Comprehensive Study of Multiple Stages Progressing to Nonalcoholic Steatohepatitis with Subsequent Fibrosis in SD Rats

Abstract
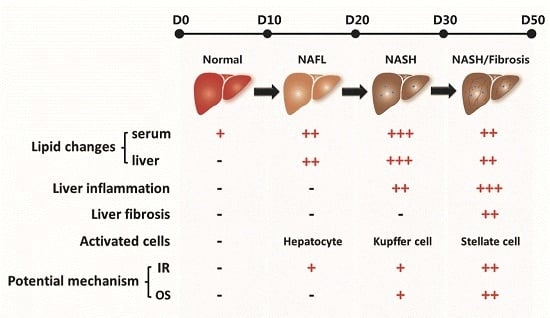
:
1. Introduction
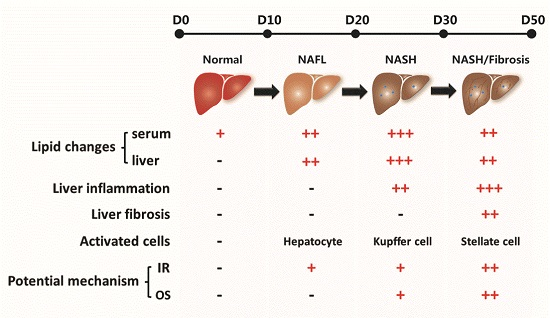
2. Results
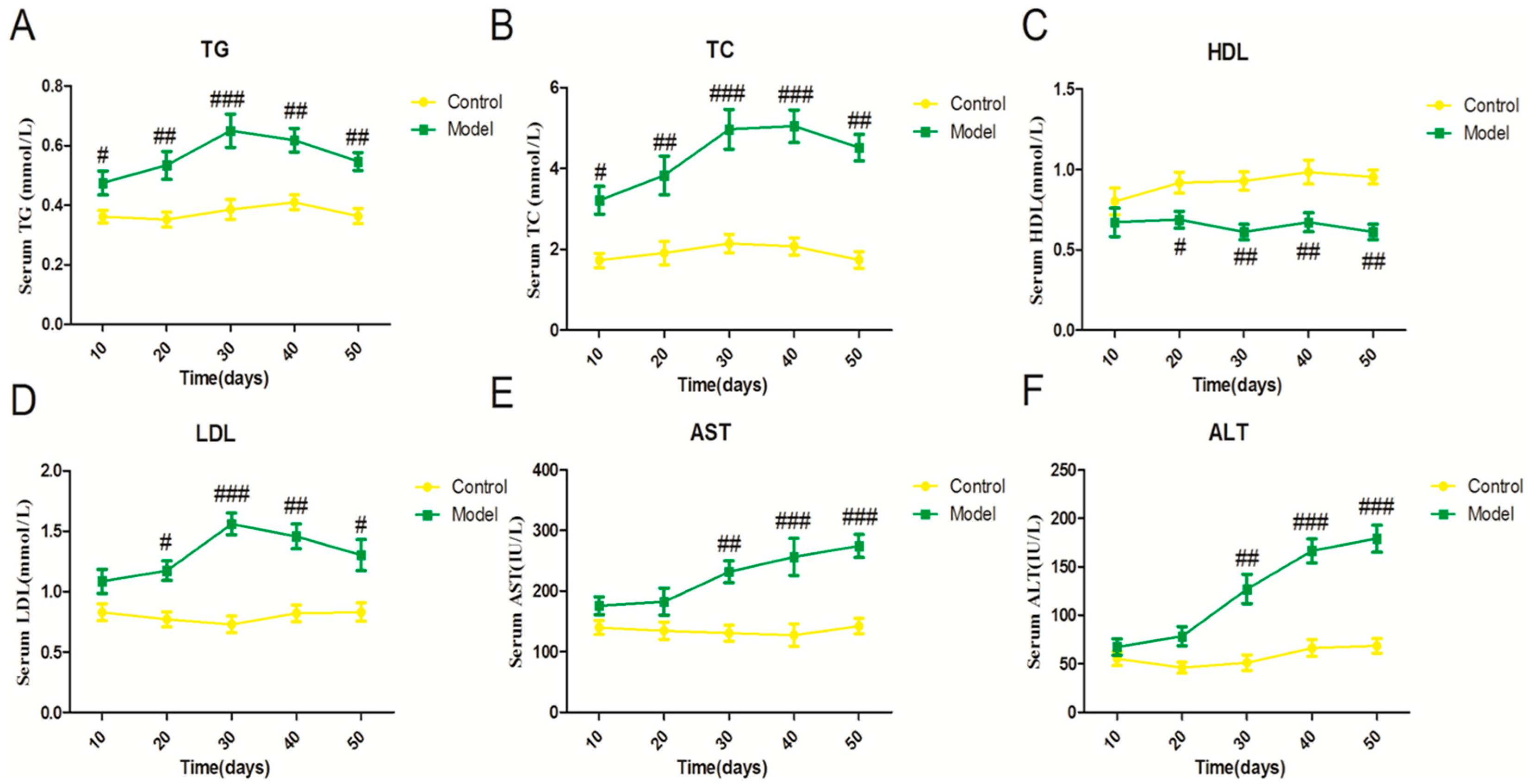
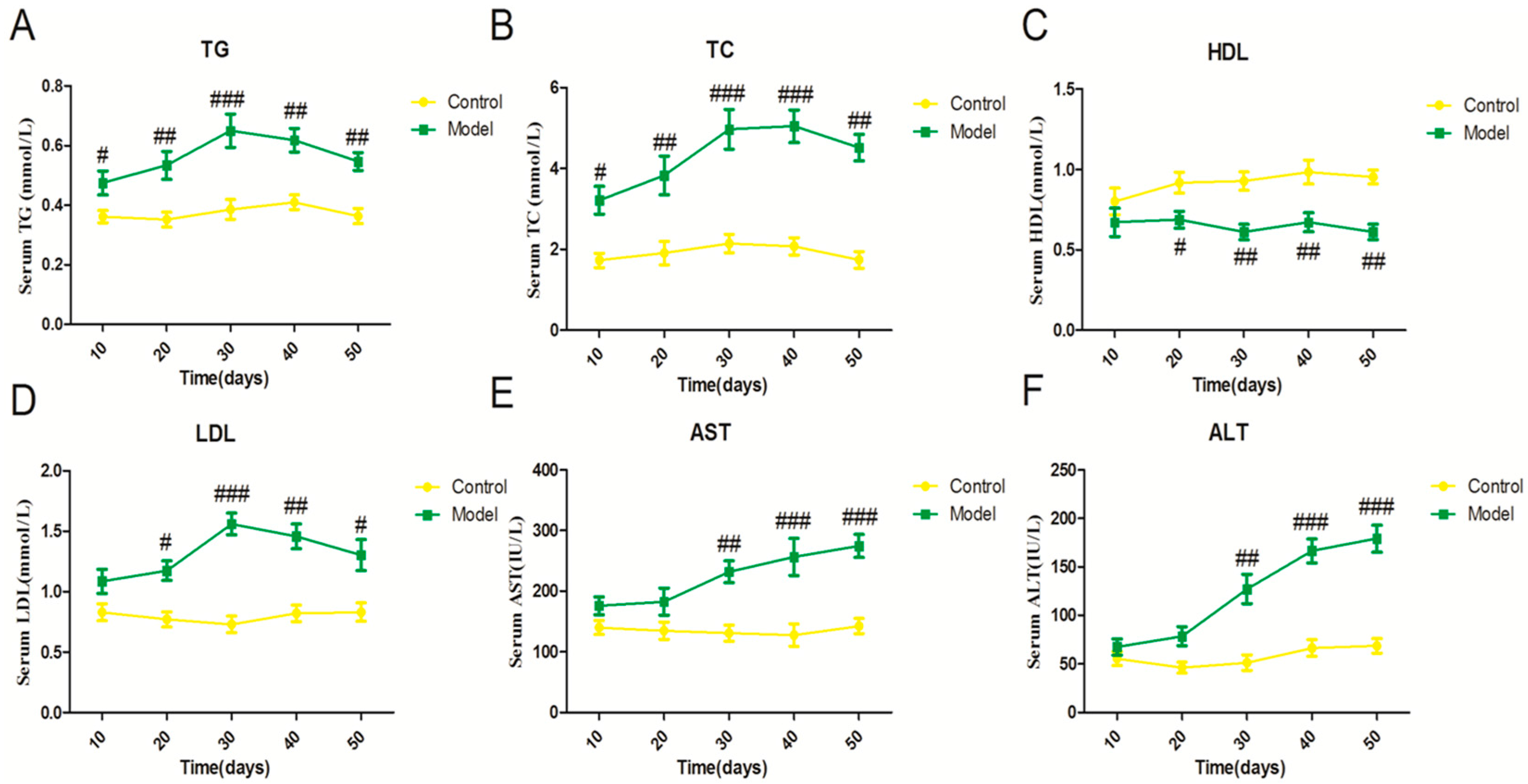
2.1. Effect of a High Fat-Sucrose Diet on Metabolic Profiles in Sprague-Dawley (SD) Rats
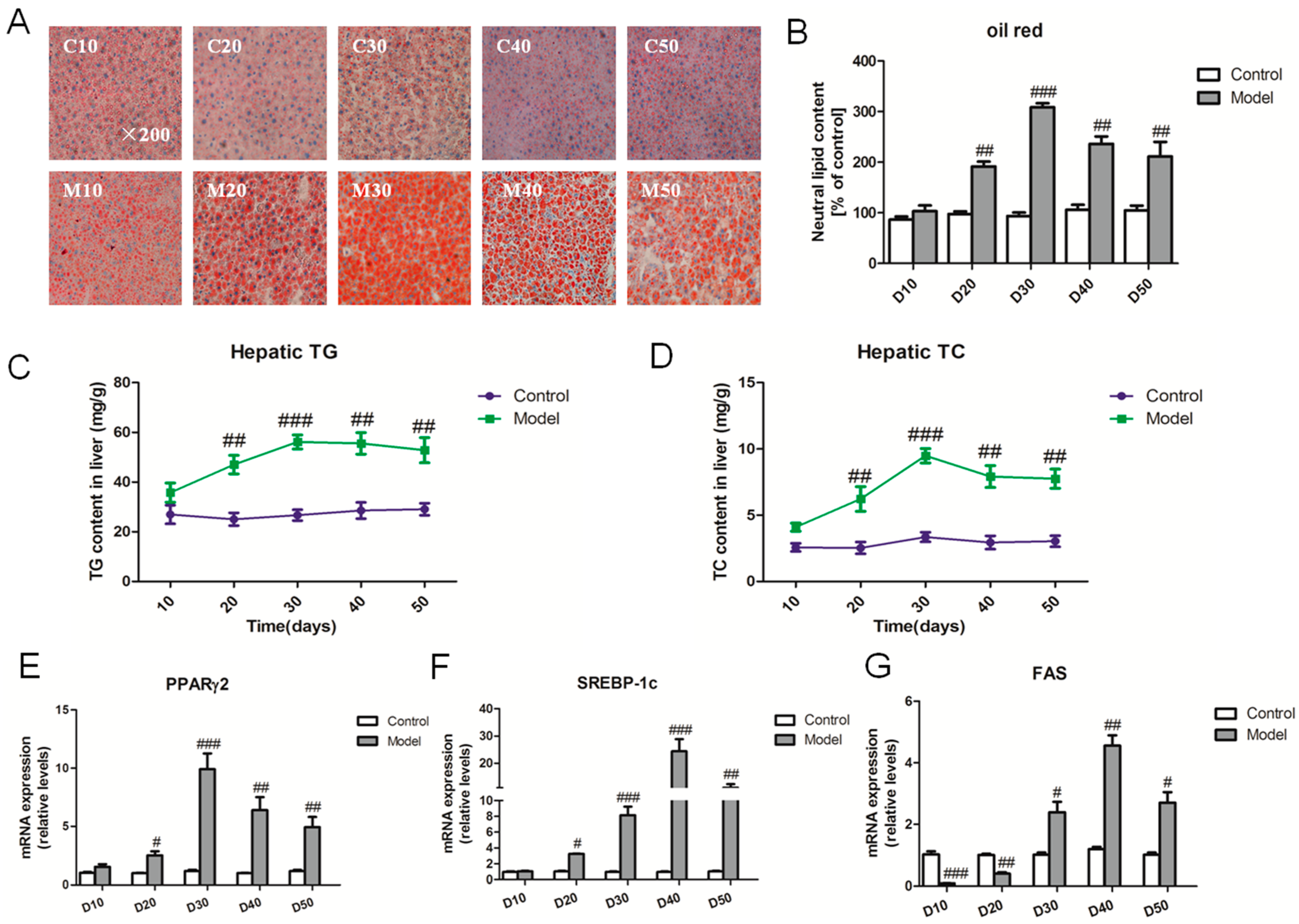
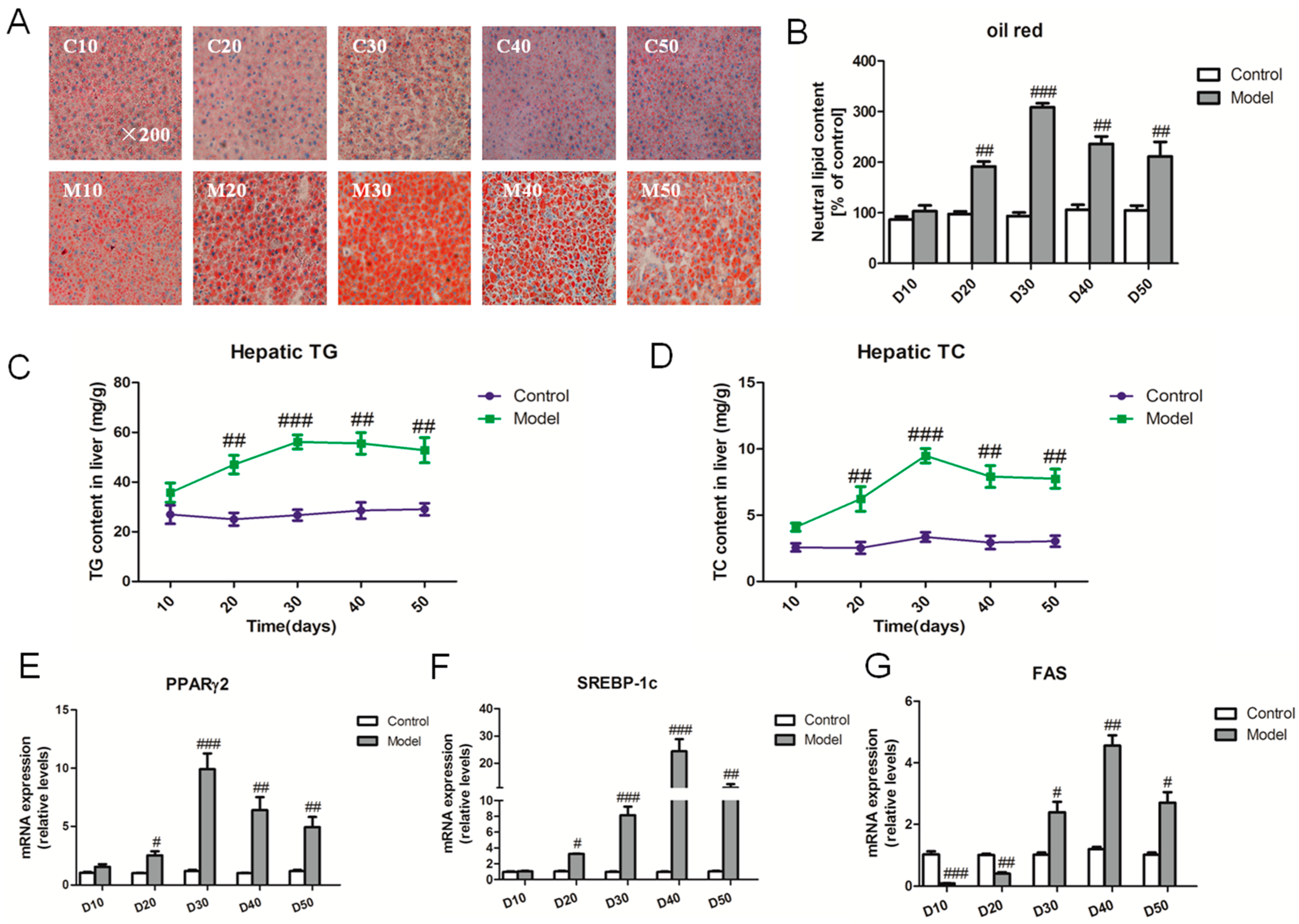
2.2. Effect of a High Fat-Sucrose Diet on Hepatic Lipid Accumulation
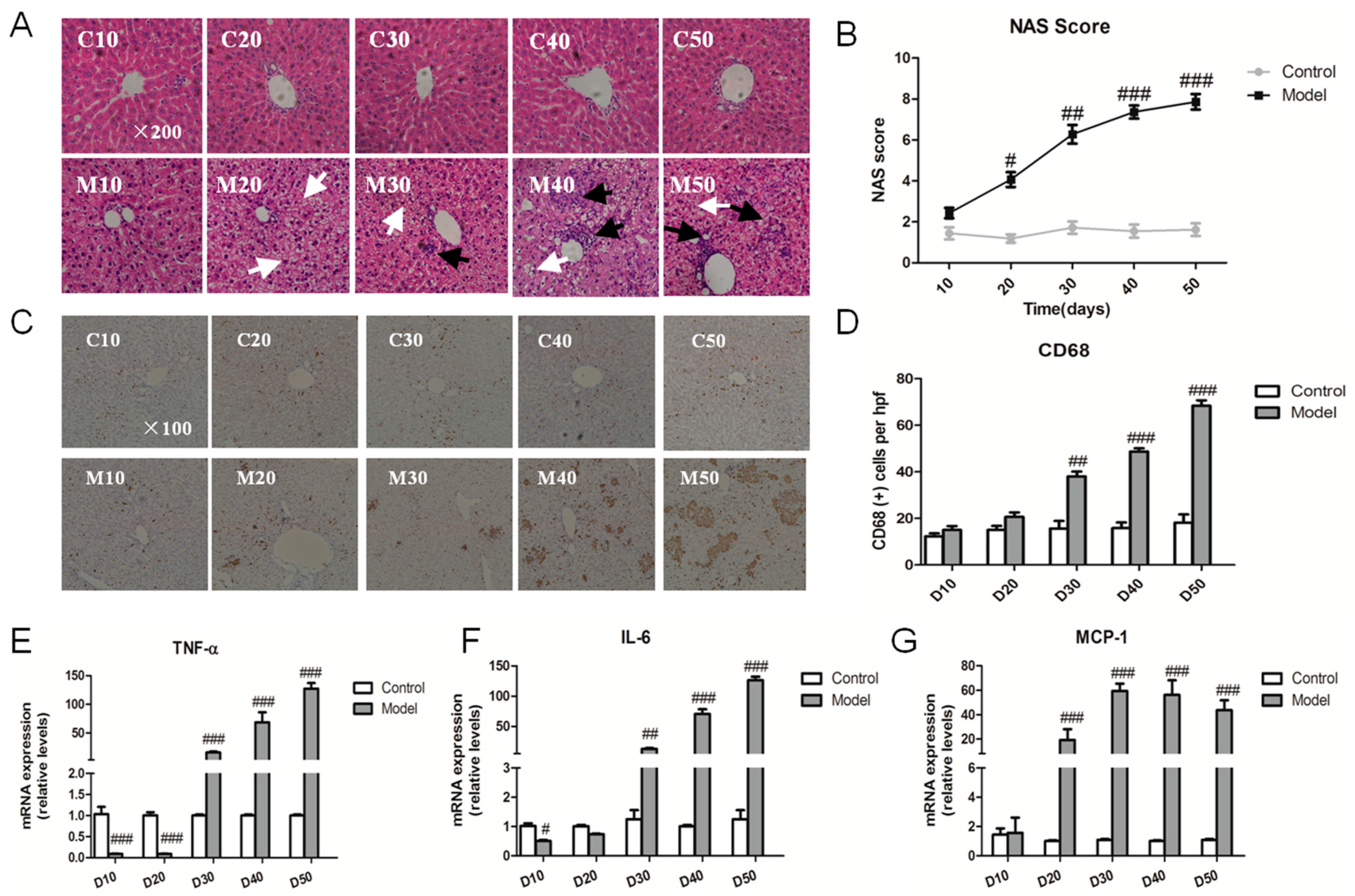
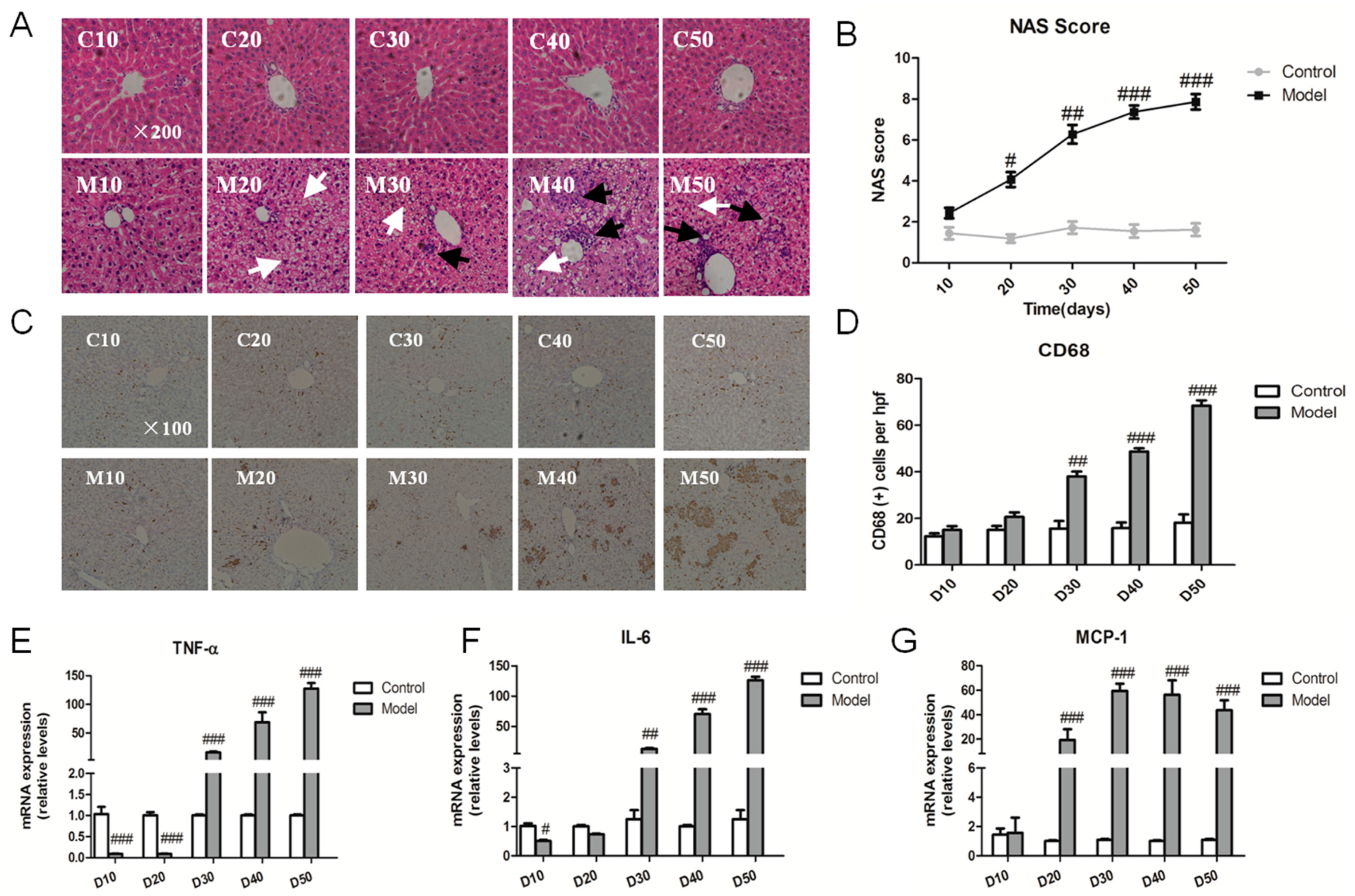
2.3. Effect of the High Fat-Sucrose Diet on the Hepatic Inflammation Response
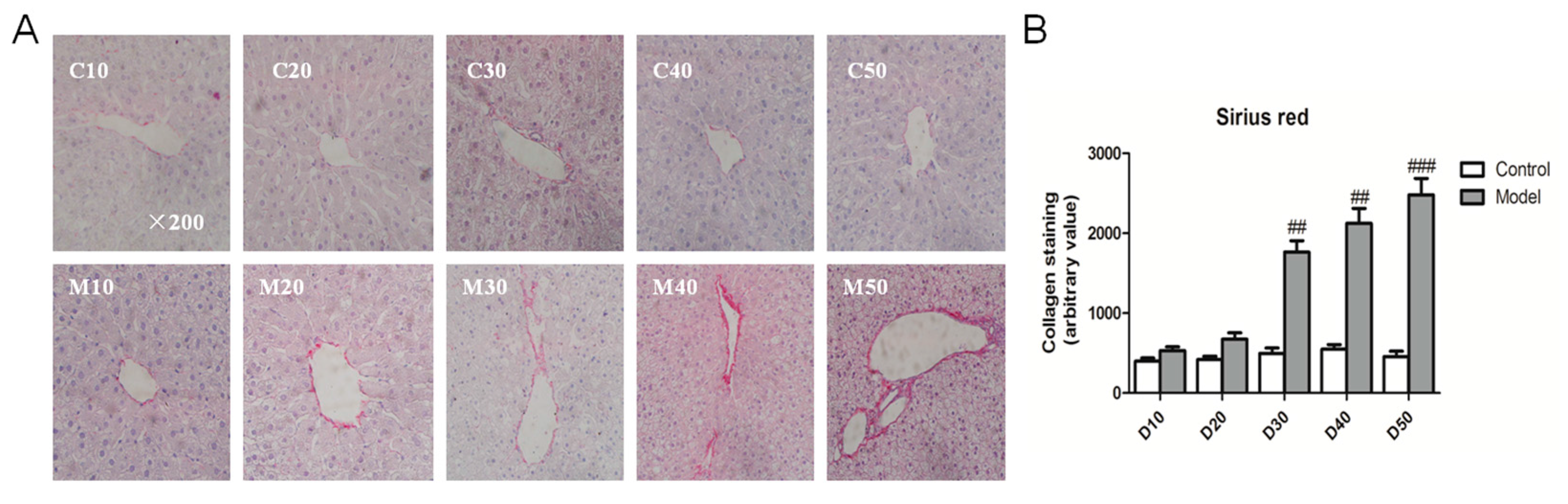
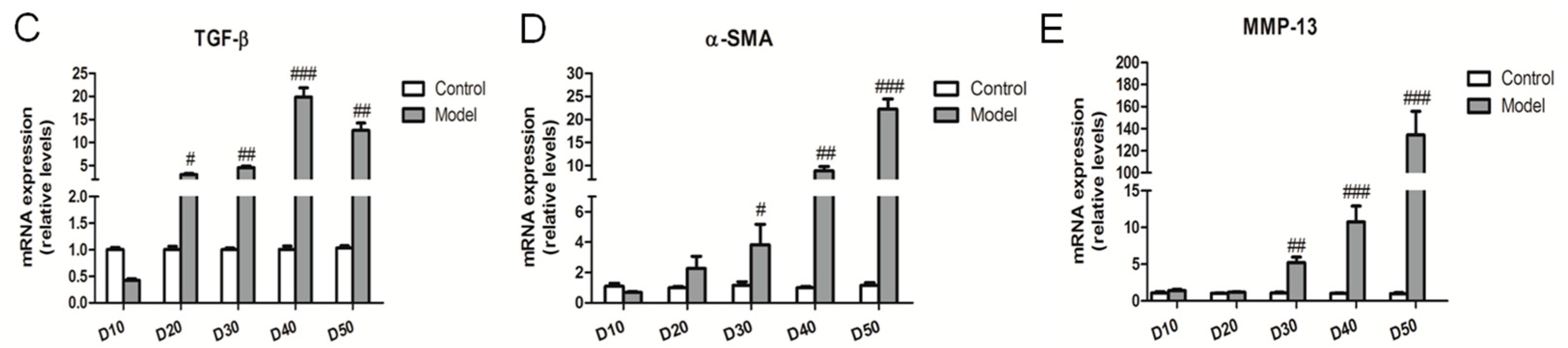
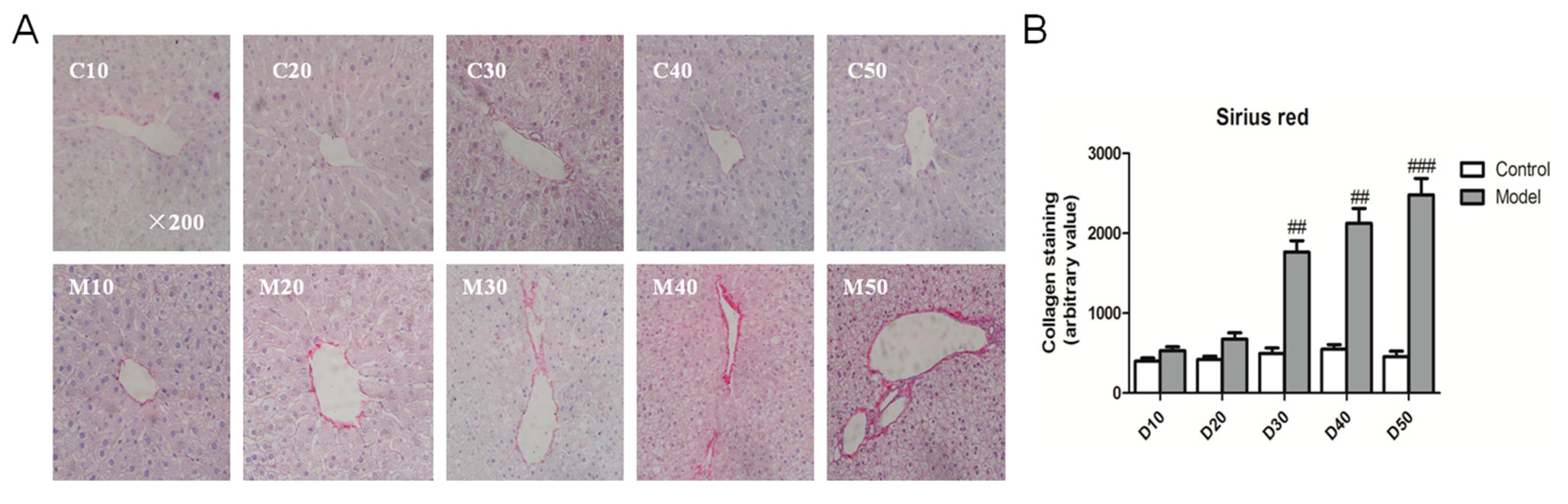
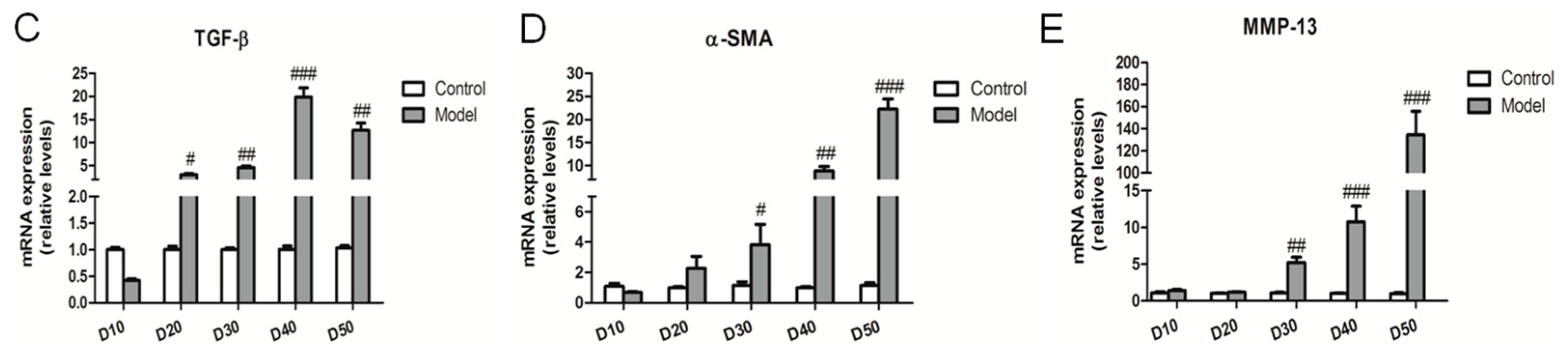
2.4. Effect of the High Fat-Sucrose Diet on Hepatic Fibrosis
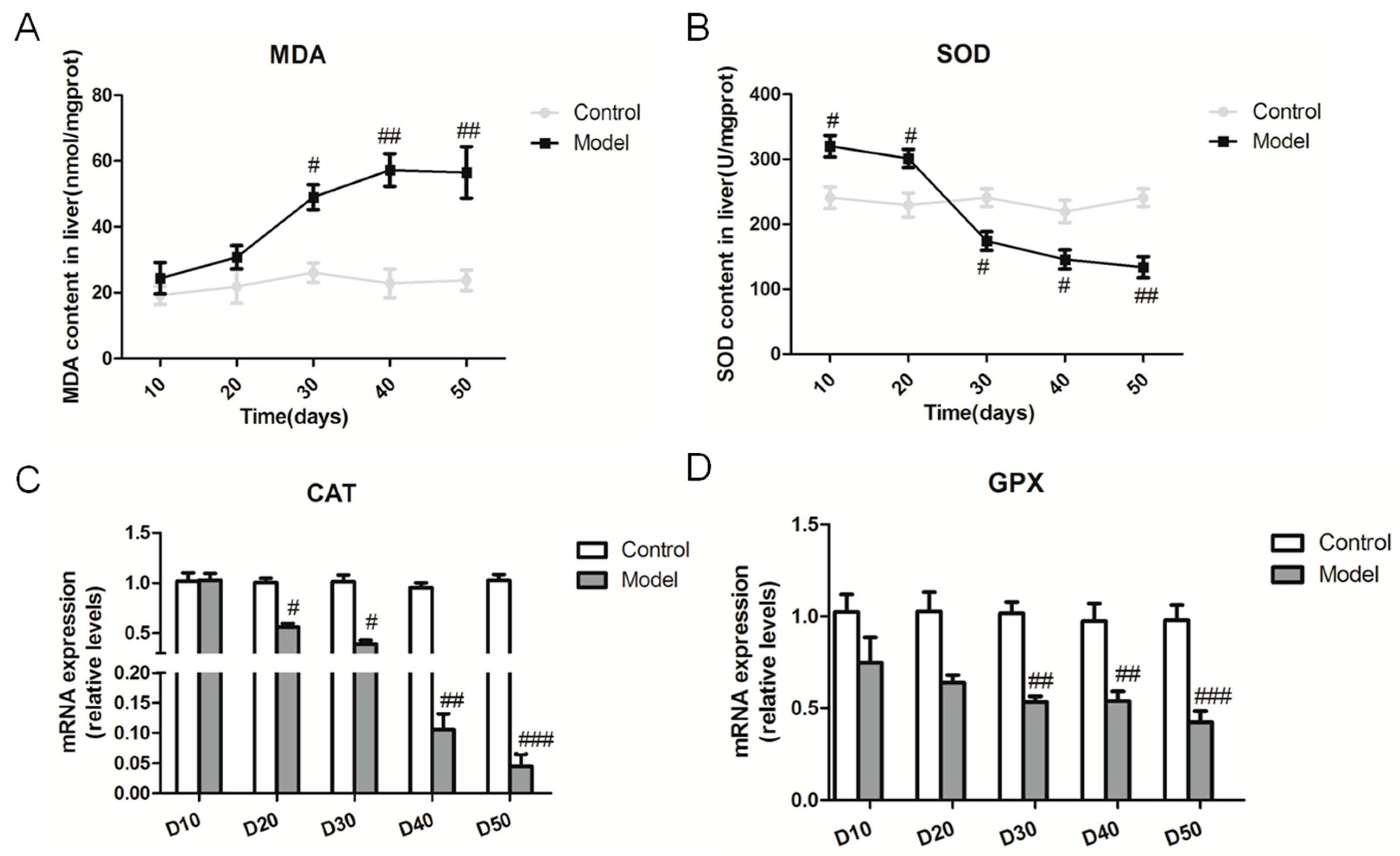
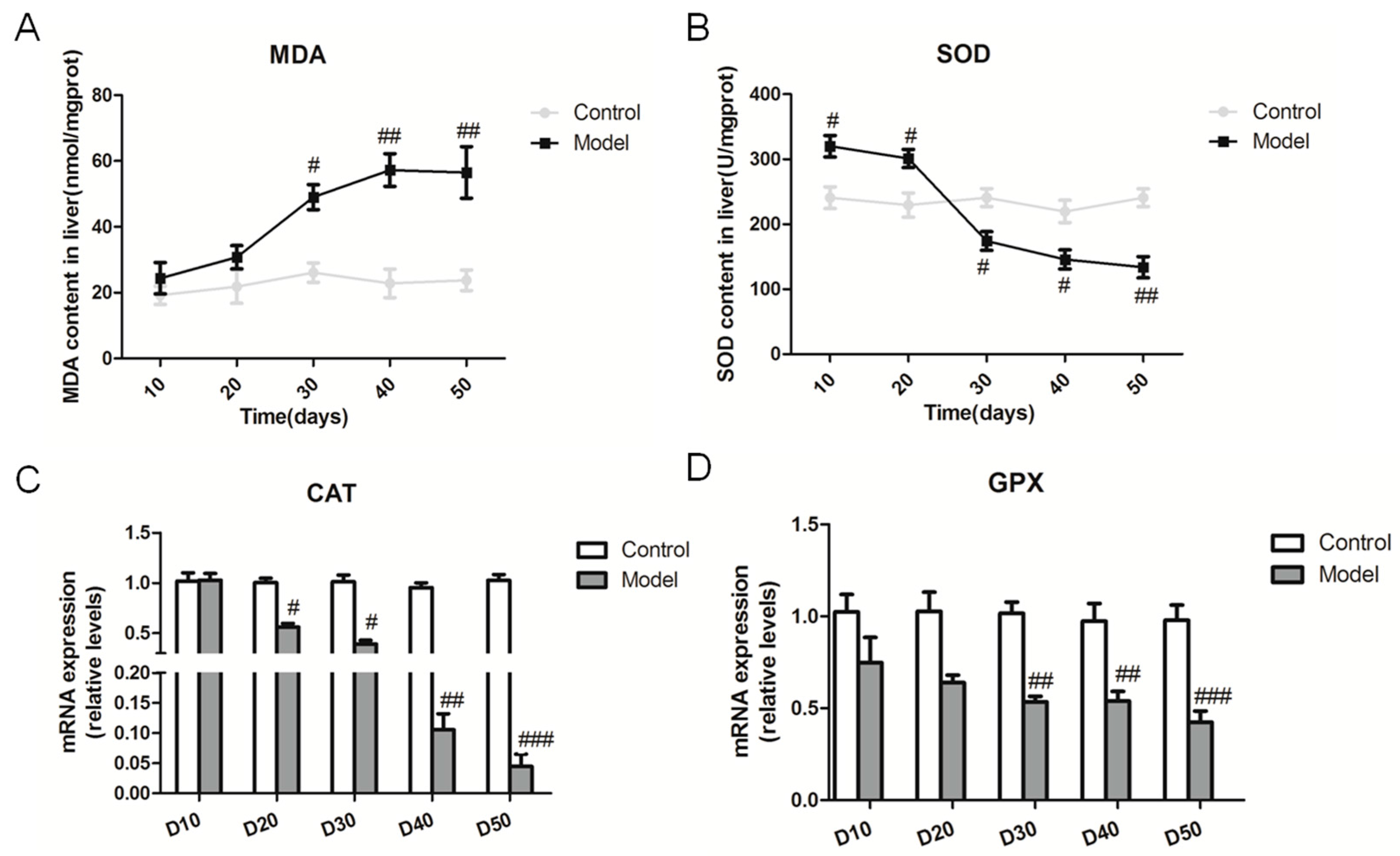
2.5. Effect of the High Fat-Sucrose Diet on the Hepatic Oxidative Stress Response
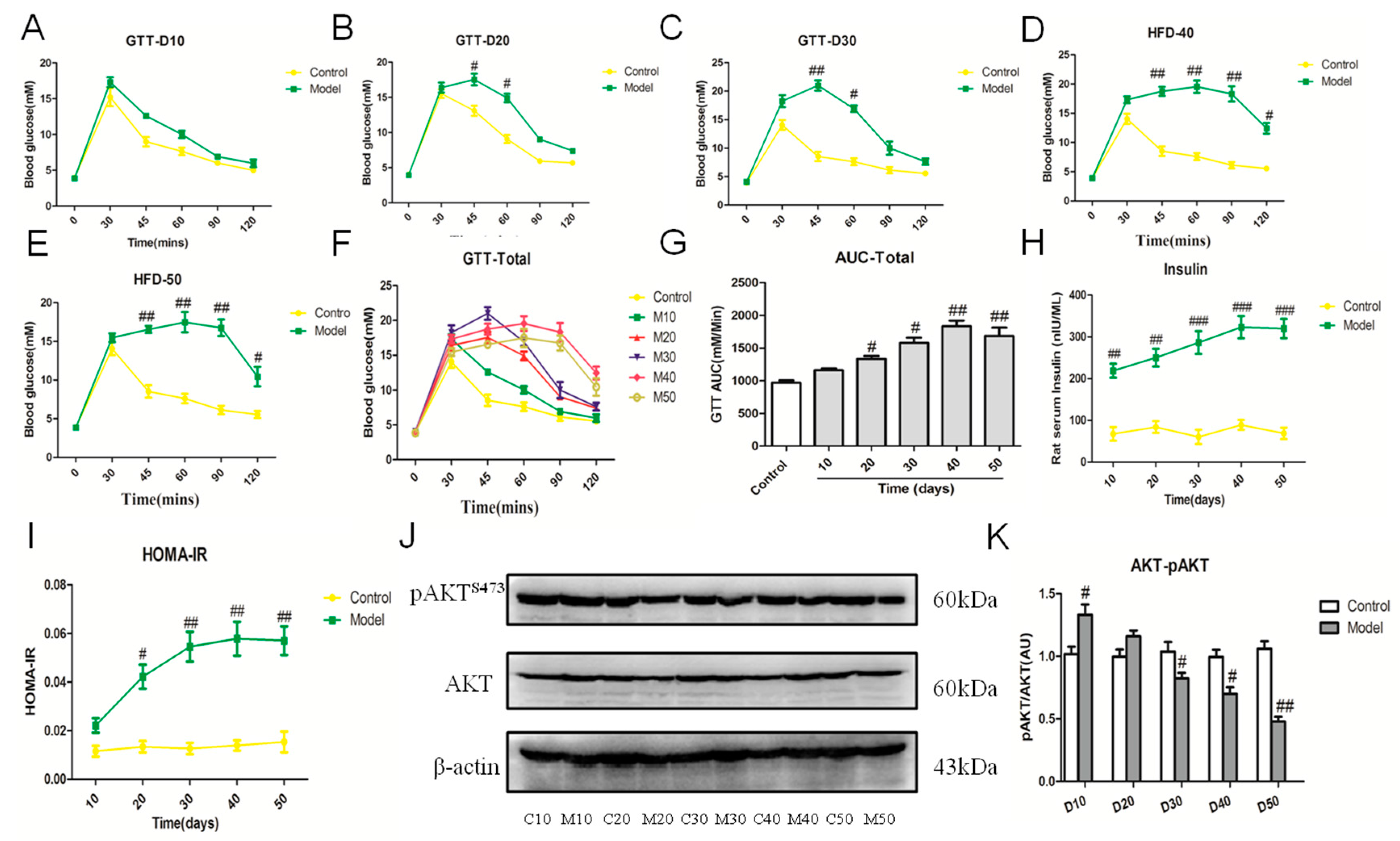
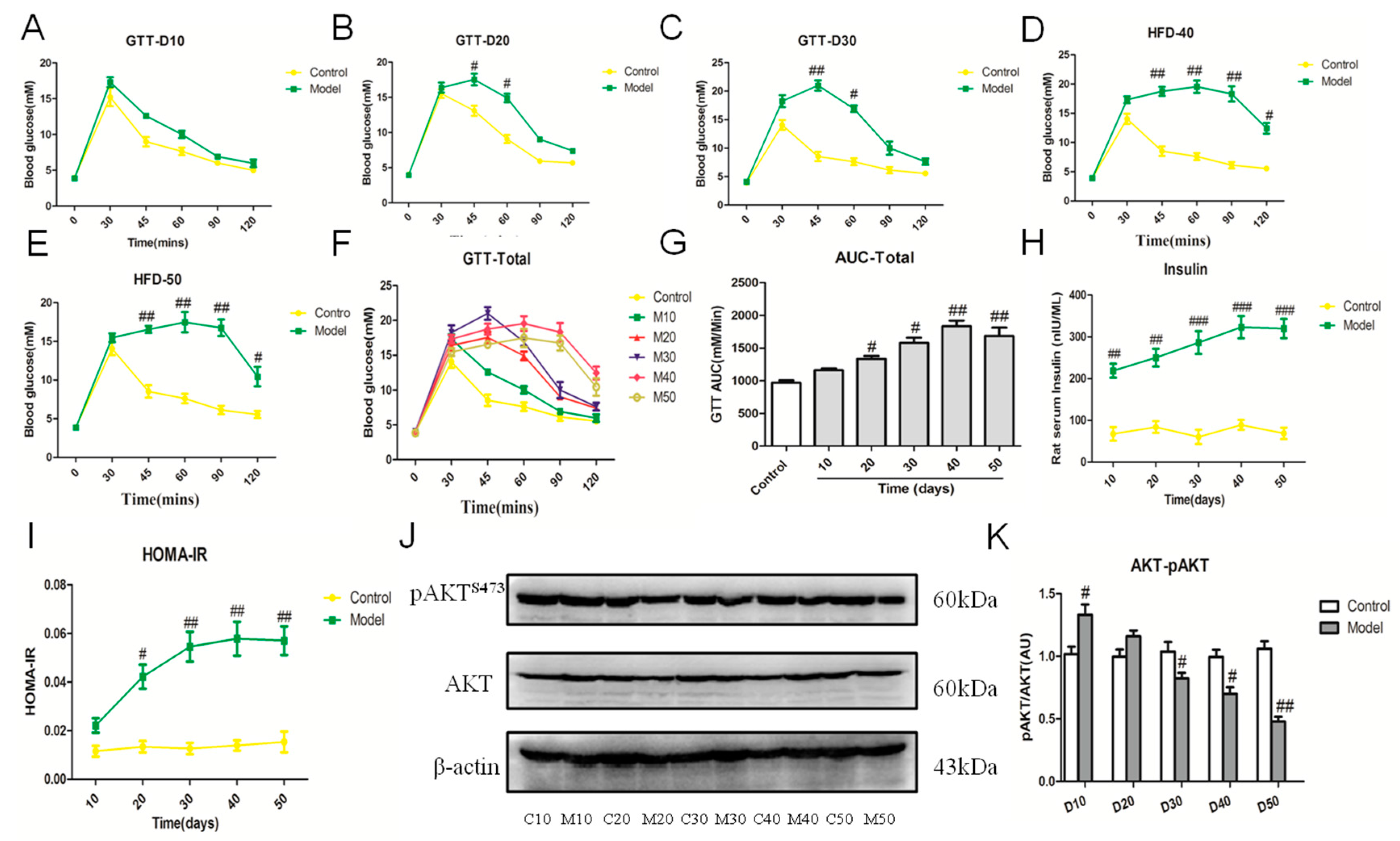
2.6. Effect of the High Fat-Sucrose Diet on Hepatic Insulin Resistance
3. Discussion
4. Materials and Methods
4.1. Animal Studies
4.2. Serum Biochemical Analysis
4.3. Liver Histopathological Analysis
