The Role of Microglia in Diabetic Retinopathy: Inflammation, Microvasculature Defects and Neurodegeneration


Molecular Signal Transduction Laboratories, Institute for Microscopic Anatomy and Neurobiology, Johannes Gutenberg University, School of Medicine, 55131 Mainz, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(1), 110; https://doi.org/10.3390/ijms19010110
Submission received: 5 December 2017
/
Revised: 23 December 2017
/
Accepted: 25 December 2017
/
Published: 1 January 2018
(This article belongs to the Special Issue Microglia in Aging and Neurodegenerative Disease)
Abstract
:Diabetic retinopathy is a common complication of diabetes mellitus, which appears in one third of all diabetic patients and is a prominent cause of vision loss. First discovered as a microvascular disease, intensive research in the field identified inflammation and neurodegeneration to be part of diabetic retinopathy. Microglia, the resident monocytes of the retina, are activated due to a complex interplay between the different cell types of the retina and diverse pathological pathways. The trigger for developing diabetic retinopathy is diabetes-induced hyperglycemia, accompanied by leukostasis and vascular leakages. Transcriptional changes in activated microglia, mediated via the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) and extracellular signal–regulated kinase (ERK) signaling pathways, results in release of various pro-inflammatory mediators, including cytokines, chemokines, caspases and glutamate. Activated microglia additionally increased proliferation and migration. Among other consequences, these changes in microglia severely affected retinal neurons, causing increased apoptosis and subsequent thinning of the nerve fiber layer, resulting in visual loss. New potential therapeutics need to interfere with these diabetic complications even before changes in the retina are diagnosed, to prevent neuronal apoptosis and blindness in patients.
1. Introduction
Diabetic retinopathy (DR) is one of the most common complications of diabetes [1] and the main cause of vision impairment and loss in individuals from 20 to 74 years of age [2,3]. After suffering 20 years with diabetes, nearly all patients with type 1 and more than 60% of patients with type 2 diabetes develop a retinopathy [4,5,6] and the number of patients extensively increases every year [7]. The main cause of the complication remains unclear. Symptoms of DR range from hemorrhages, micro-aneurysms, cotton-wool spots, lipid exudates, macular edema, capillary occlusion and neovascularization and ultimately blindness [8].
Clinically, DR can be divided into two forms: non-proliferating diabetic retinopathy (NPDR) and proliferating diabetic retinopathy (PDR) [9]. NPDR is the first stage of DR and is characterized by damage to retinal vasculature, increased vascular permeability, thickening of the basement membrane, loss of pericytes and the beginning of mobilization of blood vessels. It can be divided into mild (micro-aneurysms), moderate (micro-aneurysms, retinal hemorrhages or hard exudates), severe (20 hemorrhages in each of the four quadrants, venous beading in two quadrants or intra-retinal microvascular anomalies) and very severe (combined complications) phases [9,10,11]. NPDR can further progress into PDR, which is defined by pathological neovascular growth (angiogenesis), vitreous hemorrhage, retinal scars and detachment, resulting in irreversible vision loss and total blindness [11,12]. Diabetic macular edema (DME), a side effect of both NPDR and PDR, can occur at any stage of DR and is caused by increased vascular permeability and leakage of proteins and lipids into the extracellular space. The functional damage of the retinal vascular epithelium results in an increase of extracellular fluid, which, collected by Müller cells, leads to swelling of the retina and increased cell death, further resulting in visual impairment and loss [13,14,15].
DR was first described as a classically microvascular disease, based on the discovery of the microvascular changes in the retina and for an extended period, DR was solely a microvascular abnormality. It was gradually discovered that all major cell types of the retina are altered and today DR is believed to result from interplay between endothelial cells, microglia, astrocytes, Müller cells and neurons. Thus, DR is a microvascular disease as well as a chronic inflammation and retinal neurodegeneration, which is not only influenced by local changes, but also by systemic metabolic and cardiovascular parameters. Degeneration, inflammation and vascular alternations occur and operate parallel and in close relation.
In this review, the inflammatory component of the disease is examined carefully and summarized regarding the role of microglia cells in neurodegeneration of retinal neurons. Inflammation means tissue invasion of immune cells to clear after an infection or damage of tissue. The retina was long believed to be an immune-privileged tissue. However, it was discovered that several cytokines and chemokines are increased in the diabetic retina [16], which resulted in growing evidence that inflammation and microglia play a major role in the development and progression of DR, as well as in retinal neurodegeneration [17,18,19]. Microglia are the resident inflammatory cells of the central nervous system (CNS), which upon activation can modulate inflammatory processes and are involved in a variety of other neuroinflammatory and neurodegenerative diseases, such as Alzheimer’s, Parkinson’s and Huntington’s diseases [20,21].
