Various Mechanisms Involve the Nuclear Factor (Erythroid-Derived 2)-Like (NRF2) to Achieve Cytoprotection in Long-Term Cisplatin-Treated Urothelial Carcinoma Cell Lines

 , , , and
, , , and 
Abstract
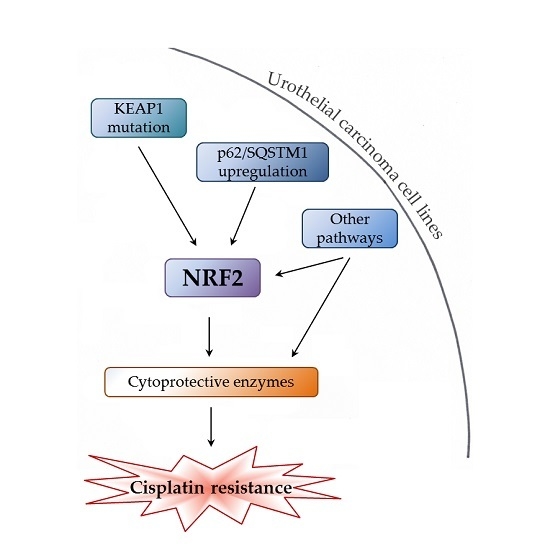
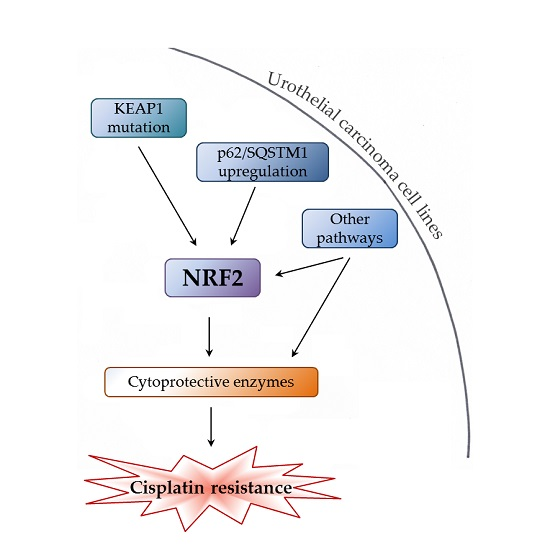
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
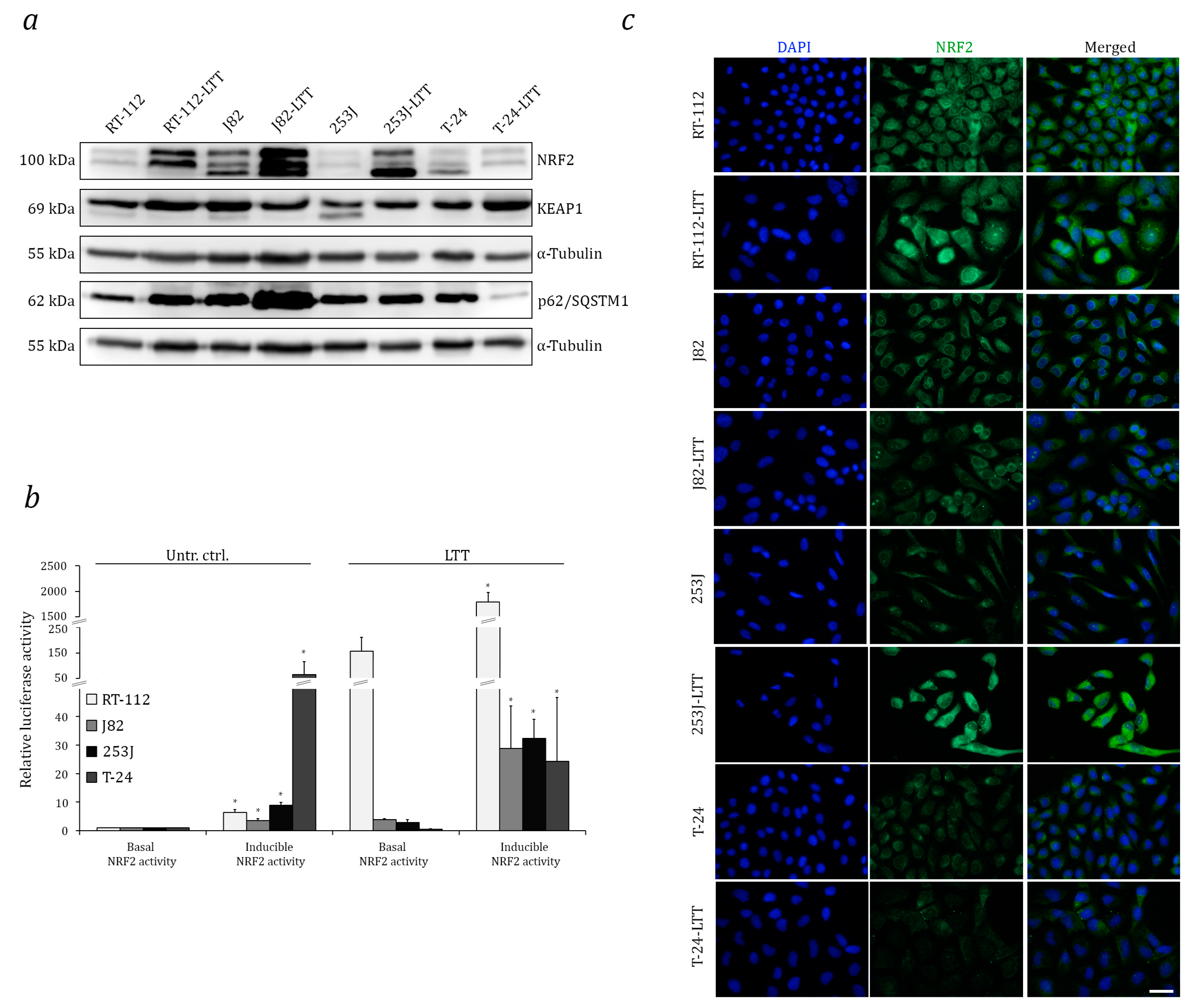
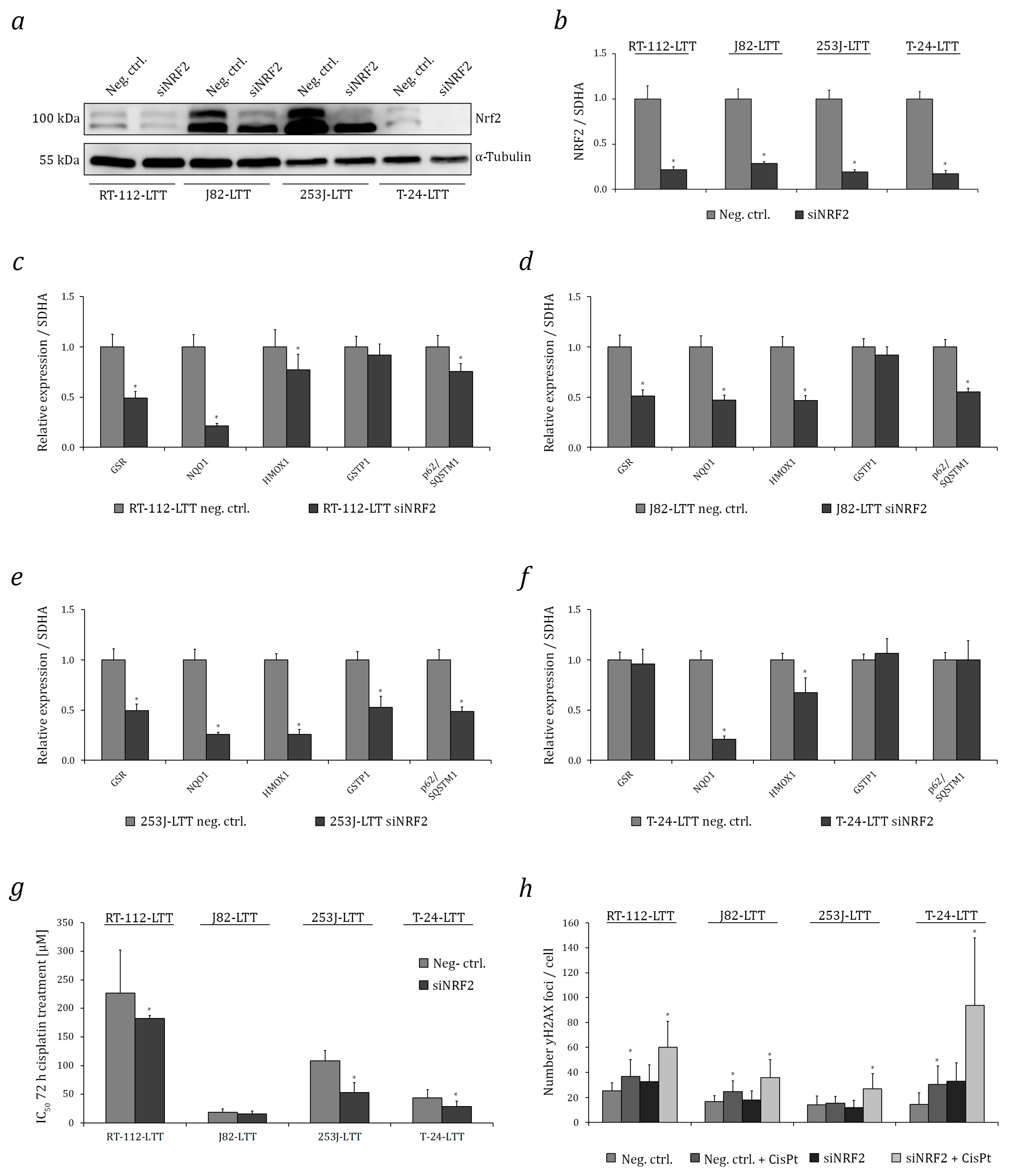
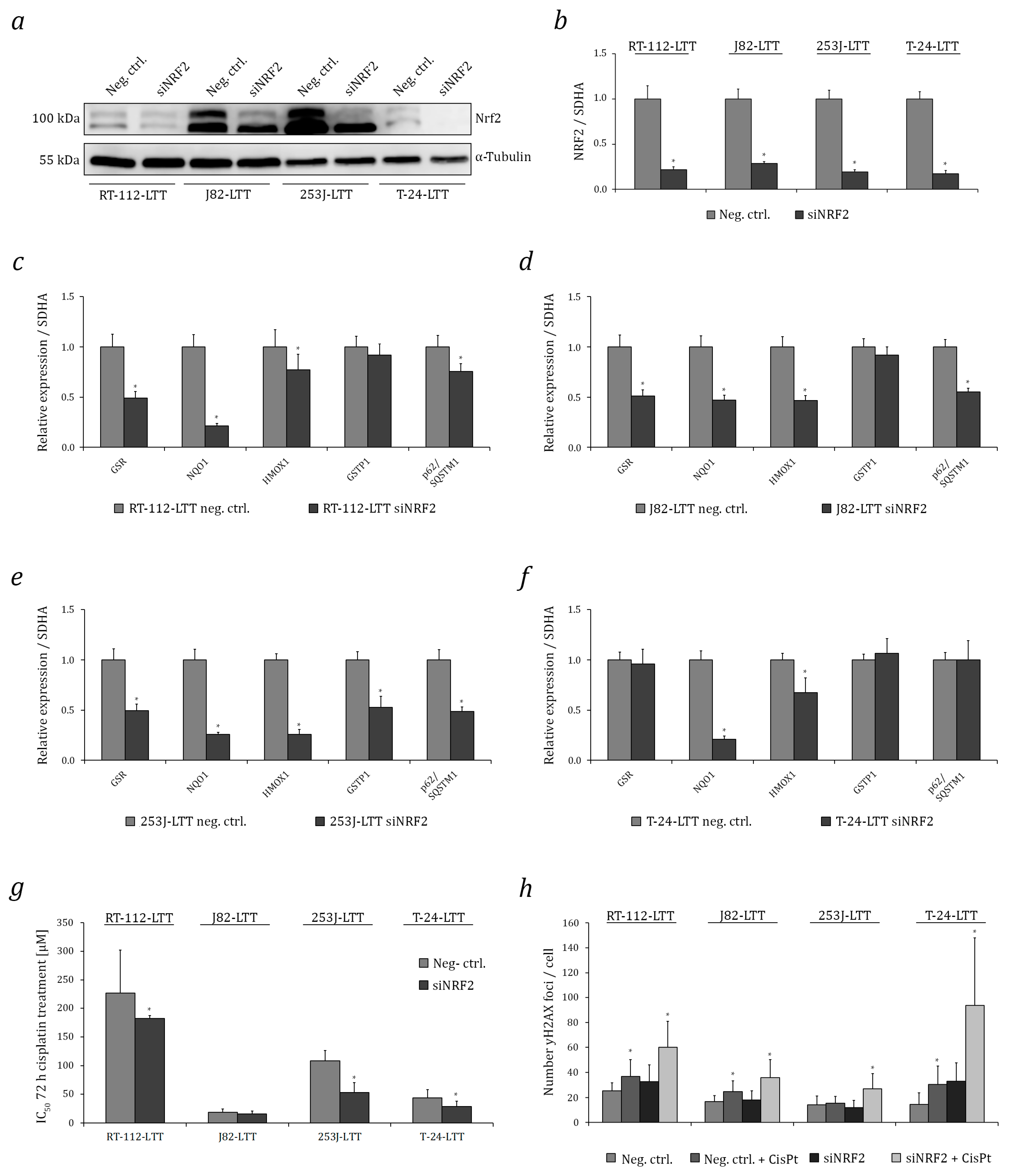
2.1. Increased Expression and Transcriptional Activity of Nuclear Factor Erythroid 2-Related Factor 2 (NRF2) in Long-Term Cisplatin Treated Cell Lines (LTTs)
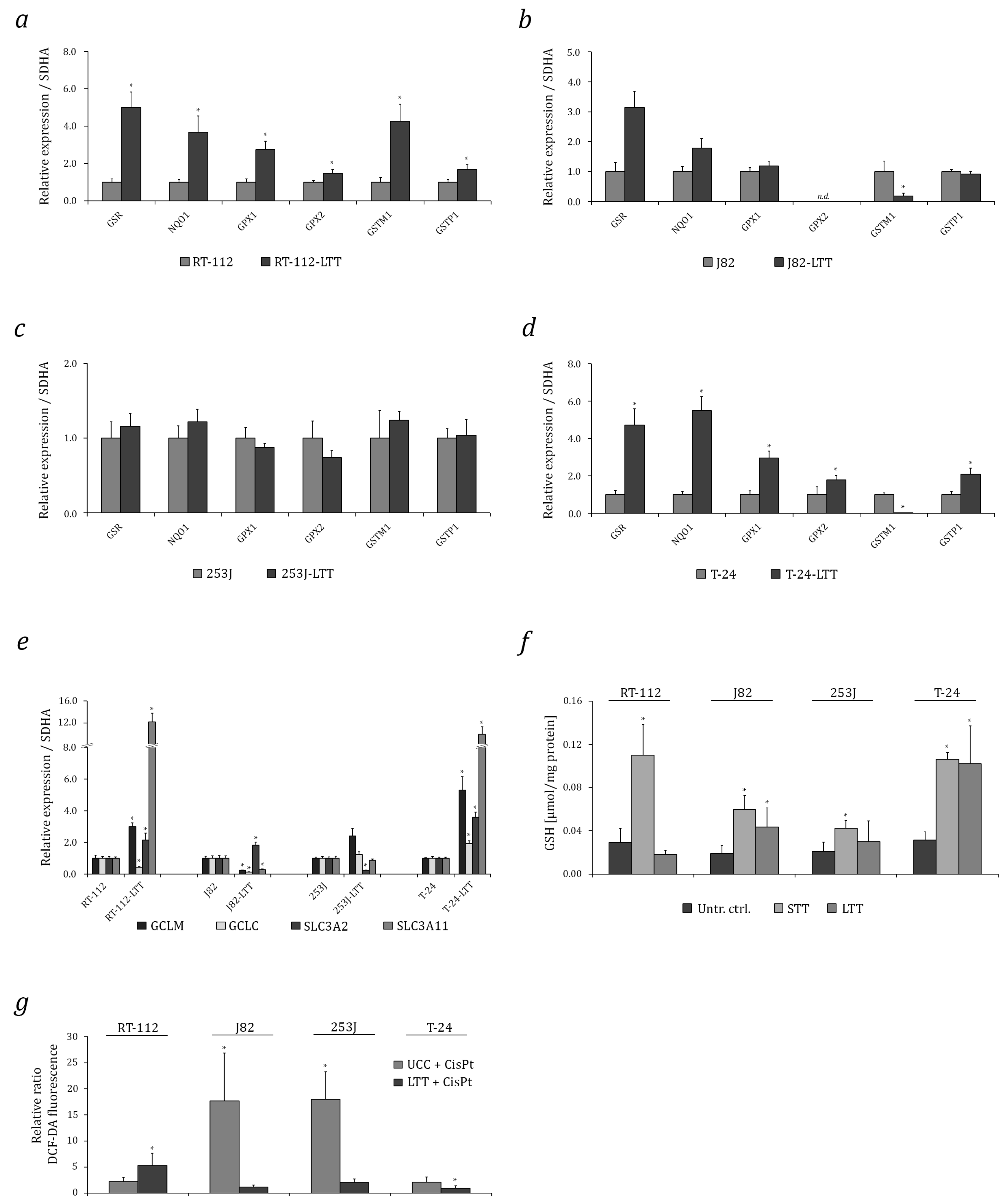
2.2. Induction of Cytoprotective Enzymes and Elevated Glutathione (GSH) Levels in LTTs
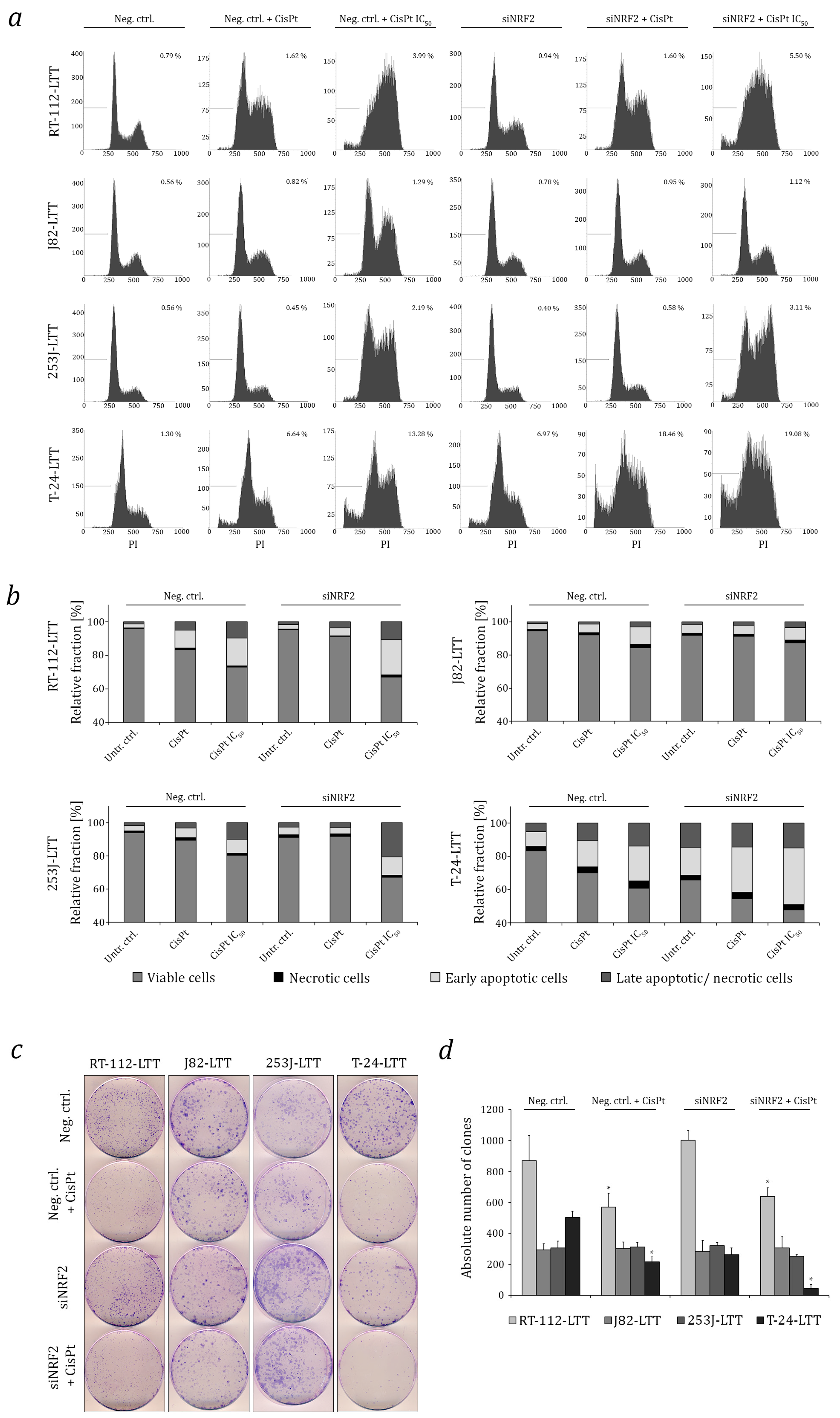
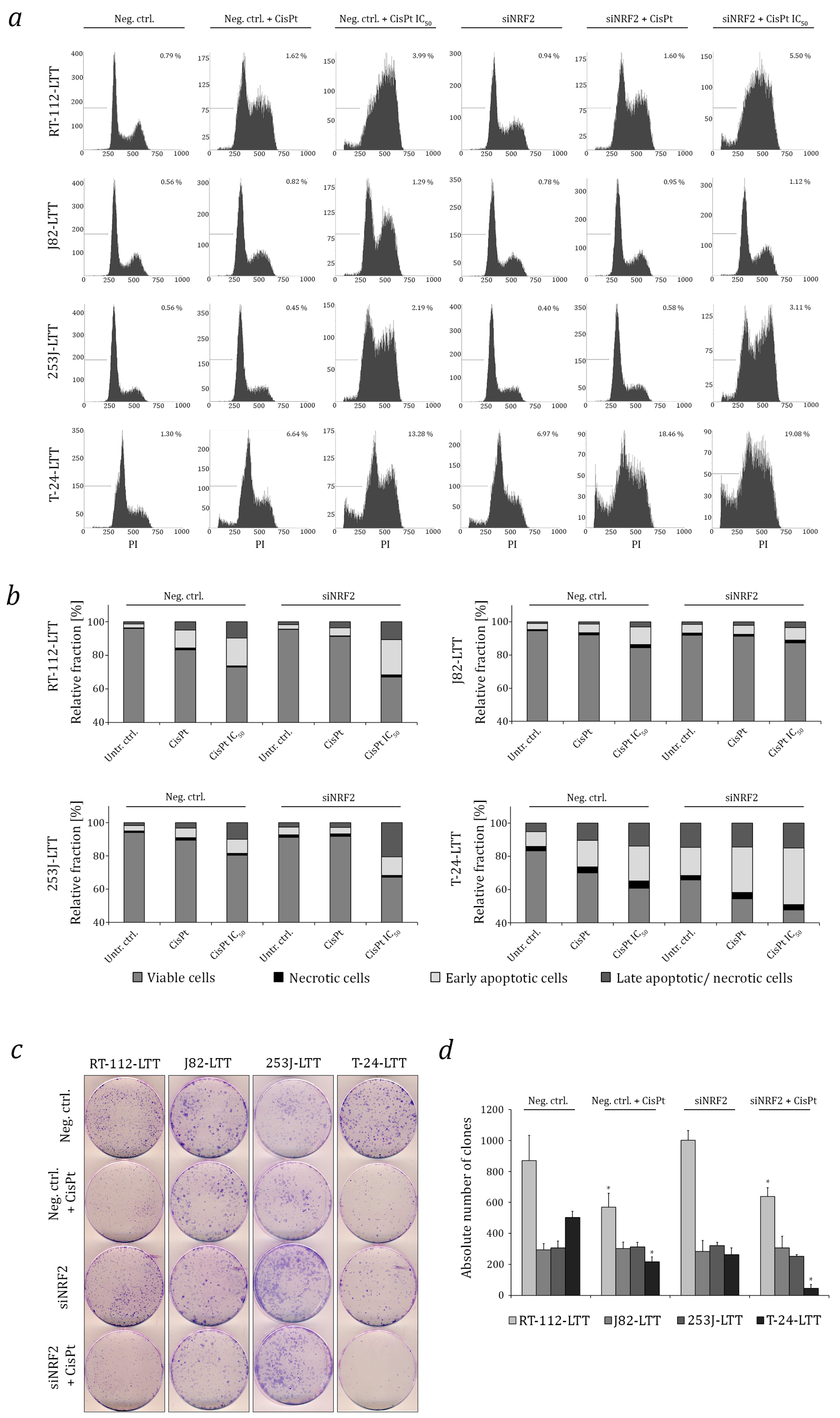
2.3. NRF2 as A Target to Sensitise Cisplatin-Resistant Urothelial Carcinoma Cell Lines (UCCs)
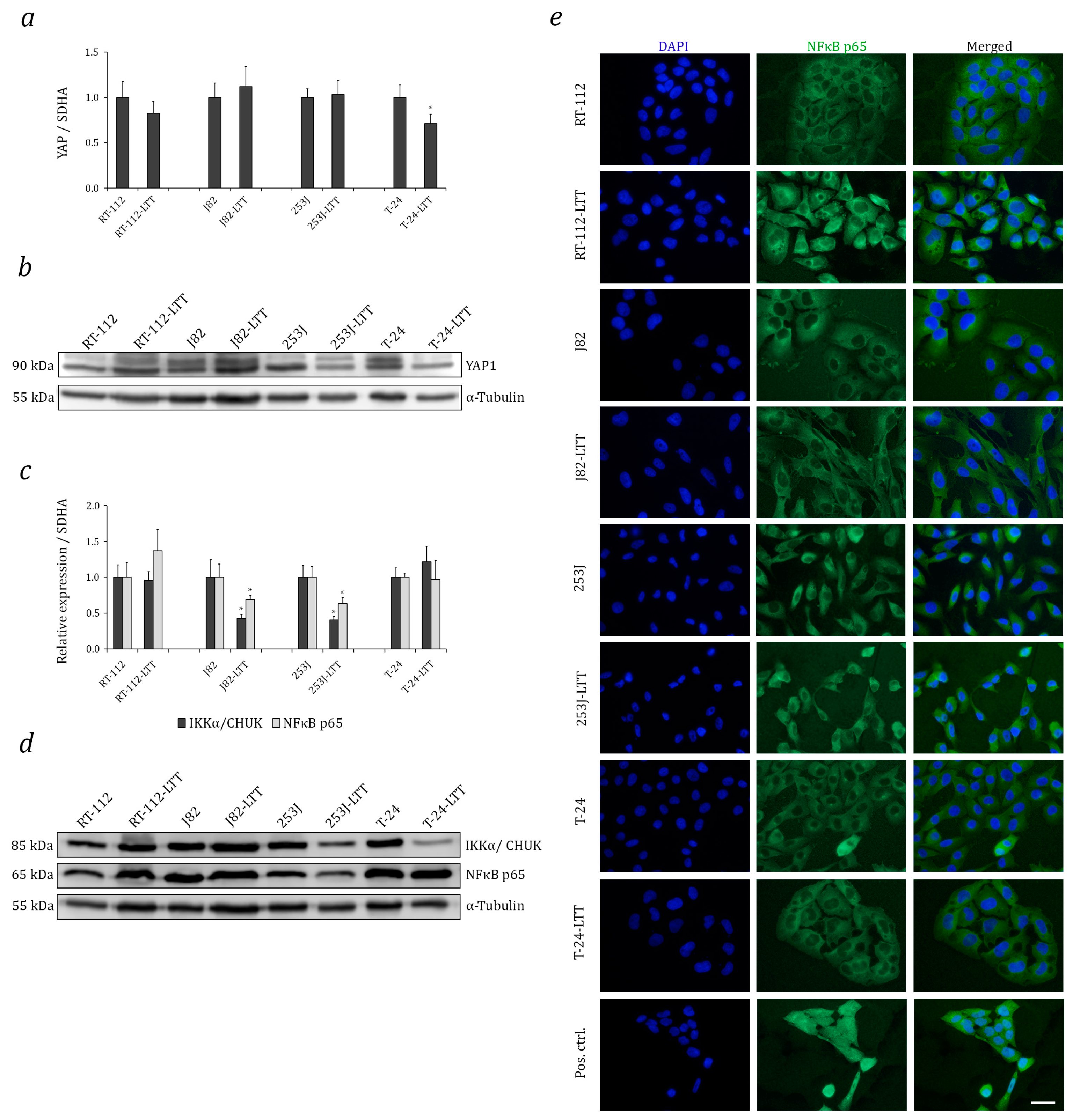
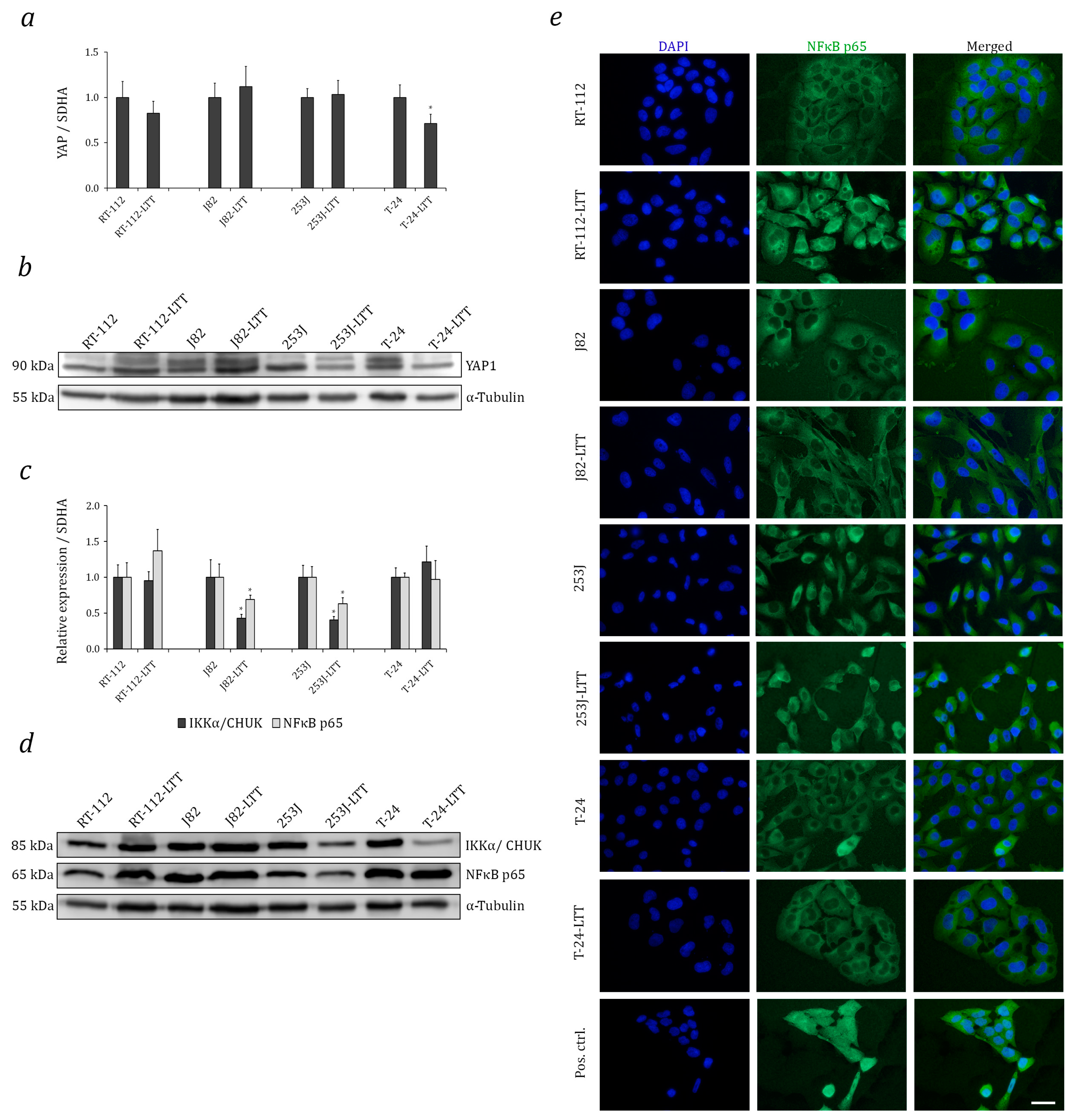
2.4. Relation of Increased NRF2 Expression to Hippo and Nuclear Factor Kappa B (NF-κB) Pathways in LTTs
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Treatment, Plasmid Transfection, and siRNA-Mediated Knockdown
4.2. Molecular Analyses
4.3. Mutation Analysis by Sequencing
4.4. Measurement of Cell Viability
4.5. Flow Cytometry
4.6. Immunofluorescence
4.7. Western Blot
4.8. GSH Assay
4.9. Use of the cBioPortal Data Base
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| LTT | Long-term cisplatin treated |
| LTTs | Long-term cisplatin treated cell lines |
| STT | Short-term cisplatin treated |
| UC | Urothelial carcinoma |
| UCCs | Urothelial carcinoma cell lines |
| SDHA | Succinate dehydrogenase complex |
| NRF2/NFEL2L | Nuclear factor (erythroid-derived 2)-like 2 |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| p62/SQSTM1 | Sequestosome-1 |
| ROS | Reactive oxygen species |
| GSH | Glutathione |
| GSR | Glutathione reductase |
| NQO1 | NAD(P)H quinone dehydrogenase 1 |
| HMOX1 | Heme oxygenase 1 |
| GPX1/2 | Glutathione peroxidase 1/2 |
| GST | Glutathione S-transferase |
| GCLC | Glutamate–cystein ligase catalytic subunit |
| GCLM | Glutamate–cystein ligase modifier subunit |
| SLC3A2 | 4F2 cell-surface antigen heavy chain |
| SLC7A11 | Cystine/glutamate transporter |
| YAP1 | Yes associated protein 1 |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| SCC | Squamous cell carcinoma |
| CHUK/IKKα | IκB kinase |
References