4.4. Quantitative Real-Time Polymerase Chain Reaction (RT-qPCR)
4.5. Glucose Tolerance Test (GTT)—HOMA-IR
4.6. Western Blotting Analysis
4.7. Lipid and Hepatocellular Oxidative Stress Analysis of the Liver
4.8. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Demir, M.; Lang, S.; Steffen, H.M. Nonalcoholic fatty liver disease—Current status and future directions. J. Dig. Dis. 2015, 16, 541–557. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, B.; Meng, X.; Yao, S.; Jin, L.; Yang, J.; Wang, J.; Zhang, H.; Zhang, Z.; Cai, D.; et al. Berberine prevents progression from hepatic steatosis to steatohepatitis and fibrosis by reducing endoplasmic reticulum stress. Sci. Rep. 2016, 6, 20848. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.M.; Oh, S.W.; Hwang, S.S.; Lee, C.; Kwon, H.; Chung, G.E. Association of nonalcoholic fatty liver disease with components of metabolic syndrome according to body mass index in korean adults. Am. J. Gastroenterol. 2012, 107, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, H.J.; Lee, K.E.; Kim, D.J.; Kim, S.K.; Ahn, C.W.; Lim, S.K.; Kim, K.R.; Lee, H.C.; Huh, K.B. Metabolic significance of nonalcoholic fatty liver disease in nonobese, nondiabetic adults. Arch. Intern. Med. 2004, 164, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Rui, G.; Man, L.F.; Liong, E.C.; Tipoe, G.L. Therapeutic approaches to non-alcoholic fatty liver disease: Past achievements and future challenges. Hepatobiliary Pancreat. Dis. Int. 2013, 12, 125–135. [Google Scholar]
