Chemokines as a Conductor of Bone Marrow Microenvironment in Chronic Myeloid Leukemia



Abstract
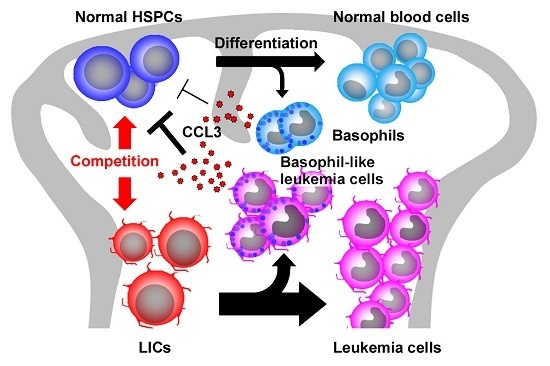
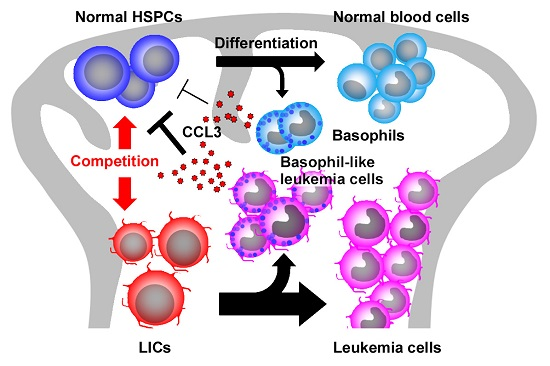
:
1. Introduction
2. Results
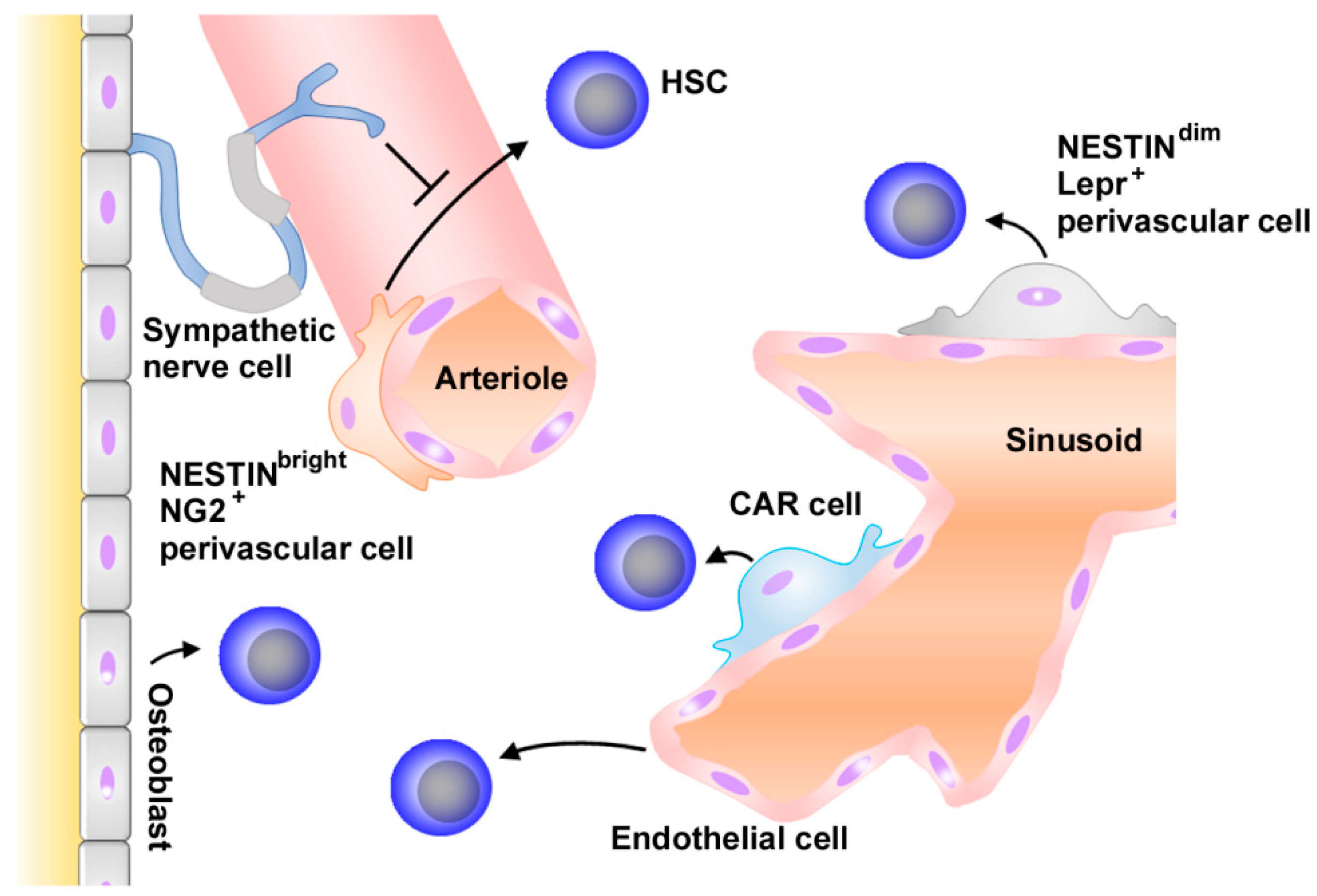
2.1. Normal Hematopoiesis and Niche
2.2. Chemokines in Normal Hematopoiesis
2.3. CML
2.4. CXCL12 in CML
2.5. CCL3 and Its Related Chemokines in CML
2.6. Other Chemokines in CML
3. Future Perspective
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Morrison, S.J.; Scadden, D.T. The bone marrow niche for haematopoietic stem cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Nitta, E.; Suda, T. Regulation of hematopoietic stem cell integrity through p53 and its related factors. Ann. N. Y. Acad. Sci. 2016, 1370, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Ugarte, F.; Forsberg, E.C. Haematopoietic stem cell niches: New insights inspire new questions. EMBO J. 2013, 32, 2535–2547. [Google Scholar] [CrossRef] [PubMed]
- Medyouf, H. The microenvironment in human myeloid malignancies: Emerging concepts and therapeutic implications. Blood 2017, 129, 1617–1626. [Google Scholar] [CrossRef] [PubMed]
- Korn, C.; Mendez-Ferrer, S. Myeloid malignancies and the microenvironment. Blood 2017, 129, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Cheshier, S.H.; Morrison, S.J.; Liao, X.; Weissman, I.L. In vivo proliferation and cell cycle kinetics of long-term self-renewing hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 1999, 96, 3120–3125. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, T. CXC chemokine ligand 12 (CXCL12) and its receptor CXCR4. J. Mol. Med. 2014, 92, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Kent, D.; Copley, M.; Benz, C.; Dykstra, B.; Bowie, M.; Eaves, C. Regulation of hematopoietic stem cells by the steel factor/KIT signaling pathway. Clin. Cancer Res. 2008, 14, 1926–1930. [Google Scholar] [CrossRef] [PubMed]
- Karanu, F.N.; Murdoch, B.; Gallacher, L.; Wu, D.M.; Koremoto, M.; Sakano, S.; Bhatia, M. The notch ligand jagged-1 represents a novel growth factor of human hematopoietic stem cells. J. Exp. Med. 2000, 192, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, G.; Murdoch, B.; Wu, D.; Baker, D.P.; Williams, K.P.; Chadwick, K.; Ling, L.E.; Karanu, F.N.; Bhatia, M. Sonic hedgehog induces the proliferation of primitive human hematopoietic cells via BMP regulation. Nat. Immunol. 2001, 2, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Duncan, A.W.; Ailles, L.; Domen, J.; Scherer, D.C.; Willert, K.; Hintz, L.; Nusse, R.; Weissman, I.L. A role for WNT signalling in self-renewal of haematopoietic stem cells. Nature 2003, 423, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Blank, U.; Karlsson, S. TGF-β signaling in the control of hematopoietic stem cells. Blood 2015, 125, 3542–3550. [Google Scholar] [CrossRef] [PubMed]
- Ema, H.; Suda, T. Two anatomically distinct niches regulate stem cell activity. Blood 2012, 120, 2174–2181. [Google Scholar] [CrossRef] [PubMed]
- Kiel, M.J.; Yilmaz, O.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. Slam family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Mendelson, A.; Frenette, P.S. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat. Med. 2014, 20, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.E.; Wagers, A.J.; Gulati, A.P.; Johnson, F.L.; Weissman, I.L. Physiological migration of hematopoietic stem and progenitor cells. Science 2001, 294, 1933–1936. [Google Scholar] [CrossRef] [PubMed]
