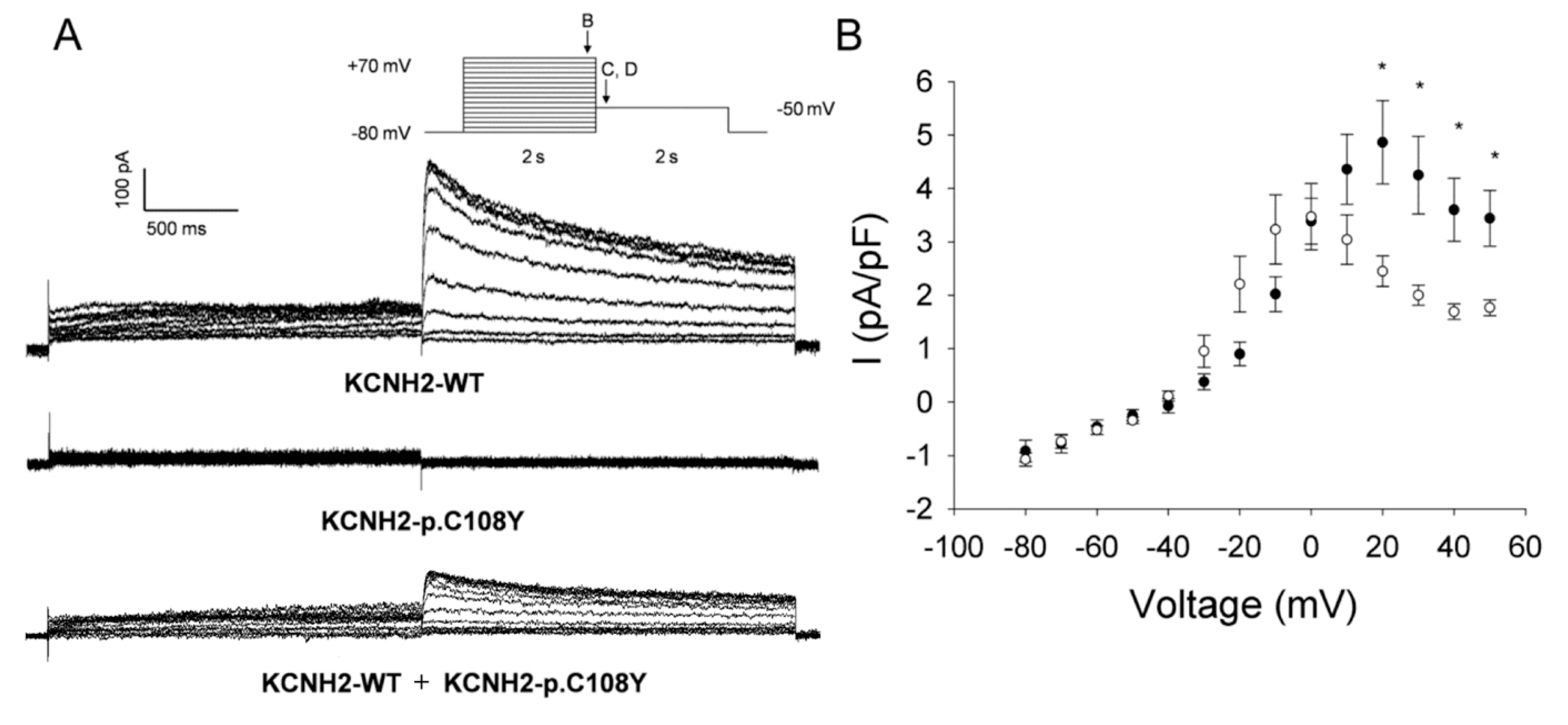
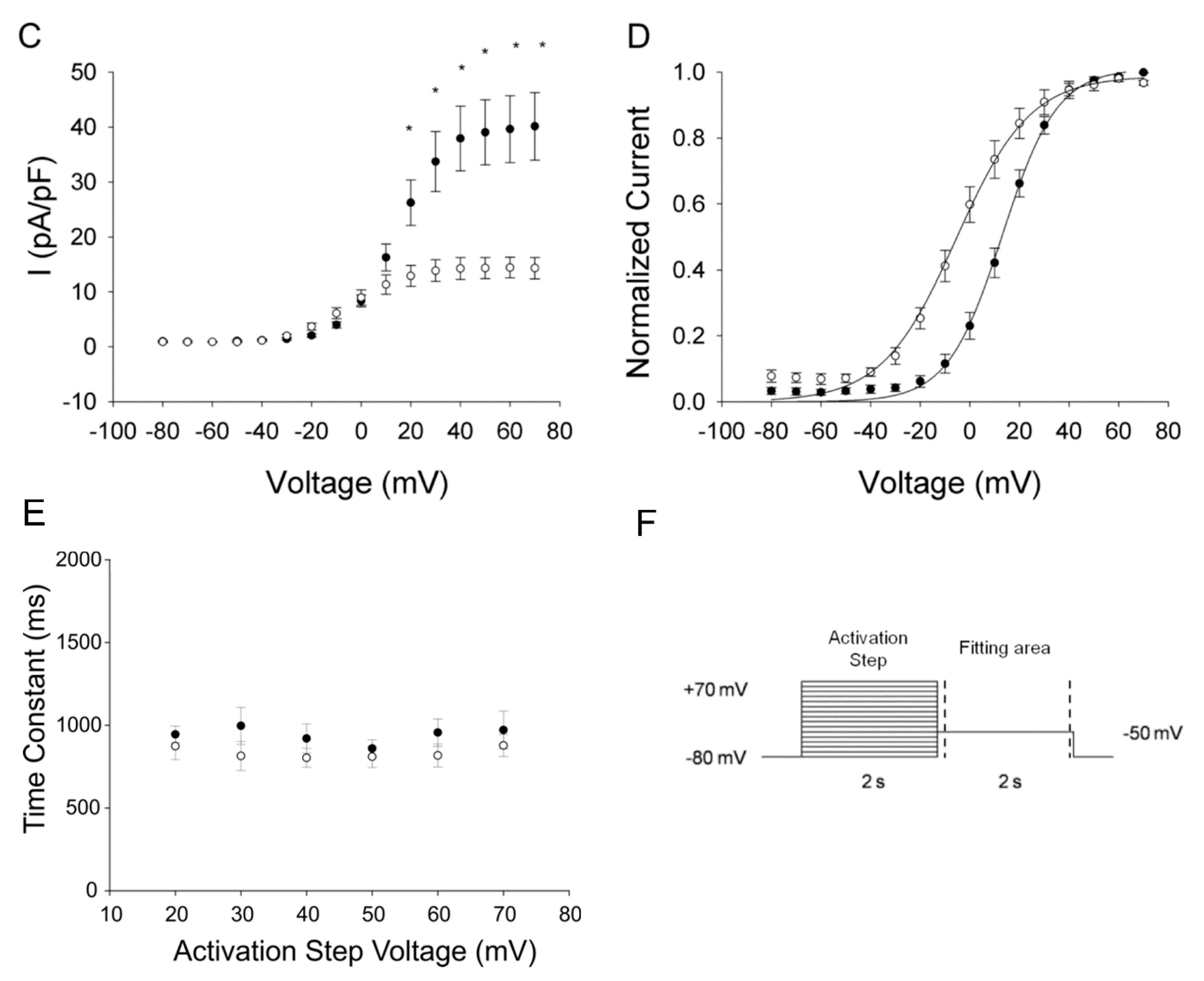
2.3. Functional Consequences of the KCNQ1-p.R583H and KCNH2-p.C108Y Variants
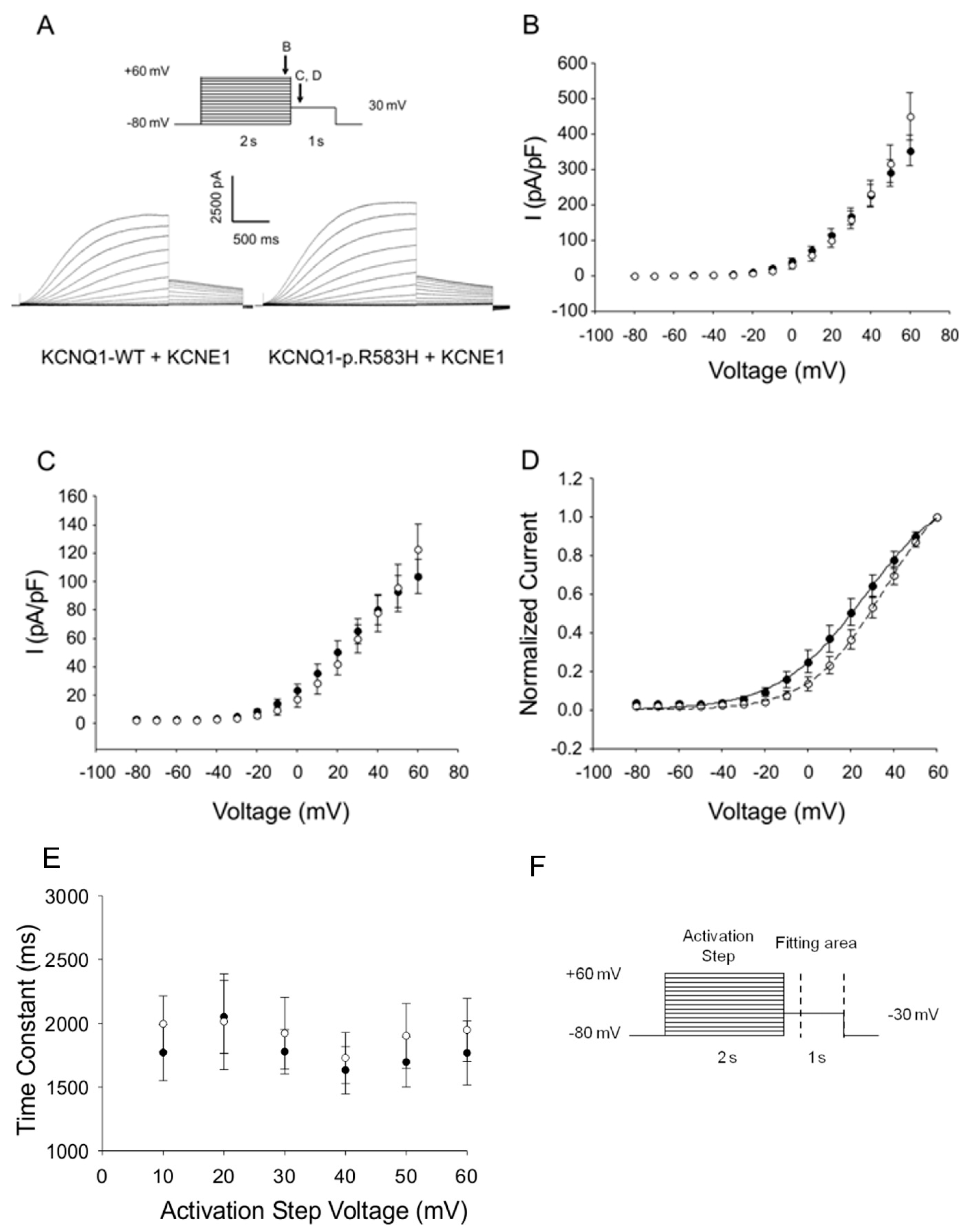
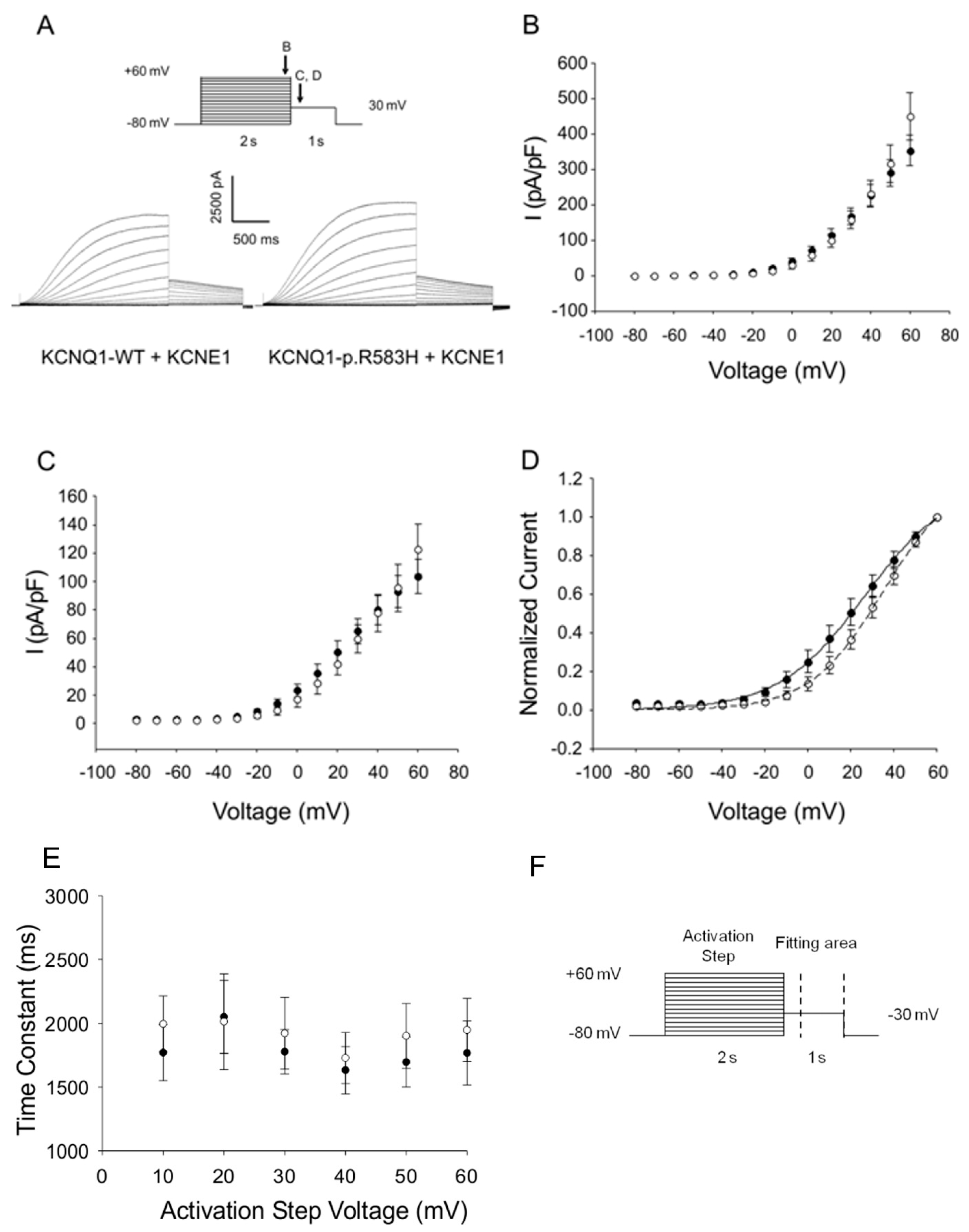
2.3.1. Functional Effects of KCNQ1-p.R583H
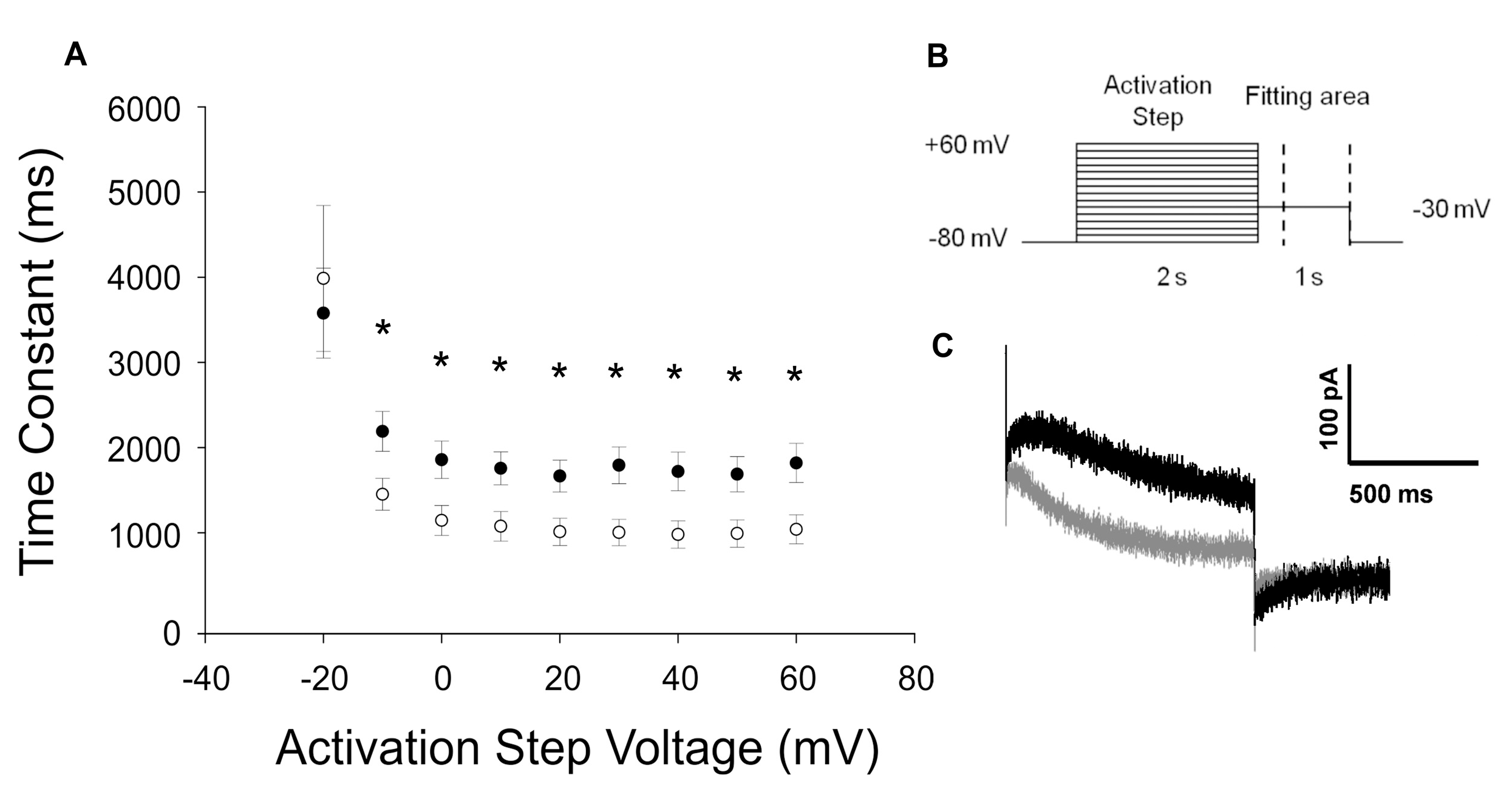
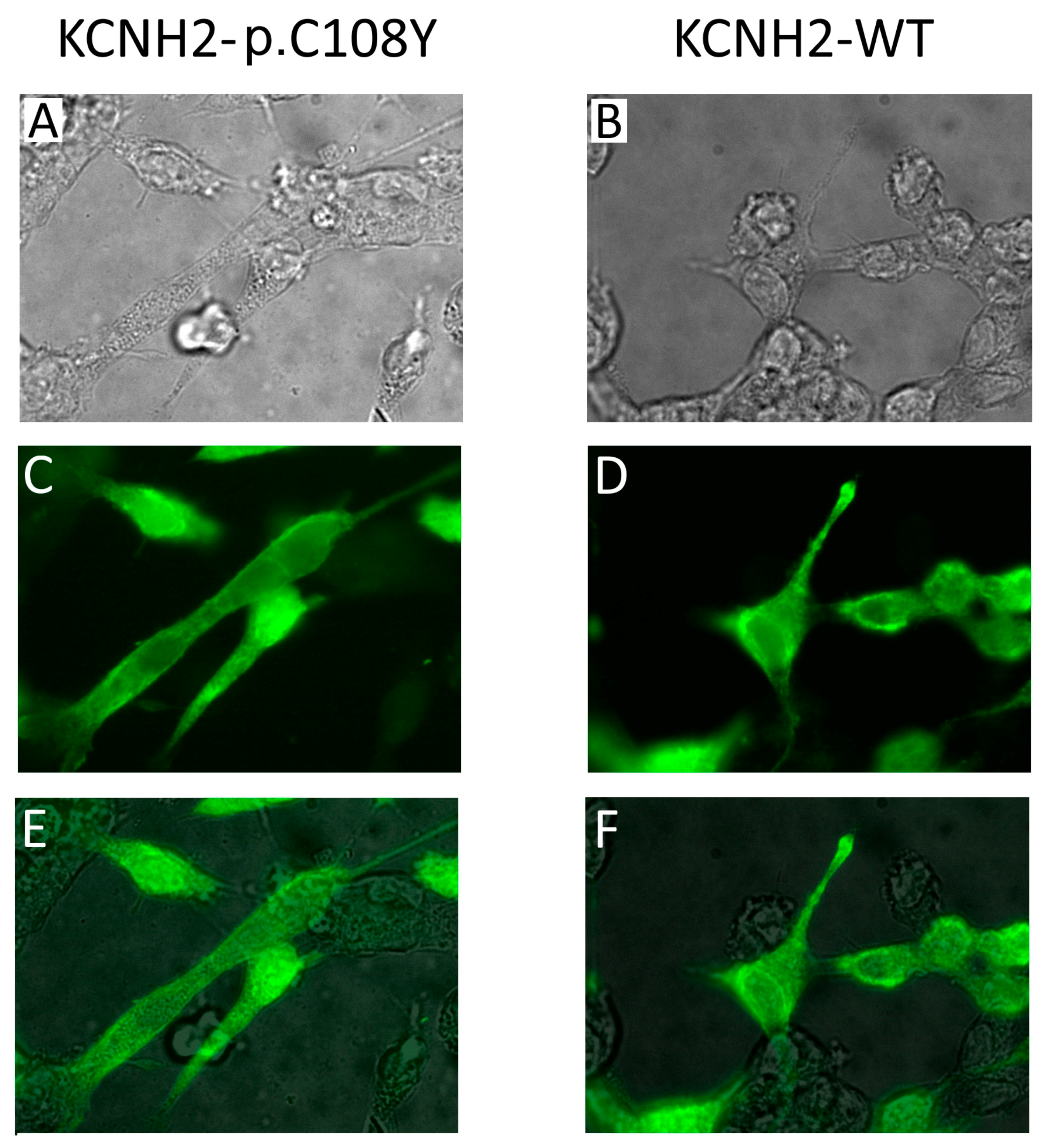
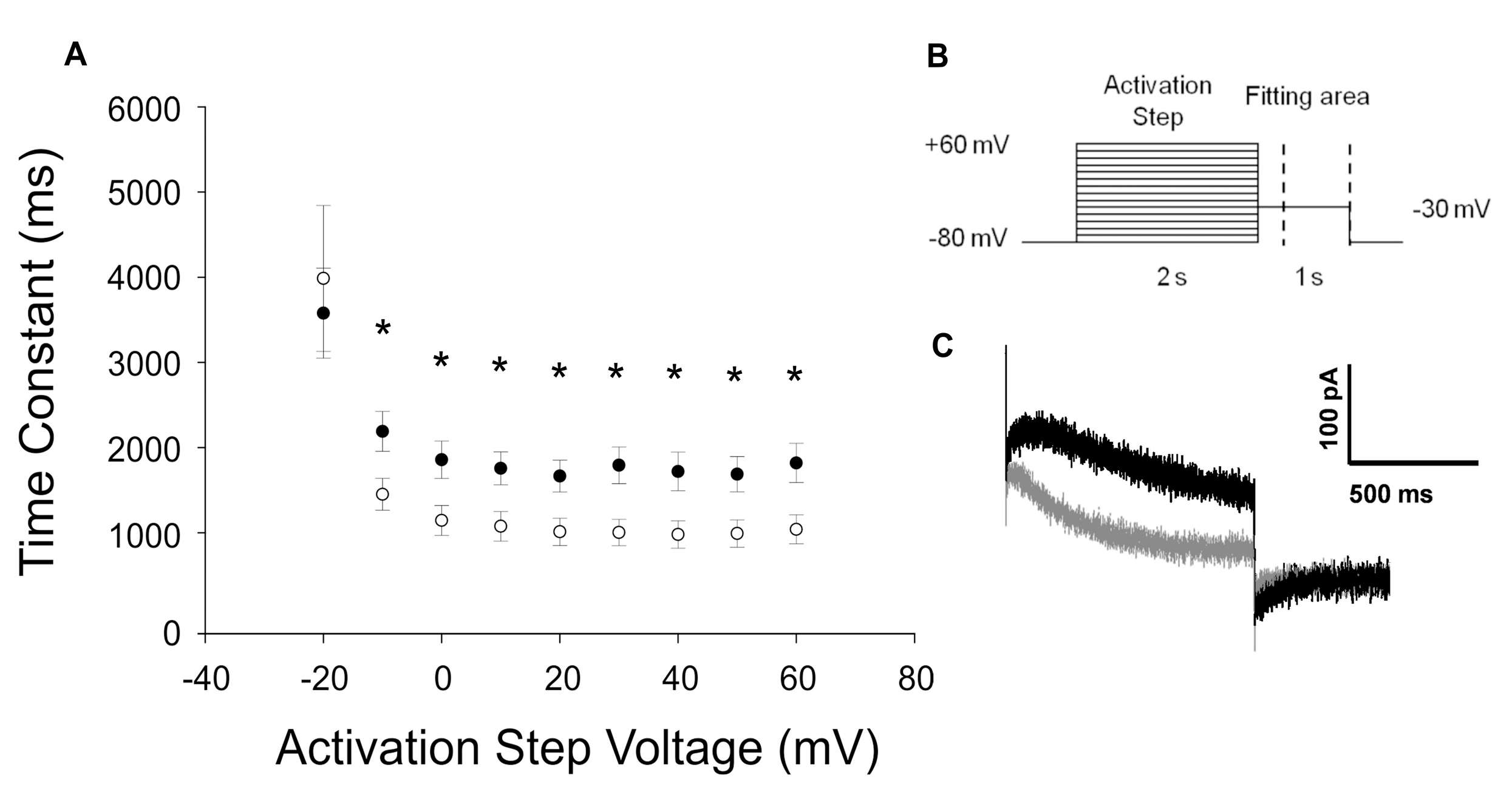
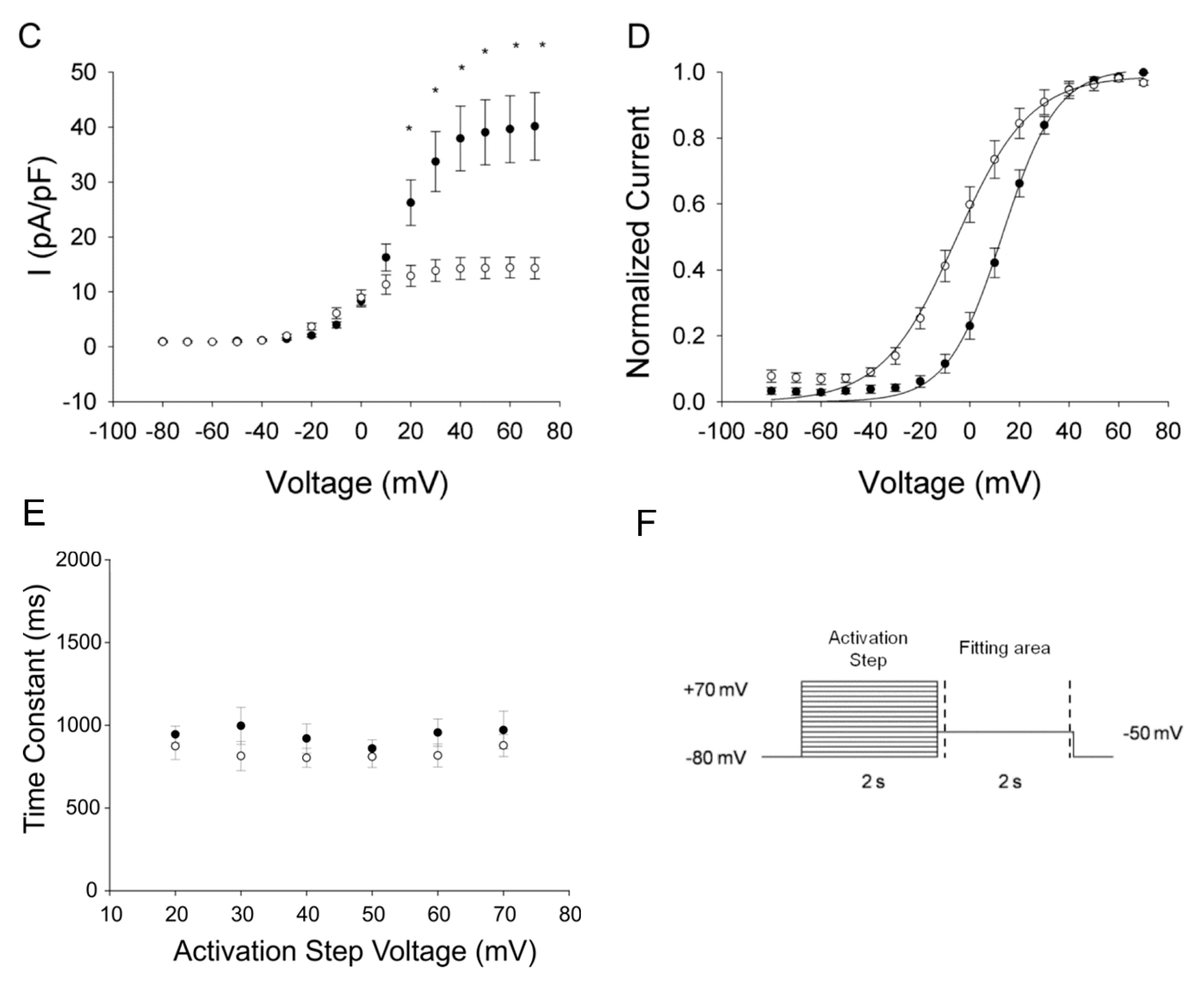
2.3.2. KCNH2-p.C108Y Exhibits a Dominant-Negative Loss-of-Function
3. Discussion