- Antoni, S.; Ferlay, J.; Soerjomataram, I.; Znaor, A.; Jemal, A.; Bray, F. Bladder cancer incidence and mortality: A global overview and recent trends. Eur. Urol. 2017, 71, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, C.N.; Calabro, F.; Pizzocaro, G.; Marini, L.; Schnetzer, S.; Sella, A. Chemotherapy with an every-2-week regimen of gemcitabine and paclitaxel in patients with transitional cell carcinoma who have received prior cisplatin-based therapy. Cancer 2001, 92, 2993–2998. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Galsky, M.D.; Pal, S.K.; Chowdhury, S.; Harshman, L.C.; Crabb, S.J.; Wong, Y.N.; Yu, E.Y.; Powles, T.; Moshier, E.L.; Ladoire, S.; et al. Comparative effectiveness of gemcitabine plus cisplatin versus methotrexate, vinblastine, doxorubicin, plus cisplatin as neoadjuvant therapy for muscle-invasive bladder cancer. Cancer 2015, 121, 2586–2593. [Google Scholar] [CrossRef] [PubMed]
- De Santis, M.; Bellmunt, J.; Mead, G.; Kerst, J.M.; Leahy, M.; Maroto, P.; Gil, T.; Marreaud, S.; Daugaard, G.; Skoneczna, I.; et al. Randomized phase II/III trial assessing gemcitabine/carboplatin and methotrexate/carboplatin/vinblastine in patients with advanced urothelial cancer who are unfit for cisplatin-based chemotherapy: EORTC study 30986. J. Clin. Oncol. 2012, 30, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Bellmunt, J.; von der Maase, H.; Mead, G.M.; Skoneczna, I.; De Santis, M.; Daugaard, G.; Boehle, A.; Chevreau, C.; Paz-Ares, L.; Laufman, L.R.; et al. Randomized phase III study comparing paclitaxel/cisplatin/gemcitabine and gemcitabine/cisplatin in patients with locally advanced or metastatic urothelial cancer without prior systemic therapy: EORTC Intergroup Study 30987. J. Clin. Oncol. 2012, 30, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.M.; Büsselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers (Basel) 2011, 3, 1351–1371. [Google Scholar] [CrossRef] [PubMed]
- Milowsky, M.I.; Rumble, R.B.; Booth, C.M.; Gilligan, T.; Eapen, L.J.; Hauke, R.J.; Boumansour, P.; Lee, C.T. Guideline on muscle-invasive and metastatic bladder cancer (European association of urology guideline): American society of clinical oncology clinical practice guideline endorsement. J. Clin. Oncol. 2016, 34, 1945–1952. [Google Scholar] [CrossRef] [PubMed]
- Buttigliero, C.; Tucci, M.; Vignani, F.; Scagliotti, G.V.; Di Maio, M. Molecular biomarkers to predict response to neoadjuvant chemotherapy for bladder cancer. Cancer Treat. Rev. 2017, 54, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Drayton, R.M.; Catto, J.W.F. Molecular mechanisms of cisplatin resistance in bladder cancer. Expert Rev. Anticancer Ther. 2012, 12, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, S.; Ohmichi, M.; Nishio, Y.; Hayasaka, T.; Kimura, A.; Ohta, T.; Saito, M.; Kawagoe, J.; Takahashi, K.; Yada-Hashimoto, N.; et al. Inhibition of NFkappaB increases the efficacy of cisplatin in in vitro and in vivo ovarian cancer models. J. Biol. Chem. 2004, 279, 23477–23485. [Google Scholar] [CrossRef] [PubMed]