- Sipkins, D.A.; Wei, X.; Wu, J.W.; Runnels, J.M.; Cote, D.; Means, T.K.; Luster, A.D.; Scadden, D.T.; Lin, C.P. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature 2005, 435, 969–973. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, P.A.; Gorlin, R.J.; Lukens, J.N.; Taniuchi, S.; Bohinjec, J.; Francois, F.; Klotman, M.E.; Diaz, G.A. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat. Genet. 2003, 34, 70–74. [Google Scholar] [CrossRef] [PubMed]
- McDermott, D.H.; Gao, J.L.; Liu, Q.; Siwicki, M.; Martens, C.; Jacobs, P.; Velez, D.; Yim, E.; Bryke, C.R.; Hsu, N.; et al. Chromothriptic cure of WHIM syndrome. Cell 2015, 160, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Han, Y.C.; Zou, Y.R. CXCR4 is required for the quiescence of primitive hematopoietic cells. J. Exp. Med. 2008, 205, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Sacchetti, B.; Funari, A.; Michienzi, S.; di Cesare, S.; Piersanti, S.; Saggio, I.; Tagliafico, E.; Ferrari, S.; Robey, P.G.; Riminucci, M.; et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 2007, 131, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCL12–CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Saunders, T.L.; Enikolopov, G.; Morrison, S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Asada, N.; Kunisaki, Y.; Pierce, H.; Wang, Z.; Fernandez, N.F.; Birbrair, A.; Ma’ayan, A.; Frenette, P.S. Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat. Cell Biol. 2017, 19, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, T.; Hirota, S.; Tachibana, K.; Takakura, N.; Nishikawa, S.; Kitamura, Y.; Yoshida, N.; Kikutani, H.; Kishimoto, T. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 1996, 382, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, K.; Hirota, S.; Iizasa, H.; Yoshida, H.; Kawabata, K.; Kataoka, Y.; Kitamura, Y.; Matsushima, K.; Yoshida, N.; Nishikawa, S.; et al. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 1998, 393, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.R.; Kottmann, A.H.; Kuroda, M.; Taniuchi, I.; Littman, D.R. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 1998, 393, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Jones, D.; Borghesani, P.R.; Segal, R.A.; Nagasawa, T.; Kishimoto, T.; Bronson, R.T.; Springer, T.A. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 9448–9453. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Morrison, S.J. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013, 495, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Omatsu, Y.; Sugiyama, T.; Kohara, H.; Kondoh, G.; Fujii, N.; Kohno, K.; Nagasawa, T. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity 2010, 33, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, A.; Hsu, Y.M.; Day, R.B.; Schuettpelz, L.G.; Christopher, M.J.; Borgerding, J.N.; Nagasawa, T.; Link, D.C. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 2013, 495, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Ferrer, S.; Lucas, D.; Battista, M.; Frenette, P.S. Haematopoietic stem cell release is regulated by circadian oscillations. Nature 2008, 452, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, A.; Yoshie, O. The chemokine superfamily revisited. Immunity 2012, 36, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, E.J.; Lolis, E. Structure, function, and inhibition of chemokines. Ann. Rev. Pharmacol. Toxicol. 2002, 42, 469–499. [Google Scholar] [CrossRef] [PubMed]