- Mahady, S.E.; George, J. Management of nonalcoholic steatohepatitis: An evidence-based approach. Clin. Liv. Dis. 2012, 16, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; van Rooyen, D.; Gan, L.; Chitturi, S. NASH is an inflammatory disorder: Pathogenic, prognostic and therapeutic implications. Gut Liv. 2012, 6, 149–171. [Google Scholar] [CrossRef] [PubMed]
- Bril, F.; Ntim, K.; Lomonaco, R.; Cusi, K. Treatment of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH). In International Textbook of Diabetes Mellitus, 4th ed.; DeFronzo, R.A., Ferrannini, E., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2015; pp. 49–56. [Google Scholar]
- Mazen Noureddin, M.D.; Alexanian, D.; Kaplowitz, N. Non-alcoholic fatty liver disease (NAFLD): The lipid disease of the liver and the effect of statins. Lipid Manag. 2015, 149–173. [Google Scholar] [CrossRef]
- Fagone, P.; Mangano, K.; Pesce, A.; Portale, T.R.; Puleo, S.; Nicoletti, F. Emerging therapeutic targets for the treatment of hepatic fibrosis. Drug Discov. Today 2015, 21, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Diehl, A.M. Hepatic triglyceride synthesis and nonalcoholic fatty liver disease. Curr. Opin. Lipidol. 