- Ciamporcero, E.; Shen, H.; Ramakrishnan, S.; Yu Ku, S.; Chintala, S.; Shen, L.; Adelaiye, R.; Miles, K.M.; Ullio, C.; Pizzimenti, S.; et al. YAP activation protects urothelial cell carcinoma from treatment-induced DNA damage. Oncogene 2016, 35, 1541–1553. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.K.; Khatri, R.; Jaiswal, A.K. Regulation of Nrf2-an update. Free Radic. Biol. Med. 2014, 66, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Slocum, S.L.; Skoko, J.J.; Shin, S.; Kensler, T.W. When NRF2 talks, who’s listening? Antioxid. Redox Signal. 2010, 13, 1649–1663. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Yamamoto, M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv. Enzyme Regul. 2006, 46, 113–140. [Google Scholar] [CrossRef] [PubMed]
- Hayden, A.; Douglas, J.; Sommerlad, M.; Andrews, L.; Gould, K.; Hussain, S.; Thomas, G.J.; Packham, G.; Crabb, S.J. The Nrf2 transcription factor contributes to resistance to cisplatin in bladder cancer. Urol. Oncol. 2014, 32, 806–814. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Geismann, C.; Arlt, A.; Sebens, S.; Schafer, H. Cytoprotection “gone astray”: Nrf2 and its role in cancer. Onco. Targets Ther. 2014, 7, 1497–1518. [Google Scholar] [PubMed]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [PubMed]
- Na, H.K.; Surh, Y.J. Oncogenic potential of Nrf2 and its principal target protein heme oxygenase-1. Free Radic. Biol. Med. 2014, 67, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Baird, L.; Dinkova-Kostova, A.T. The cytoprotective role of the Keap1-Nrf2 pathway. Arch. Toxicol. 2011, 85, 241–272. [Google Scholar] [CrossRef] [PubMed]
- Zoja, C.; Benigni, A.; Remuzzi, G. The Nrf2 pathway in the progression of renal disease. Nephrol. Dial. Transplant. 2014, 29, i19–i24. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Yamachika, S.; He, F.; Karin, M. p62/SQSTM1-Dr. Jekyll and Mr. Hyde that prevents oxidative stress but promotes liver cancer. FEBS Lett. 2016, 590, 2375–2397. [Google Scholar] [CrossRef] [PubMed]
- Homma, S.; Ishii, Y.; Morishima, Y.; Yamadori, T.; Matsuno, Y.; Haraguchi, N.; Kikuchi, N.; Satoh, H.; Sakamoto, T.; Hizawa, N.; et al. Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clin. Cancer Res. 2009, 15, 3423–3432. [Google Scholar] [CrossRef] [PubMed]
- Nezis, I.P.; Stenmark, H. p62 at the interface of autophagy, oxidative stress signaling, and cancer. Antioxid. Redox Signal. 2012, 17, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Praslicka, B.J.; Kerins, M.J.; Ooi, A. The complex role of NRF2 in cancer: A genomic view. Curr. Opin. Toxicol. 2016, 1, 37–45. [Google Scholar] [CrossRef]