4. Materials and Methods
4.1. Clinical Investigations
4.2. Molecular Genetics
4.3. Mutagenesis
4.4. Cell Culture and Heterologous Expression
4.5. Cellular Electrophysiology
4.6. Cell Culture and Immunocytochemistry
4.7. Data Analysis and Statistics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Cm | Cell membrane capacitance |
| ECG | Electrocardiogram |
| HR | Heart rate |
| IHERG | Potassium current conducted by HERG channel |
| IKCNQ1 | Potassium current conducted by KCNQ1 channel |
| IKs | Slow delayed rectifier potassium current |
| IKr | Rapid delayed rectifier potassium current |
| k | Slope factor |
| LQTS | Long QT syndrome |
| MAF | Minor allele frequency |
| mV | Millivolt |
| ms | Millisecond |
| MΩ | Megaohm |
| pA | Picoampere |
| pF | Picofarad |
| QTc | QT interval corrected for heart rate |
| s | Seconds |
| SEM | Standard error of the mean |
| V1/2 | Half-maximal activation voltage |
| yrs | Years |
References
- Viskin, S. Long QT syndromes and torsade de pointes. Lancet 1999, 354, 1625–1633. [Google Scholar] [CrossRef]
- Detta, N.; Frisso, G.; Salvatore, F. The multi-faceted aspects of the complex cardiac Nav1.5 protein in membrane function and pathophysiology. Biochim. Biophys. Acta 2015, 1854, 1502–1509. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J. Introduction to the arrhythmogenic disorders of genetic origin series. Circ. Arrhythm. Electrophysiol. 2012, 5, 604–605. [Google Scholar] [CrossRef] [PubMed]
- Vyas, B.; Puri, R.D.; Namboodiri, N.; Nair, M.; Sharma, D.; Movva, S.; Saxena, R.; Bohora, S.; Aggarwal, N.; Vora, A.; et al. KCNQ1 mutations associated with Jervell and Lange-Nielsen syndrome and autosomal recessive Romano-Ward syndrome in India-expanding the spectrum of long QT syndrome type 1. Am. J. Med. Genet. 2016, 170, 1510–1519. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Ackerman, M.J.; George, A.L., Jr.; Wilde, A.A. Impact of genetics on the clinical management of channelopathies. J. Am. Coll. Cardiol. 2013, 62, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Human Gene Mutation Database. Available online: http://www.hgmd.cf.ac.uk/ac/index/php (accessed on 10 January 2017).