2008, 19, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Ko, E.H.; Ji, E.K.; Kim, E.; Lee, H.; Choi, H.; Yu, J.H.; Kim, H.J.; Seong, J.K.; Kim, K.S. Nuclear receptor PPARγ-regulated monoacylglycerol O-acyltransferase 1 (MGAT1) expression is responsible for the lipid accumulation in diet-induced hepatic steatosis. Proc. Natl. Acad. Sci. USA 2012, 109, 13656–13661. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Bo, S.; Cassader, M.; De, M.F.; Gambino, R. Impact of sterol regulatory element-binding factor-1c polymorphism on incidence of nonalcoholic fatty liver disease and on the severity of liver disease and of glucose and lipid dysmetabolism. Am. J. Clin. Nutr. 2013, 98, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Tosello-Trampont, A.-C.; Landes, S.G.; Nguyen, V.; Novobrantseva, T.I.; Hahn, Y.S. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-α production. J. Biol. Chem. 2012, 287, 40161–40172. [Google Scholar] [CrossRef] [PubMed]
- Gressner, A.M.; Weiskirchen, R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-β as major players and therapeutic targets. J. Cell. Mol. Med. 2006, 10, 76–99. [Google Scholar] [CrossRef] [PubMed]
- Videla, L.A.; Rodrigo, R.; Araya, J.; Poniachik, J. Insulin resistance and oxidative stress interdependency in non-alcoholic fatty liver disease. Trends Mol. Med. 2006, 12, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Uebanso, T.; Maekawa, K.; Ishikawa, M.; Taguchi, R.; Nammo, T.; Nishimakimogami, T.; Udagawa, H.; Fujii, M.; Shibazaki, Y. Characterization of hepatic lipid profiles in a mouse model with nonalcoholic steatohepatitis and subsequent fibrosis. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Larter, C.Z.; Yeh, M.M. Animal models of NASH: Getting both pathology and metabolic context right. J. Gastroenterol. Hepatol. 2008, 23, 1635–1648. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.M.; Levy, J.C.; Matthews, D.R. Use and abuse of homa modeling. Diabetes Care 2004, 27, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; Larter, C.Z. Nonalcoholic fatty liver disease: From steatosis to cirrhosis. Hepatology 2006, 43, S99–S112. [Google Scholar] [CrossRef] [PubMed]
- Cheung, O.; Sanyal, A.J. Recent advances in nonalcoholic fatty liver disease. Curr. Opin. Gastroenterol. 2010, 26, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Postic, C.; Girard, J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: Lessons from genetically engineered mice. J. Clin. Investig. 2008, 118, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Nagaya, T.; Tanaka, N.; Suzuki, T.; Sano, K. Down-regulation of SREBP-1c is associated with the development of burned-out nash. J. Hepatol. 2010, 53, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Magkos, F. Hepatic steatosis as a marker of metabolic dysfunction. Nutrients 2015, 7, 4995–5019. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Chew, G.T.; Watts, G.F. New peroxisome proliferator-activated receptor agonists: Potential treatments for atherogenic dyslipidemia and non-alcoholic fatty liver disease. Expert Opin. Pharmacother. 