- Oshimori, N.; Oristian, D.; Fuchs, E. TGF-β promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 2015, 160. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Kuo, M.T. Role of glutathione in the regulation of Cisplatin resistance in cancer chemotherapy. Met. Based Drugs 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Menegon, S.; Columbano, A.; Giordano, S. The dual roles of NRF2 in cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef] [PubMed]
- Peklak-Scott, C.; Smitherman, P.K.; Townsend, A.J.; Morrow, C.S. Role of glutathione S-transferase P1–1 in the cellular detoxification of cisplatin. Mol. Cancer Ther. 2008, 7, 3247–3255. [Google Scholar] [CrossRef] [PubMed]
- Rocha, C.R.; Garcia, C.C.; Vieira, D.B.; Quinet, A.; de Andrade-Lima, L.C.; Munford, V.; Belizario, J.E.; Menck, C.F. Glutathione depletion sensitizes cisplatin- and temozolomide-resistant glioma cells in vitro and in vivo. Cell Death Dis. 2014, 5, e1505. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Koppula, S.; Kim, I.S.; More, S.V.; Kim, B.W.; Choi, D.K. Nuclear factor erythroid 2-related factor 2 signaling in Parkinson disease: A promising multi therapeutic target against oxidative stress, neuroinflammation and cell death. CNS Neurol. Disord. Drug Targets 2012, 11, 1015–1029. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Sadee, W. Membrane transporters and channels in chemoresistance and -sensitivity of tumor cells. Cancer Lett. 2006, 239, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Dai, Z.; Barbacioru, C.; Sadee, W. Cystine-glutamate transporter SLC7A11 in cancer chemosensitivity and chemoresistance. Cancer Res. 2005, 65, 7446–7454. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.; Kwak, M. Shadows of NRF2 in cancer: Resistance to chemotherapy. Curr. Opin. Toxicol. 2016, 1, 20–28. [Google Scholar] [CrossRef]
- Byun, S.S.; Kim, S.W.; Choi, H.; Lee, C.; Lee, E. Augmentation of cisplatin sensitivity in cisplatin-resistant human bladder cancer cells by modulating glutathione concentrations and glutathione-related enzyme activities. BJU Int. 2005, 95, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Savic-Radojevic, A.; Mimic-Oka, J.; Pljesa-Ercegovac, M.; Opacic, M.; Dragicevic, D.; Kravic, T.; Djokic, M.; Micic, S.; Simic, T. Glutathione S-transferase-P1 expression correlates with increased antioxidant capacity in transitional cell carcinoma of the urinary bladder. Eur. Urol. 2007, 52, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Dadhania, V.; Zhang, M.; Zhang, L.; Bondaruk, J.; Majewski, T.; Siefker-Radtke, A.; Guo, C.C.; Dinney, C.; Cogdell, D.E.; Zhang, S.; et al. Meta-analysis of the luminal and basal subtypes of bladder cancer and the identification of signature immunohistochemical markers for clinical use. EBioMedicine 2016, 12, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar]