- Vandercappellen, J.; van Damme, J.; Struyf, S. The role of CXC chemokines and their receptors in cancer. Cancer Lett. 2008, 267, 226–244. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Bonecchi, R.; Locati, M. Tuning inflammation and immunity by chemokine sequestration: Decoys and more. Nat. Rev. Immunol. 2006, 6, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Toksoz, D.; Dexter, T.M.; Lord, B.I.; Wright, E.G.; Lajtha, L.G. The regulation of hemopoiesis in long-term bone marrow cultures. II. Stimulation and inhibition of stem cell proliferation. Blood 1980, 55, 931–936. [Google Scholar] [PubMed]
- Graham, G.J.; Wright, E.G.; Hewick, R.; Wolpe, S.D.; Wilkie, N.M.; Donaldson, D.; Lorimore, S.; Pragnell, I.B. Identification and characterization of an inhibitor of haemopoietic stem cell proliferation. Nature 1990, 344, 442–444. [Google Scholar] [CrossRef] [PubMed]
- Broxmeyer, H.E.; Sherry, B.; Lu, L.; Cooper, S.; Oh, K.O.; Tekamp-Olson, P.; Kwon, B.S.; Cerami, A. Enhancing and suppressing effects of recombinant murine macrophage inflammatory proteins on colony formation in vitro by bone marrow myeloid progenitor cells. Blood 1990, 76, 1110–1116. [Google Scholar] [PubMed]
- Lord, B.I.; Dexter, T.M.; Clements, J.M.; Hunter, M.A.; Gearing, A.J. Macrophage-inflammatory protein protects multipotent hematopoietic cells from the cytotoxic effects of hydroxyurea in vivo. Blood 1992, 79, 2605–2609. [Google Scholar] [PubMed]
- Verfaillie, C.M.; Catanzarro, P.M.; Li, W.N. Macrophage inflammatory protein 1α, interleukin 3 and diffusible marrow stromal factors maintain human hematopoietic stem cells for at least eight weeks in vitro. J. Exp. Med. 1994, 179, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Broxmeyer, H.E.; Sherry, B.; Lu, L.; Cooper, S.; Carow, C.; Wolpe, S.D.; Cerami, A. Myelopoietic enhancing effects of murine macrophage inflammatory proteins 1 and 2 on colony formation in vitro by murine and human bone marrow granulocyte/macrophage progenitor cells. J. Exp. Med. 1989, 170, 1583–1594. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Tanabe, Y.; Yoshikawa, S.; Yamanishi, Y.; Morishita, S.; Komatsu, N.; Karasuyama, H.; Hirao, A.; Mukaida, N. MIP-1α/CCL3-expressing basophil-lineage cells drive the leukemic hematopoiesis of chronic myeloid leukemia in mice. Blood 2016, 127, 2607–2617. [Google Scholar] [CrossRef] [PubMed]
- Broxmeyer, H.E.; Sherry, B.; Cooper, S.; Lu, L.; Maze, R.; Beckmann, M.P.; Cerami, A.; Ralph, P. Comparative analysis of the human macrophage inflammatory protein family of cytokines (chemokines) on proliferation of human myeloid progenitor cells. Interacting effects involving suppression, synergistic suppression, and blocking of suppression. J. Immunol. 1993, 150, 3448–3458. [Google Scholar] [PubMed]
- Broxmeyer, H.E.; Kim, C.H. Regulation of hematopoiesis in a sea of chemokine family members with a plethora of redundant activities. Exp. Hematol. 1999, 27, 1113–1123. [Google Scholar] [CrossRef]
- Papayannopoulou, T. Current mechanistic scenarios in hematopoietic stem/progenitor cell mobilization. Blood 2004, 103, 1580–1585. [Google Scholar] [CrossRef] [PubMed]
- Lord, B.I.; Woolford, L.B.; Wood, L.M.; Czaplewski, L.G.; McCourt, M.; Hunter, M.G.; Edwards, R.M. Mobilization of early hematopoietic progenitor cells with BB-10010: A genetically engineered variant of human macrophage inflammatory protein-1α. Blood 1995, 85, 3412–3415. [Google Scholar] [PubMed]
- Broxmeyer, H.E.; Cooper, S.; Hangoc, G.; Gao, J.L.; Murphy, P.M. Dominant myelopoietic effector functions mediated by chemokine receptor CCR1. J. Exp. Med. 1999, 189, 1987–1992. [Google Scholar] [CrossRef] [PubMed]
- Laterveer, L.; Lindley, I.J.; Hamilton, M.S.; Willemze, R.; Fibbe, W.E. Interleukin-8 induces rapid mobilization of hematopoietic stem cells with radioprotective capacity and long-term myelolymphoid repopulating ability. Blood 1995, 85, 2269–2275. [Google Scholar] [PubMed]