2014, 15, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Baffy, G. Kupffer cells in non-alcoholic fatty liver disease: The emerging view. J. Hepatol. 2009, 51, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van, N.M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalparida, A. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Koek, G.H.; Liedorp, P.R.; Bast, A. The role of oxidative stress in non-alcoholic steatohepatitis. Clin. Chim. Acta 2011, 412, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Mitsuyoshi, H.; Itoh, Y.; Okanoue, T. Role of oxidative stress in non-alcoholic steatohepatitis. Nihon Rinsho. 2006, 64, 1077–1082. [Google Scholar] [PubMed]
- Tang, T.; Sui, Y.; Lian, M.; Li, Z.; Hua, J. Pro-inflammatory activated Kupffer cells by lipids induce hepatic NKT cells deficiency through activation-induced cell death. PLoS ONE 2013, 8, e81949. [Google Scholar] [CrossRef] [PubMed]
- Österreicher, C.H.; Lemberger, U.J.; Mahon, R.; Rülicke, T.; Trauner, M.; Casanova, E. O147 hepatic stellate cells are the major source of collagen in murine models of liver fibrosis. J. Hepatol. 2014, 60, S61. [Google Scholar] [CrossRef]
- Angulo, P. Gi epidemiology: Nonalcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2007, 25, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Voican, C.S.; Perlemuter, G. Insulin resistance and oxidative stress: Two therapeutic targets in non-alcoholic steatohepatitis. J. Hepatol. 2011, 54, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhou, G.; Li, M.; Li, W.; Lü, J.; Xiong, L.; Liang, L.; Zhao, Y.; Xu, D.; Yu, J. A novel non-alcoholic steatohepatitis animal model featured with insulin resistance, hepatic inflammation and fibrosis. Scand. J. Gastroenterol. 2010, 45, 1360–1371. [Google Scholar] [CrossRef] [PubMed]
- Adkins, Y.; Schie, I.W.; Fedor, D.; Reddy, A.; Nguyen, S.; Zhou, P.; Kelley, D.S.; Wu, J. A novel mouse model of nonalcoholic steatohepatitis with significant insulin resistance. Lab. Investig. J. Tech. Methods Pathol. 2013, 93, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.K.; Gornicka, A.; Eguchi, A.; Povero, D.; Wree, A.; Feldstein, A.E. Tu2046 early compensatory mechanism of the liver after the initial accumulation of fat in a diet-induced nafld model. Gastroenterology 2015, 148, S-969. [Google Scholar] [CrossRef]
- Meroni, M.; Dongiovanni, P.; Baselli, G.A.; Rametta, R.; Gatti, S.; Maggioni, M.; Valenti, L. Impact and mechanisms of insulin resistance on liver damage progression in a mouse model of non-alcoholic steatohepatitis (NASH). Dig. Liv. Dis. 2016, 48, e11. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Body Weight (g) | Liver Index | Food Intake (g/day) |
|---|---|---|---|
| C10 | 323.75 ± 13.54 | 0.033 ± 0.003 | 26 ± 0.3 |