- Skowron, M.A.; Niegisch, G.; Fritz, G.; Arent, T.; van Roermund, J.G.; Romano, A.; Albers, P.; Schulz, W.A.; Hoffmann, M.J. Phenotype plasticity rather than repopulation from CD90/CK14+ cancer stem cells leads to cisplatin resistance of urothelial carcinoma cell lines. J. Exp. Clin. Cancer Res. 2015, 34, 144. [Google Scholar] [CrossRef] [PubMed]
- Hohn, A.; Kruger, K.; Skowron, M.A.; Bormann, S.; Schumacher, L.; Schulz, W.A.; Hoffmann, M.J.; Niegisch, G.; Fritz, G. Distinct mechanisms contribute to acquired cisplatin resistance of urothelial carcinoma cells. Oncotarget 2016, 7, 41320–41335. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, L.D.; Lee, J.; Gnad, F.; Klijn, C.; Schaub, A.; Reeder, J.; Daemen, A.; Bakalarski, C.E.; Holcomb, T.; Shames, D.S.; et al. Recurrent loss of NFE2L2 exon 2 is a mechanism for Nrf2 pathway activation in human cancers. Cell Rep. 2016, 16, 2605–2617. [Google Scholar] [CrossRef] [PubMed]
- Yoo, N.J.; Kim, H.R.; Kim, Y.R.; An, C.H.; Lee, S.H. Somatic mutations of the KEAP1 gene in common solid cancers. Histopathology 2012, 60, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Wu, K.; Liu, Q.; Han, N.; Zhang, L.; Chu, Q.; Chen, Y. Modification of platinum sensitivity by KEAP1/NRF2 signals in non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 83. [Google Scholar] [CrossRef] [PubMed]
- Siegsmund, M.J.; Marx, C.; Seemann, O.; Schummer, B.; Steidler, A.; Toktomambetova, L.; Kohrmann, K.U.; Rassweiler, J.; Alken, P. Cisplatin-resistant bladder carcinoma cells: Enhanced expression of metallothioneins. Urol. Res. 1999, 27, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Wang, M.; Shen, Y.; Liao, J.; Li, A.; Wang, M. PKIS: Computational identification of protein kinases for experimentally discovered protein phosphorylation sites. BMC Bioinform. 2013, 14, 247. [Google Scholar] [CrossRef] [PubMed]
- Adamson, R.; Krojer, T.; Bartual, S.G.; Burgess-Brown, N.A.; Borkowska, O.; Chalk, R.; Newman, J.A.; Kopec, J.; Dixon-Clarke, S.E.; Mathea, S.; et al. Crystal structure of human CUL3 N-terminal domain bound to KEAP1 BTB and 3-box. 2017. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Su, J.; Xu, Y.; Kang, J.; Li, H.; Zhang, L.; Yi, H.; Xiang, X.; Liu, F.; Sun, L. p62/SQSTM1 involved in cisplatin resistance in human ovarian cancer cells by clearing ubiquitinated proteins. Eur. J. Cancer 2011, 47, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Yu, H.; Gu, S.; Xu, Y.; Su, J.; Li, H.; Kang, J.; Cui, M. p62/SQSTM1 is involved in cisplatin resistance in human ovarian cancer cells via the Keap1-Nrf2-ARE system. Int. J. Oncol. 2014, 45, 2341–2348. [Google Scholar] [CrossRef] [PubMed]