- Amin, A.S.; Tan, H.L.; Wilde, A.A. Cardiac ion channels in health and disease. Heart Rhythm 2010, 7, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Riuro, H.; Campuzano, O.; Berne, P.; Arbelo, E.; Iglesias, A.; Perez-Serra, A.; Coll-Vidal, M.; Partemi, S.; Mademont-Soler, I.; Pico, F.; et al. Genetic analysis, in silico prediction, and family segregation in long QT syndrome. Eur. J. Hum. Genet. 2015, 23, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, I.; Moss, A.J. Long QT syndrome. J. Am. Coll. Cardiol. 2008, 51, 2291–2300. [Google Scholar] [CrossRef] [PubMed]
- Westenskow, P.; Splawski, I.; Timothy, K.W.; Keating, M.T.; Sanguinetti, M.C. Compound mutations: A common cause of severe long-QT syndrome. Circulation 2004, 109, 1834–1841. [Google Scholar] [CrossRef] [PubMed]
- Itoh, H.; Shimizu, W.; Hayashi, K.; Yamagata, K.; Sakaguchi, T.; Ohno, S.; Makiyama, T.; Akao, M.; Ai, T.; Noda, T.; et al. Long QT syndrome with compound mutations is associated with a more severe phenotype: A Japanese multicenter study. Heart Rhythm 2010, 7, 1411–1418. [Google Scholar] [CrossRef] [PubMed]
- Kapplinger, J.D.; Tester, D.J.; Salisbury, B.A.; Carr, J.L.; Harris-Kerr, C.; Pollevick, G.D.; Wilde, A.A.; Ackerman, M.J. Spectrum and prevalence of mutations from the first 2500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm 2009, 6, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Detta, N.; Frisso, G.; Zullo, A.; Sarubbi, B.; Cozzolino, C.; Romeo, E.; Wang, D.W.; Calabro, R.; Salvatore, F.; George, A.L., Jr. Novel deletion mutation in the cardiac sodium channel inactivation gate causes long QT syndrome. Int. J. Cardiol. 2013, 165, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Giudicessi, J.R.; Ackerman, M.J. Determinants of incomplete penetrance and variable expressivity in heritable cardiac arrhythmia syndromes. Transl. Res. 2013, 161, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kanters, J.K.; Fanoe, S.; Larsen, L.A.; Bloch Thomsen, P.E.; Toft, E.; Christiansen, M. T wave morphology analysis distinguishes between KvLQT1 and HERG mutations in long QT syndrome. Heart Rhythm 2004, 1, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, C.; Priori, S.G.; Schwartz, P.J.; Bloise, R.; Ronchetti, E.; Nastoli, J.; Bottelli, G.; Cerrone, M.; Leonardi, S. Genetic testing in the long QT syndrome: Development and validation of an efficient approach to genotyping in clinical practice. JAMA 2005, 294, 2975–2980. [Google Scholar] [CrossRef] [PubMed]
- Goldenberg, I.; Horr, S.; Moss, A.J.; Lopes, C.M.; Barsheshet, A.; McNitt, S.; Zareba, W.; Andrews, J.M.L.; Robinson, L.; Locati, E.H.; et al. Risk for life-threatening cardiac events in patients with genotype-confirmed long-QT syndrome and normal-range corrected QT intervals. J. Am. Coll. Cardiol. 2011, 57, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Linna, E.H.; Perkiomaki, J.S.; Karsikas, M.; Seppanen, T.; Savolainen, M.; Kesaniemi, Y.A.; Makikallio, T.; Huikuri, H.V. Functional significance of KCNH2 (HERG) K897T polymorphism for cardiac repolarization assessed by analysis of T-wave morphology. Ann. Noninvasive Electrocardiol. 2006, 11, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Paavonen, K.J.; Chapman, H.; Laitinen, P.J.; Fodstad, H.; Piippo, K.; Swan, H.; Toivonen, L.; Viitasalo, M.; Kontula, K.; Pasternack, M. Functional characterization of the common amino acid 897 polymorphism of the cardiac potassium channel KCNH2 (HERG). Cardiovasc. Res. 2003, 59, 603–611, PMID:14499861. [Google Scholar] [CrossRef]
- Anson, B.D.; Ackerman, M.J.; Tester, D.J.; Will, M.L.; Delisle, B.P.; Anderson, C.L.; January, C.T. Molecular and functional characterization of common polymorphisms in HERG (KCNH2) potassium channels. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H2434–H2441. [Google Scholar] [CrossRef] [PubMed]
- Bezzina, C.R.; Verkerk, A.O.; Busjahn, A.; Jeron, A.; Erdmann, J.; Koopmann, T.T.; Bhuiyan, Z.A.; Wilders, R.; Mannens, M.M.; Tan, H.L.; et al. A common polymorphism in KCNH2 (HERG) hastens cardiac repolarization. Cardiovasc. Res. 2003, 59, 27–36, PMID:12829173. [Google Scholar] [CrossRef]
- Glinka, A.; Polak, S. Wild type and K897T polymorphisms of the hERG gene: Modeling the APD in Caucasians. Bioinformation 2012, 8, 1062–1065. [Google Scholar] [CrossRef] [PubMed]