- Laterveer, L.; Lindley, I.J.; Heemskerk, D.P.; Camps, J.A.; Pauwels, E.K.; Willemze, R.; Fibbe, W.E. Rapid mobilization of hematopoietic progenitor cells in rhesus monkeys by a single intravenous injection of interleukin-8. Blood 1996, 87, 781–788. [Google Scholar] [PubMed]
- Wang, J.; Mukaida, N.; Zhang, Y.; Ito, T.; Nakao, S.; Matsushima, K. Enhanced mobilization of hematopoietic progenitor cells by mouse MIP-2 and granulocyte colony-stimulating factor in mice. J. Leukoc. Biol. 1997, 62, 503–509. [Google Scholar] [PubMed]
- Pelus, L.M.; Bian, H.; King, A.G.; Fukuda, S. Neutrophil-derived MMP-9 mediates synergistic mobilization of hematopoietic stem and progenitor cells by the combination of G-CSF and the chemokines GROβ/CXCL2 and GROβT/CXCL2Δ4. Blood 2004, 103, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Y.; Kasahara, T.; Harada, A.; Matsushima, K.; Mukaida, N. Detection of mouse IL-8 receptor homologue expression on peripheral blood leukocytes and mature myeloid lineage cells in bone marrow. J. Leukoc. Biol. 1996, 60, 372–381. [Google Scholar] [PubMed]
- Pelus, L.M.; Bian, H.; Fukuda, S.; Wong, D.; Merzouk, A.; Salari, H. The CXCR4 agonist peptide, CTCE-0021, rapidly mobilizes polymorphonuclear neutrophils and hematopoietic progenitor cells into peripheral blood and synergizes with granulocyte colony-stimulating factor. Exp. Hematol. 2005, 33, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Broxmeyer, H.E.; Orschell, C.M.; Clapp, D.W.; Hangoc, G.; Cooper, S.; Plett, P.A.; Liles, W.C.; Li, X.; Graham-Evans, B.; Campbell, T.B.; et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with amd3100, a CXCR4 antagonist. J. Exp. Med. 2005, 201, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Horuk, R. The duffy antigen receptor for chemokines DARC/ACKR1. Front. Immunol. 2015, 6, 279. [Google Scholar] [CrossRef] [PubMed]
- Ergen, A.V.; Boles, N.C.; Goodell, M.A. Rantes/ccl5 influences hematopoietic stem cell subtypes and causes myeloid skewing. Blood 2012, 119, 2500–2509. [Google Scholar] [CrossRef] [PubMed]
- Duchene, J.; Novitzky-Basso, I.; Thiriot, A.; Casanova-Acebes, M.; Bianchini, M.; Etheridge, S.L.; Hub, E.; Nitz, K.; Artinger, K.; Eller, K.; et al. Atypical chemokine receptor 1 on nucleated erythroid cells regulates hematopoiesis. Nat. Immunol. 2017, 18, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Apperley, J.F. Chronic myeloid leukaemia. Lancet 2015, 385, 1447–1459. [Google Scholar] [CrossRef]
- Deininger, M.W.N.; Goldman, J.M.; Melo, J.V. The molecular biology of chronic myeloid leukemia. Blood 2000, 96, 3343–3356. [Google Scholar] [PubMed]
- Mahon, F.X.; Rea, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B.; et al. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre stop imatinib (stim) trial. Lancet Oncol. 2010, 11, 1029–1035. [Google Scholar] [CrossRef]
- Holyoake, T.L.; Vetrie, D. The chronic myeloid leukemia stem cell: Stemming the tide of persistence. Blood 2017, 129, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Naka, K.; Hoshii, T.; Muraguchi, T.; Tadokoro, Y.; Ooshio, T.; Kondo, Y.; Nakao, S.; Motoyama, N.; Hirao, A. TGF-β-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 2010, 463, 676–680. [Google Scholar] [CrossRef] [PubMed]