- Zavattari, P.; Perra, A.; Menegon, S.; Kowalik, M.A.; Petrelli, A.; Angioni, M.M.; Follenzi, A.; Quagliata, L.; Ledda-Columbano, G.M.; Terracciano, L.; et al. Nrf2, but not beta-catenin, mutation represents an early event in rat hepatocarcinogenesis. Hepatology 2015, 62, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Rebouissou, S.; Zucman Rossi, J. NRF2/KEAP1 and Wnt/β-catenin in the multistep process of liver carcinogenesis in humans and rats. Hepatology 2015, 62, 677–679. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, N.; Chaunsali, L.; Deshmukh, P.; Padmanabhan, B. Analysis of dimerization of BTB-IVR domains of Keap1 and its interaction with Cul3, by molecular modeling. Bioinformation 2013, 9, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Tan, Y.X.; Yang, G.Z.; Zhang, J.; Pan, Y.F.; Liu, C.; Fu, J.; Chen, Y.; Ding, Z.W.; Dong, L.W.; et al. Gankyrin has an antioxidative role through the feedback regulation of Nrf2 in hepatocellular carcinoma. J. Exp. Med. 2016, 213, 859–875. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Cheng, L. Gankyrin as a potential therapeutic target for cancer. Investig. New Drugs 2017, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, N.F.; Sun, Z.; Chen, W.; Zhang, D.D. Nrf2 and p21 regulate the fine balance between life and death by controlling ROS levels. Cell Cycle 2009, 8, 3255–3256. [Google Scholar] [CrossRef] [PubMed]
- Toledano, M.B. The guardian recruits cops: The p53-p21 axis delegates prosurvival duties to the Keap1-Nrf2 stress pathway. Mol. Cell 2009, 34, 637–639. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Carroll, K.S. Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem. Biol. 2010, 5, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Jeddi, F.; Soozangar, N.; Sadeghi, M.R.; Somi, M.H.; Samadi, N. Contradictory roles of Nrf2/Keap1 signaling pathway in cancer prevention/promotion and chemoresistance. DNA Repair (Amst) 2017, 54, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Mohell, N.; Alfredsson, J.; Fransson, A.; Uustalu, M.; Bystrom, S.; Gullbo, J.; Hallberg, A.; Bykov, V.J.; Bjorklund, U.; Wiman, K.G. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 2015, 6, e1794. [Google Scholar] [CrossRef] [PubMed]
- Jamali, B.; Nakhjavani, M.; Hosseinzadeh, L.; Amidi, S.; Nikounezhad, N.; Shirazi, F.H. Intracellular GSH alterations and its relationship to level of resistance following exposure to cisplatin in cancer cells. Iran. J. Pharm. Res. 2015, 14, 513–519. [Google Scholar] [PubMed]