- Nof, E.; Cordeiro, J.M.; Perez, G.J.; Scornik, F.S.; Calloe, K.; Love, B.; Burashnikov, E.; Caceres, G.; Gunsburg, M.; Antzelevitch, C. A common single nucleotide polymorphism can exacerbate long-QT type 2 syndrome leading to sudden infant death. Circ. Cardiovasc. Genet. 2010, 3, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Husser, D.; Stridh, M.; Sornmo, L.; Roden, D.M.; Darbar, D.; Bollmann, A. A genotype-dependent intermediate ECG phenotype in patients with persistent lone atrial fibrillation genotype ECG-phenotype correlation in atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2009, 2, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, J.R.; Zicha, S.; Coutu, P.; Hebert, T.E.; Nattel, S. Atrial fibrillation-associated minK38G/S polymorphism modulates delayed rectifier current and membrane localization. Cardiovasc. Res. 2005, 67, 520–552. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Mizumaki, K.; Hata, Y.; Inoue, H. Abnormal repolarization dynamics in a patient with KCNE1(G38S) who presented with torsades de pointes. J. Electrocardiol. 2016, 49, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Nishide, K.; Kato, M.; Hata, Y.; Mizumaki, K.; Kinoshita, K.; Nonobe, Y.; Tabata, T.; Sakamoto, T.; Kataoka, N.; et al. Glycine/Serine polymorphism at position 38 influences KCNE1 subunit’s modulatory actions on rapid and slow delayed rectifier K+ currents. Circ. J. 2014, 78, 610–618, PMID:24419801. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Mizumaki, K.; Hata, Y.; Sakamoto, T.; Nakatani, Y.; Kataoka, N.; Ichida, F.; Inoue, H.; Nishida, N. Latent pathogenicity of the G38S polymorphism of KCNE1 K+ channel modulator. Heart Vessels 2016, 32, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.N.; Ackerman, M.J. QTc: How long is too long? Br. J. Sports Med. 2009, 43, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.C.; Curran, M.E.; Zou, A.; Shen, J.; Spector, P.S.; Atkinson, D.L.; Keating, M.T. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 1996, 384, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Barhanin, J.; Lesage, F.; Guillemare, E.; Fink, M.; Lazdunski, M.; Romey, G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 1996, 384, 78–80. [Google Scholar] [CrossRef] [PubMed]
- Adaixo, R.; Harley, C.A.; Castro-Rodrigues, A.F.; Morais-Cabral, J.H. Structural properties of PAS domains from the KCNH potassium channels. PLoS ONE 2013, 8, e59265. [Google Scholar] [CrossRef] [PubMed]
- Gianulis, E.C.; Trudeau, M.C. Rescue of aberrant gating by a genetically encoded PAS (Per-Arnt-Sim) domain in several long QT syndrome mutant human ether-á-go-go-related gene potassium channels. J. Biol. Chem. 2011, 286, 22160–22169. [Google Scholar] [CrossRef] [PubMed]
- Crotti, L.; Lundquist, A.L.; Insolia, R.; Pedrazzini, M.; Ferrandi, C.; de Ferrari, G.M.; Vicentini, A.; Yang, P.; Roden, D.M.; George, A.L., Jr.; et al. KCNH2-K897T is a genetic modifier of latent congenital long-QT syndrome. Circulation 2005, 112, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Pietila, E.; Fodstad, H.; Niskasaari, E.; Laitinen, P.P.; Swan, H.; Savolainen, M.; Kesaniemi, Y.A.; Kontula, K.; Huikuri, H.V. Association between HERG K897T polymorphism and QT interval in middle-aged Finnish women. J. Am. Coll. Cardiol. 2002, 40, 511–514, PMID:12142119. [Google Scholar] [CrossRef]
- Crotti, L.; Monti, M.C.; Insolia, R.; Peljto, A.; Goosen, A.; Brink, P.A.; Greenberg, D.A.; Schwartz, P.J.; George, A.L., Jr. NOS1AP is a genetic modifier of the long-QT syndrome. Circulation 2009, 120, 1657–1663. [Google Scholar] [CrossRef] [PubMed]
- Sotoodehnia, N.; Isaacs, A.; de Bakker, P.I.; Dörr, M.; Newton-Cheh, C.; Nolte, I.M.; van der Harst, P.; Müller, M.; Eijgelsheim, M.; Alonso, A.; et al. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat. Genet. 2010, 42, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Tomás, M.; Napolitano, C.; de Giuli, L.; Bloise, R.; Subirana, I.; Malovini, A.; Bellazzi, R.; Arking, D.E.; Marban, E.; Chakravarti, A.; et al. Polymorphisms in the NOS1AP gene modulate QT interval duration and risk of arrhythmias in the Long QT Syndrome. J. Am. Coll. Cardiol. 2010, 55, 2745–2752. [Google Scholar] [CrossRef] [PubMed]
- World Medical Association declaration of Helsinki—ethical principles for medical research involving human subjects. JAMA 2013, 310, 2191–2194, PMID:25951678.
- Hoppe, K.; Hack, G.; Lehmann–Horn, F.; Jurkat–Rott, K.; Wearing, S.; Zullo, A.; Carsana, A.; Klingler, W. Hypermetabolism in B-lymphocytes from malignant hyperthermia susceptible individuals. Sci. Rep. 2016, 6, 33372. [Google Scholar] [CrossRef] [PubMed]
- Frisso, G.; Limongelli, G.; Pacileo, G.; del Giudice, A.; Forgione, L.; Calabro, P.; Iacomino, M.; Detta, N.; diFonzo, L.M.; Maddaloni, V.; et al. A child cohort study from southern Italy enlarges the genetic spectrum of hypertrophic cardiomyopathy. Clin. Gen. 2009, 76, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed]
- ExAC Browser (Beta), Exome Aggregation Consortium. Available online: http://exac.broadinstitute.org/ (accessed on 10 January 2017).