- MacLean, A.L.; Filippi, S.; Stumpf, M.P. The ecology in the hematopoietic stem cell niche determines the clinical outcome in chronic myeloid leukemia. Proc. Natl. Acad. Sci. USA 2014, 111, 3883–3888. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Fulzele, K.; Catic, A.; Sun, C.C.; Dombkowski, D.; Hurley, M.P.; Lezeau, S.; Attar, E.; Wu, J.Y.; Lin, H.Y.; et al. Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat. Med. 2013, 19, 1513–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laperrousaz, B.; Jeanpierre, S.; Sagorny, K.; Voeltzel, T.; Ramas, S.; Kaniewski, B.; Ffrench, M.; Salesse, S.; Nicolini, F.E.; Maguer-Satta, V. Primitive CML cell expansion relies on abnormal levels of BMPS provided by the niche and on BMPRIb overexpression. Blood 2013, 122, 3767–3777. [Google Scholar] [CrossRef] [PubMed]
- Krause, D.S.; Lazarides, K.; Lewis, J.B.; von Andrian, U.H.; Van Etten, R.A. Selectins and their ligands are required for homing and engraftment of BCR-ABL1+ leukemic stem cells in the bone marrow niche. Blood 2014, 123, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Ho, Y.W.; Huang, Q.; Maeda, T.; Lin, A.; Lee, S.U.; Hair, A.; Holyoake, T.L.; Huettner, C.; Bhatia, R. Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell 2012, 21, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Taverna, S.; Amodeo, V.; Saieva, L.; Russo, A.; Giallombardo, M.; de Leo, G.; Alessandro, R. Exosomal shuttling of MIR-126 in endothelial cells modulates adhesive and migratory abilities of chronic myelogenous leukemia cells. Mol. Cancer 2014, 13, 169. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Sadovnik, I.; Cerny-Reiterer, S.; Rulicke, T.; Stefanzl, G.; Willmann, M.; Hoermann, G.; Bilban, M.; Blatt, K.; Herndlhofer, S.; et al. Dipeptidylpeptidase iv (CD26) defines leukemic stem cells (LSC) in chronic myeloid leukemia. Blood 2014, 123, 3951–3962. [Google Scholar] [CrossRef] [PubMed]
- Durig, J.; Rosenthal, C.; Elmaagacli, A.; Heyworth, C.; Halfmeyer, K.; Kasper, C.; Novotny, J.; Duhrsen, U. Biological effects of stroma-derived factor-1α on normal and CML CD34+ haemopoietic cells. Leukemia 2000, 14, 1652–1660. [Google Scholar] [CrossRef] [PubMed]
- Peled, A.; Hardan, I.; Trakhtenbrot, L.; Gur, E.; Magid, M.; Darash-Yahana, M.; Cohen, N.; Grabovsky, V.; Franitza, S.; Kollet, O.; et al. Immature leukemic CD34+CXCR4+ cells from CML patients have lower integrin-dependent migration and adhesion in response to the chemokine SDF-1. Stem Cells 2002, 20, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Kronenwett, R.; Butterweck, U.; Steidl, U.; Kliszewski, S.; Neumann, F.; Bork, S.; Blanco, E.D.; Roes, N.; Graf, T.; Brors, B.; et al. Distinct molecular phenotype of malignant CD34+ hematopoietic stem and progenitor cells in chronic myelogenous leukemia. Oncogene 2005, 24, 5313–5324. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Tien, S.C.; Tien, H.F.; Zhang, H.; Bokoch, G.M.; Chang, Z.F. p210(BCR-ABL) desensitizes CDC42 gtpase signaling for SDF-1α-directed migration in chronic myeloid leukemia cells. Oncogene 2009, 28, 4105–4115. [Google Scholar] [CrossRef] [PubMed]
- Apperley, J.F. Part I: Mechanisms of resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol. 2007, 8, 1018–1029. [Google Scholar] [CrossRef]