- Cho, J.M.; Manandhar, S.; Lee, H.R.; Park, H.M.; Kwak, M.K. Role of the Nrf2-antioxidant system in cytotoxicity mediated by anticancer cisplatin: Implication to cancer cell resistance. Cancer Lett. 2008, 260, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Drayton, R.M.; Dudziec, E.; Peter, S.; Bertz, S.; Hartmann, A.; Bryant, H.E.; Catto, J.W. Reduced expression of miRNA-27a modulates cisplatin resistance in bladder cancer by targeting the cystine/glutamate exchanger SLC7A11. Clin. Cancer Res. 2014, 20, 1990–2000. [Google Scholar] [CrossRef] [PubMed]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohe, R.; Kipp, A.P. Selenium in the redox regulation of the Nrf2 and the Wnt pathway. Methods Enzymol. 2013, 527, 65–86. [Google Scholar] [PubMed]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Samatiwat, P.; Prawan, A.; Senggunprai, L.; Kukongviriyapan, U.; Kukongviriyapan, V. Nrf2 inhibition sensitizes cholangiocarcinoma cells to cytotoxic and antiproliferative activities of chemotherapeutic agents. Tumour. Biol. 2016, 37, 11495–11507. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Chowdhry, S.; Dinkova-Kostova, A.T.; Sutherland, C. Dual regulation of transcription factor Nrf2 by Keap1 and by the combined actions of beta-TrCP and GSK-3. Biochem. Soc. Trans. 2015, 43, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Lü, F.; Stewart, D.; Zhang, Y. Mechanisms underlying chemopreventive effects of flavonoids via multiple signaling nodes within Nrf2-ARE and AhR-XRE gene regulatory networks. Curr. Chem. Biol. 2013, 7, 151–176. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; McMahon, M. Cross-talk between transcription factors AhR and Nrf2: lessons for cancer chemoprevention from dioxin. Toxicol. Sci. 2009, 111, 199–201. [Google Scholar] [CrossRef] [PubMed]
- Nioi, P.; Hayes, J.D. Contribution of NAD(P)H: Quinone oxidoreductase 1 to protection against carcinogenesis, and regulation of its gene by the Nrf2 basic-region leucine zipper and the arylhydrocarbon receptor basic helix-loop-helix transcription factors. Mutat. Res. 2004, 555, 149–171. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.J. Ubiquitin signalling in the NF-κB pathway. Nat. Cell Biol. 2005, 7, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, Z.; Dong, J. P62: An emerging oncotarget for osteolytic metastasis. J. Bone Oncol. 2016, 5, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Camp, N.D.; James, R.G.; Dawson, D.W.; Yan, F.; Davison, J.M.; Houck, S.A.; Tang, X.; Zheng, N.; Major, M.B.; Moon, R.T. Wilms tumor gene on X chromosome (WTX) inhibits degradation of NRF2 protein through competitive binding to KEAP1 protein. J. Biol. Chem. 2012, 287, 6539–6550. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Hayes, J.D.; Wolf, C.R. Generation of a stable antioxidant response element-driven reporter gene cell line and its use to show redox-dependent activation of nrf2 by cancer chemotherapeutic agents. Cancer Res. 2006, 66, 10983–10994. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, I.; Migliorati, G.; Pagliacci, M.C.; Grignani, F.; Riccardi, C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 1991, 139, 271–279. [Google Scholar] [CrossRef]