- D’Emmanuele di Villa Bianca, R.; Mitidieri, E.; Esposito, D.; Donnarumma, E.; Russo, A.; Fusco, F.; Ianaro, A.; Mirone, V.; Cirino, G.; Russo, G.; et al. Human cystathionine-β-synthase phosphorylation on serine227 modulates hydrogen sulfide production in human urothelium. PLoS ONE 2015, 10, e0136859. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
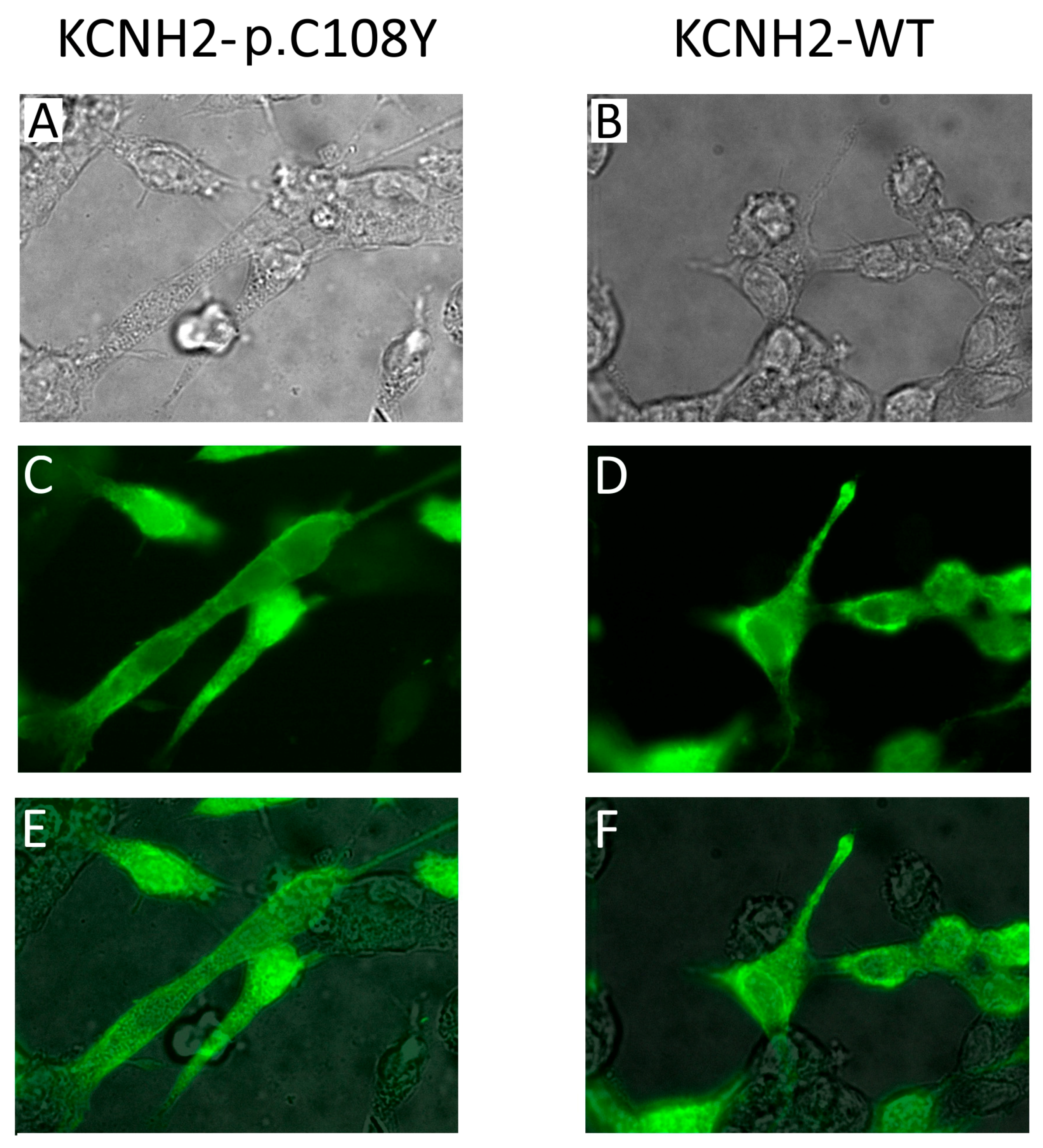{kind=link}
| Subjects * | II-1 | II-2 | II-3 | |
|---|---|---|---|---|
| Age at diagnosis | 12 years | 9 years | 6 years | |
| QTc † | at diagnosis | 560 | 530 | 470 |
| after therapy | 470 | 470–490 | 420 | |
| HR | after therapy | 56 | 58 | 80 # |
| Syncope | Aged 10 years | Aged nine years | Aged 10 years, after 4 s sinus pause | |
| Aged 11 years | ||||
| Sinus pauses | No pause | No pause | Aged 9 years | |
| Aged 10 years | ||||
| Therapy | Propanolol | Propanolol | Propanolol | |
| DDD-pacemaker | ||||
| Other clinical information | None | None | Down syndrome | |
| Gene | Nucleotide Variation | Amino Acid Variation | MAF | Bioinformatic Tools | |
|---|---|---|---|---|---|
| Polyphen | SIFT | ||||
| KCNE1 | G112A | G38S | 0.352 | Benign | Tolerated |
| KCNH2 | G323A | C108Y | N.D. | Probably damaging | Damaging |
| KCNH2 | A2690C | K897T | 0.187 | Benign | Not tolerated |
| KCNQ1 | G1748A | R583H | 0.000016 | Benign | Not tolerated |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zullo, A.; Frisso, G.; Detta, N.; Sarubbi, B.; Romeo, E.; Cordella, A.; Vanoye, C.G.; Calabrò, R.; George, A.L., Jr.; Salvatore, F. Allelic Complexity in Long QT Syndrome: A Family-Case Study. Int. J. Mol. Sci. 2017, 18, 1633. https://doi.org/10.3390/ijms18081633
Zullo A, Frisso G, Detta N, Sarubbi B, Romeo E, Cordella A, Vanoye CG, Calabrò R, George AL Jr., Salvatore F. Allelic Complexity in Long QT Syndrome: A Family-Case Study. International Journal of Molecular Sciences. 2017; 18(8):1633. https://doi.org/10.3390/ijms18081633
Chicago/Turabian StyleZullo, Alberto, Giulia Frisso, Nicola Detta, Berardo Sarubbi, Emanuele Romeo, Angela Cordella, Carlos G. Vanoye, Raffaele Calabrò, Alfred L. George, Jr., and Francesco Salvatore. 2017. "Allelic Complexity in Long QT Syndrome: A Family-Case Study" International Journal of Molecular Sciences 18, no. 8: 1633. https://doi.org/10.3390/ijms18081633