- Jin, L.; Tabe, Y.; Konoplev, S.; Xu, Y.; Leysath, C.E.; Lu, H.; Kimura, S.; Ohsaka, A.; Rios, M.B.; Calvert, L.; et al. CXCR4 up-regulation by imatinib induces chronic myelogenous leukemia (CML) cell migration to bone marrow stroma and promotes survival of quiescent CML cells. Mol. Cancer Ther. 2008, 7, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Tabe, Y.; Jin, L.; Iwabuchi, K.; Wang, R.Y.; Ichikawa, N.; Miida, T.; Cortes, J.; Andreeff, M.; Konopleva, M. Role of stromal microenvironment in nonpharmacological resistance of CML to imatinib through Lyn/CXCR4 interactions in lipid rafts. Leukemia 2012, 26, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Azab, A.K.; Manley, P.W.; Kung, A.L.; Christie, A.L.; Bronson, R.; Ghobrial, I.M.; Griffin, J.D. Inhibition of CXCR4 in CML cells disrupts their interaction with the bone marrow microenvironment and sensitizes them to nilotinib. Leukemia 2012, 26, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Miao, H.; Li, W.; Yao, J.; Sun, Y.; Li, Z.; Zhao, L.; Guo, Q. CXCL12/CXCR4 axis confers adriamycin resistance to human chronic myelogenous leukemia and oroxylin A improves the sensitivity of K562/ADM cells. Biochem. Pharmacol. 2014, 90, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Beider, K.; Darash-Yahana, M.; Blaier, O.; Koren-Michowitz, M.; Abraham, M.; Wald, H.; Wald, O.; Galun, E.; Eizenberg, O.; Peled, A.; et al. Combination of imatinib with CXCR4 antagonist BKT140 overcomes the protective effect of stroma and targets CML in vitro and in vivo. Mol. Cancer Ther. 2014, 13, 1155–1169. [Google Scholar] [CrossRef] [PubMed]
- Onishi, I.; Nakagawa, Y.; Murayama, T.; Hidaka, M.; Yamamoto, K.; Abe-Suzuki, S.; Abe, S.; Kurata, M.; Kitagawa, M. Expression of multidrug resistance 1 gene in association with CXCL12 in chronic myelogenous leukaemia. Pathology 2014, 46, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Fleischman, A.G.; Petersen, C.L.; MacKenzie, R.; Luty, S.; Loriaux, M.; Druker, B.J.; Woltjer, R.L.; Deininger, M.W. Effects of plerixafor in combination with BCR-ABL kinase inhibition in a murine model of CML. Blood 2012, 120, 2658–2668. [Google Scholar] [CrossRef] [PubMed]
- Holyoake, T.L.; Freshney, M.G.; Sproul, A.M.; Richmond, L.J.; Alcorn, M.J.; Steward, W.P.; Fitzsimons, E.; Dunlop, D.J.; Franklin, I.M.; Pragnell, I.B. Contrasting effects of rh-MIP-1α and TGF-β 1 on chronic myeloid leukemia progenitors in vitro. Stem Cells 1993, 11, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Eaves, C.J.; Cashman, J.D.; Zoumbos, N.C.; Barnett, M.J.; Eaves, A.C. Biological strategies for the selective manipulation of normal and leukemic stem cells. Stem Cells 1993, 11, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Eaves, C.J.; Cashman, J.D.; Wolpe, S.D.; Eaves, A.C. Unresponsiveness of primitive chronic myeloid leukemia cells to macrophage inflammatory protein 1α, an inhibitor of primitive normal hematopoietic cells. Proc. Natl. Acad. Sci. USA 1993, 90, 12015–12019. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.E.; Lucas, G.; Graham, G.J.; Russell, N.H.; Mottram, R.; Whetton, A.D.; Buckle, A.M. Macrophage-inflammatory protein-1α receptor expression on normal and chronic myeloid leukemia CD34+ cells. J. Immunol. 1999, 162, 6191–6199. [Google Scholar] [PubMed]
- Chasty, R.C.; Lucas, G.S.; Owen-Lynch, P.J.; Pierce, A.; Whetton, A.D. Macrophage inflammatory protein-1α receptors are present on cells enriched for CD34 expression from patients with chronic myeloid leukemia. Blood 1995, 86, 4270–4277. [Google Scholar] [PubMed]
- Durig, J.; Testa, N.G.; Lord, B.I.; Kasper, C.; Chang, J.; Telford, N.; Dexter, T.M.; Heyworth, C.M. Characterisation of the differential response of normal and CML haemopoietic progenitor cells to macrophage inflammatory protein-1α. Leukemia 1999, 13, 2012–2022. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Naka, K.; Morishita, S.; Komatsu, N.; Hirao, A.; Mukaida, N. MIP-1α/CCL3-mediated maintenance of leukemia-initiating cells in the initiation process of chronic myeloid leukemia. J. Exp. Med. 2013, 210, 2661–2673. [Google Scholar] [CrossRef] [PubMed]