- Oeck, S.; Malewicz, N.M.; Hurst, S.; Rudner, J.; Jendrossek, V. The Focinator—A new open-source tool for high-throughput foci evaluation of DNA damage. Radiat. Oncol. 2015, 10, 163. [Google Scholar] [CrossRef] [PubMed]
- Pinkerneil, M.; Hoffmann, M.J.; Deenen, R.; Kohrer, K.; Arent, T.; Schulz, W.A.; Niegisch, G. Inhibition of class I histone deacetylases 1 and 2 promotes urothelial carcinoma cell death by various mechanisms. Mol. Cancer Ther. 2016, 15, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, P.; Bouchachia, I.; Goebels, N.; Henke, N.; Hofstetter, H.H.; Issberner, A.; Kovacs, Z.; Lewerenz, J.; Lisak, D.; Maher, P.; et al. Effects of dimethyl fumarate on neuroprotection and immunomodulation. J. Neuroinflammation 2012, 9, 163. [Google Scholar] [CrossRef] [PubMed]
- Tietze, F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: Applications to mammalian blood and other tissues. Anal. Biochem. 1969, 27, 502–522. [Google Scholar] [CrossRef]
- Maher, P.; Hanneken, A. The molecular basis of oxidative stress-induced cell death in an immortalized retinal ganglion cell line. Investig. Ophthalmol. Vis. Sci. 2005, 46, 749–757. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skowron, M.A.; Niegisch, G.; Albrecht, P.; Van Koeveringe, G.; Romano, A.; Albers, P.; Schulz, W.A.; Hoffmann, M.J. Various Mechanisms Involve the Nuclear Factor (Erythroid-Derived 2)-Like (NRF2) to Achieve Cytoprotection in Long-Term Cisplatin-Treated Urothelial Carcinoma Cell Lines. Int. J. Mol. Sci. 2017, 18, 1680. https://doi.org/10.3390/ijms18081680
Skowron MA, Niegisch G, Albrecht P, Van Koeveringe G, Romano A, Albers P, Schulz WA, Hoffmann MJ. Various Mechanisms Involve the Nuclear Factor (Erythroid-Derived 2)-Like (NRF2) to Achieve Cytoprotection in Long-Term Cisplatin-Treated Urothelial Carcinoma Cell Lines. International Journal of Molecular Sciences. 2017; 18(8):1680. https://doi.org/10.3390/ijms18081680
Chicago/Turabian StyleSkowron, Margaretha A., Günter Niegisch, Philipp Albrecht, Gommert Van Koeveringe, Andrea Romano, Peter Albers, Wolfgang A. Schulz, and Michèle J. Hoffmann. 2017. "Various Mechanisms Involve the Nuclear Factor (Erythroid-Derived 2)-Like (NRF2) to Achieve Cytoprotection in Long-Term Cisplatin-Treated Urothelial Carcinoma Cell Lines" International Journal of Molecular Sciences 18, no. 8: 1680. https://doi.org/10.3390/ijms18081680