| M10 | 264.28 ± 11.35 ### | 0.039 ± 0.003 | 24 ± 0.8 |
| C20 | 347.93 ± 9.40 | 0.031 ± 0.004 | 27 ± 0.6 |
| M20 | 284.00 ± 11.37 ### | 0.041 ± 0.002 ## | 26 ± 0.8 |
| C30 | 408.22 ± 18.37 | 0.028 ± 0.002 | 25 ± 0.9 |
| M30 | 283.08 ± 11.80 ### | 0.045 ± 0.003 ### | 26 ± 0.8 |
| C40 | 422.22 ± 10.62 | 0.032 ± 0.005 | 25 ± 0.8 |
| M40 | 279.47 ± 11.99 ### | 0.044 ± 0.003 ## | 27 ± 0.4 |
| C50 | 435.46 ± 18.17 | 0.033 ± 0.002 | 24 ± 0.4 |
| M50 | 267.28 ± 14.51 ### | 0.043 ± 0.003 ## | 26 ± 0.6 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Wu, S.; Cai, M.; Ma, J.; Li, S.; Li, M.; Xu, Y.; Wei, L.; Shang, J. Comprehensive Study of Multiple Stages Progressing to Nonalcoholic Steatohepatitis with Subsequent Fibrosis in SD Rats. Int. J. Mol. Sci. 2017, 18, 1681. https://doi.org/10.3390/ijms18081681
Wang L, Wu S, Cai M, Ma J, Li S, Li M, Xu Y, Wei L, Shang J. Comprehensive Study of Multiple Stages Progressing to Nonalcoholic Steatohepatitis with Subsequent Fibrosis in SD Rats. International Journal of Molecular Sciences. 2017; 18(8):1681. https://doi.org/10.3390/ijms18081681
Chicago/Turabian StyleWang, Lulu, Susu Wu, Minxuan Cai, Ji Ma, Shengcun Li, Maoru Li, Yan Xu, Lixin Wei, and Jing Shang. 2017. "Comprehensive Study of Multiple Stages Progressing to Nonalcoholic Steatohepatitis with Subsequent Fibrosis in SD Rats" International Journal of Molecular Sciences 18, no. 8: 1681. https://doi.org/10.3390/ijms18081681