- Wark, G.; Heyworth, C.M.; Spooncer, E.; Czaplewski, L.; Francis, J.M.; Dexter, T.M.; Whetton, A.D. ABL protein kinase abrogates the response of multipotent haemopoietic cells to the growth inhibitor macrophage inflammatory protein-1α. Oncogene 1998, 16, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Pear, W.S.; Miller, J.P.; Xu, L.; Pui, J.C.; Soffer, B.; Quackenbush, R.C.; Pendergast, A.M.; Bronson, R.; Aster, J.C.; Scott, M.L.; et al. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving p210 BCR/ABL-transduced bone marrow. Blood 1998, 92, 3780–3792. [Google Scholar] [PubMed]
- Koschmieder, S.; Gottgens, B.; Zhang, P.; Iwasaki-Arai, J.; Akashi, K.; Kutok, J.L.; Dayaram, T.; Geary, K.; Green, A.R.; Tenen, D.G.; et al. Inducible chronic phase of myeloid leukemia with expansion of hematopoietic stem cells in a transgenic model of BCR-ABL leukemogenesis. Blood 2005, 105, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Aoki, Y.; Niihori, T.; Inoue, S.-I.; Matsubara, Y. Recent advances in rasopathies. J. Hum. Genet. 2016, 61, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Yu, W.M.; Zheng, H.; Loh, M.L.; Bunting, S.T.; Pauly, M.; Huang, G.; Zhou, M.; Broxmeyer, H.E.; Scadden, D.T.; et al. Leukaemogenic effects of Ptpn11 activating mutations in the stem cell microenvironment. Nature 2016, 539, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Hantschel, O.; Gstoettenbauer, A.; Colinge, J.; Kaupe, I.; Bilban, M.; Burkard, T.R.; Valent, P.; Superti, F.G. The chemokine interleukin-8 and the surface activation protein CD69 are markers for BCR-ABL activity in chronic myeloid leukemia. Mol. Oncol. 2008, 2, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Corrado, C.; Raimondo, S.; Saieva, L.; Flugy, A.M.; De Leo, G.; Alessandro, R. Exosome-mediated crosstalk between chronic myelogenous leukemia cells and human bone marrow stromal cells triggers an interleukin 8-dependent survival of leukemia cells. Cancer Lett. 2014, 348, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Cashman, J.D.; Eaves, C.J.; Sarris, A.H.; Eaves, A.C. MCP-1, not MIP-1α, is the endogenous chemokine that cooperates with TGF-β to inhibit the cycling of primitive normal but not leukemic (CML) progenitors in long-term human marrow cultures. Blood 1998, 92, 2338–2344. [Google Scholar] [PubMed]
- Gerber, J.M.; Gucwa, J.L.; Esopi, D.; Gurel, M.; Haffner, M.C.; Vala, M.; Nelson, W.G.; Jones, R.J.; Yegnasubramanian, S. Genome-wide comparison of the transcriptomes of highly enriched normal and chronic myeloid leukemia stem and progenitor cell populations. Oncotarget 2013, 4, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Jongen-Lavrencic, M.; Salesse, S.; Delwel, R.; Verfaillie, C.M. BCR/ABL-mediated downregulation of genes implicated in cell adhesion and motility leads to impaired migration toward CCR7 ligands CCL19 and CCL21 in primary BCR/ABL-positive cells. Leukemia 2005, 19, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, K.; Iwakami, M.; Muromoto, R.; Inagaki, T.; Kitai, Y.; Kon, S.; Sekine, Y.; Oritani, K.; Matsuda, T. CCR7 is involved in BCR-ABL/stap-2-mediated cell growth in hematopoietic Ba/F3 cells. Biochem. Biophys. Res. Commun. 2015, 463, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Hromas, R.; Cripe, L.; Hangoc, G.; Cooper, S.; Broxmeyer, H.E. The exodus subfamily of CC chemokines inhibits the proliferation of chronic myelogenous leukemia progenitors. Blood 2000, 95, 1506–1508. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Standard Name | Common Alias | Receptor | SCI Activity |
|---|---|---|---|
| CXCL1 | groα, melanoma growth stimulating activity (MGSA), KC | CXCR2 | |
| CXCL2 | groβ, macrophage inflammatory protein (MIP)-2α | CXCR2 | + |
| CXCL3 | groγ, MIP-2β | CXCR2 | |
| CXCL4 | platelet factor-4 (PF-4) | CXCR3 | + |
| CXCL5 | epithelial neutrophil activating peptide (ENA)-78 | CXCR2 > CXCR2 | + |
| CXCL6 | granulocyte chemotactic protein (GCP)-2 | CXCR1, CXCR2 | + |
| CXCL7 | neutrophil activating protein (NAP)-2 | CXCR2 | |
| CXCL8 | interleukin-8 (IL-8) | CXCR1, CXCR2 | + |
| CXCL9 | monokine induced by interferon γ (Mig) | CXCR3 | + |
| CXCL10 | interferon inducible protein (IP)-10 | CXCR3 | + |
| CXCL11 | interferon inducible T-cell α chemoattractant (I-TAC) | CXCR3, CXCR7 | |
| CXCL12 | stromal-derived factor (SDF)-1 | CXCR4, CXCR7 | + |
| CXCL13 | B lymphocyte chemoattractant (BLC) | CXCR5 | |
| CXCL14 | breast and kidney expressed chemokine (BRAK) | ? | |
| CXCL15 | lungkine | ? | |
| CXCL16 | scavenger receptor for phosphatidylserine and oxidized lipoprotein (SR-PSOX) | CXCR6 | |
| CXCL17 | ? | ||
| CCL1 | I-309 | CCR8 | + |
| CCL2 | monocyte chemoattractant (MCP)-1 | CCR2 | + |
| CCL3 | MIP-1α | CCR1, CCR5 | + |
| CCL4 | MIP-1β | CCR5 > CCR1 | |
| CCL5 | regulated upon activation normal T cell expressed and secreted (RANTES) | CCR1, CCR5, CCR3 | |
| CCL6 | C10, macrophage inflammatory protein-related protein (MRP)-1 | ? | + |
| CCL7 | MCP-3 | CCR1,CCR2, CCR3 > CCR5 | |
| CCL8 | MCP-2 | CCR2, CCR1, CCR3, CCR5 | |
| CCL9 | MRP-2, MIP-1γ | ? | + |
| CCL10 | ? | + | |
| CCL11 | eotaxin | CCR3 > CCR5 | + |
| CCL12 | MCP-5 | ? | + |
| CCL13 | MCP-4 | CCR1,CCR2, CCR3 > CCR5 | + |
| CCL14 | hemofiltrate CC chemokine (HCC)-1 | CCR1 | |
| CCL15 | HCC-2 | CCR1, CCR3 | + |
| CCL16 | HCC-4 | CCR1, CCR2, CCR5 | + |
| CCL17 | thymus and activation-regulated chemokine (TARC) | CCR4 > CCR8 | |
| CCL18 | pulmonary and activation-regulated chemokine (PARC) | ? | + |
| CCL19 | EBI1-ligand chemokine (ELC) | CCR7 | + |
| CCL20 | MIP-3α, liver and activation-related chemokine (LARC) | CCR6 | + |
| CCL21 | secondary lymphoid chemokine (SLC) | CCR7 | + |
| CCL22 | macrophage-derived chemokine (MDC) | CCR4 | |
| CCL23 | myeloid progenitor inhibitory factor-1 (MPIF-1) | CCR1 | + |
| CCL24 | eotaxin-2 | CCR3 | + |
| CCL25 | thymus-expressed chemokine (TECK) | CCR9 | + |
| CCL26 | eotaxin-3 | CCR3 | |
| CCL27 | cutaneous T-cell attracting chemokine (CTACK) | CCR10 | |
| CCL28 | mucosae-associated epithelial chemokine (MEC) | CCR3, CCR10 | |
| XCL1 | lymphotaxin-α | XCR1 | + |
| XCL2 | lymphotaxin-β | XCR1 | + |
| CX3L1 | fractalkine | CX3CR1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukaida, N.; Tanabe, Y.; Baba, T. Chemokines as a Conductor of Bone Marrow Microenvironment in Chronic Myeloid Leukemia. Int. J. Mol. Sci. 2017, 18, 1824. https://doi.org/10.3390/ijms18081824
Mukaida N, Tanabe Y, Baba T. Chemokines as a Conductor of Bone Marrow Microenvironment in Chronic Myeloid Leukemia. International Journal of Molecular Sciences. 2017; 18(8):1824. https://doi.org/10.3390/ijms18081824
Chicago/Turabian StyleMukaida, Naofumi, Yamato Tanabe, and Tomohisa Baba. 2017. "Chemokines as a Conductor of Bone Marrow Microenvironment in Chronic Myeloid Leukemia" International Journal of Molecular Sciences 18, no. 8: 1824. https://doi.org/10.3390/ijms18081824
APA StyleMukaida, N., Tanabe, Y., & Baba, T. (2017). Chemokines as a Conductor of Bone Marrow Microenvironment in Chronic Myeloid Leukemia. International Journal of Molecular Sciences, 18(8), 1824. https://doi.org/10.3390/ijms18081824