Recently, functional alterations in patients with DR were described, which occurred even before development of vascular dysfunction, indicating that diabetes-induced hyperglycemia has a vasculature-independent effect on the neuronal retina [22,23]. In patients with diabetes, but without retinopathy, defects in the electroretinogram (ERG) were detected as were neurodegeneration and a thinning of the innermost layer, the nerve fiber layer, suggesting a loss of axons [19,24,25]. The link between hyperglycemia and neurodegeneration is not fully understood, however, various cues point to altered inflammation as a link and activation of retinal microglia as an early feature of DR [26].
Despite the progress made in the last years, understanding the pathogenesis of DR, the knowledge of the underlying mechanisms, is still insufficient and therapeutics are limited. More studies are needed to provide further insight into the mechanism, which may then lead to new treatments. Especially, identifying early features of the complications is important to interrupt pathologies before neuronal damage starts, to prevent visual impairment. The upregulation of numerous mediators, both angiogenic and inflammatory, has been implicated in the pathogenesis of microvascular retinopathy and will be summarized in this review.
2. The Retina
The retina is a transparent layer of neural tissue, which can be divided into nine layers, and consists of different cell types including neurons, macroglia, microglia and vascular cells (Figure 1). The function of the retina is the conversion of captured light into electrical energy and the transmission of the resulting action potential to the occipital lobe of the brain. The retina originates from the optic cup, a part of the embryonic diencephalon and, thus, is part of the CNS [27].
The construction of the retina might contribute to its vulnerability to diabetes and to the development of DR. There are three structural conditions which are needed to ensure undisturbed light transmission, but subsequently increase the vulnerability of the retina. First, the retina contains a low density of blood vessels, because these would absorb light and interfere with the retinal function. The oxygen tension declines from the inner to the outer retina [28,29,30]. Thus, the retina relies on anaerobic respiration, which is less efficient than aerobic respiration [31]. This is especially problematic because the neurons in the retina have a relatively high metabolic demand. High amounts of ATP are consumed for phototransduction, to maintain ion gradients across the cell membrane, for neurotransmission and to maintain functional photoreceptors and their transient outer segments [32]. Second, axons in the retina are not myelinated since lipids in the myelin sheath would interfere with light transmission; however, unmyelinated nerves require even more energy to maintain their membrane potentials. Third, the number of mitochondria in the retina is relatively low, because they contain light-absorbing heme-based cytochrome proteins. The combination of high metabolic demand of the retinal neurons and low vascular supply, together with few mitochondria, increases the vulnerability of the retina and reduces the ability of the retina to acclimate to metabolic stress situations.
Lactate, a product of anaerobe respiration has been linked to metabolic signaling and retinal neurodegeneration [33,34,35]. Lactate produced in Müller cells and astrocytes is secreted into the subretinal space and can bind to Gpr81 receptors and monocarboxylate transporters, located on the membrane of retinal pigment epithelium, ganglion cells and photoreceptors [33,36,37]. In neurons, lactate is believed to increase the utilization of glucose [38] and it was shown to be neuroprotective in models of cerebral ischemia [38,39] and traumatic brain injury [40,41], whereas lack of lactate has been implicated with neurodegeneration [42]. Another vulnerable structure of the retina is the blood barrier. The retina, as part of the CNS, has a blood–retina barrier (BRB), which is formed by tight junctions between adjacent endothelial cells, protects the neural tissue from various circulating components of the blood and enables the retina to regulate its own extracellular chemical composition [43,44]. The interaction of blood vessels, astrocytes and ganglion cells induces the expression of tight junction proteins, such as occludins, claudins and zonula occludens (ZO) proteins, which are important to maintain the BRB [45,46]. Disruption has been linked to numerous diseases, such as stroke and brain tumors and can lead to edema.
3. Diabetic Retinopathy
DR is a severe ocular complication of diabetes mellitus, which is accompanied by hyperglycemia, leukostasis, microvascular damage, inflammation, vascular permeability, occlusion, ischemia and neurodegeneration, all together resulting in blindness.
3.1. Hyperglycemia in Diabetic Retinopathy
High blood sugar levels in diabetes mellitus is most probably the causative etiology for DR [47,48]. One of the first events in DR is the glucose-mediated microvascular damage, which further results in increased polyol and hexosamine pathway flux, cellular oxidative stress, activation of protein kinase C, superoxide overproduction by the mitochondrial electron transport chain as well as the increase and activation of advanced glycation end products, advanced lipoxidation end-products and oxidized low density lipoproteins and their receptors [49,50,51,52,53,54,55,56], followed by massive production of free radicals in mitochondria, which further increase oxidative stress and have severe consequences such as chronic inflammation.
Accumulation of these mediators in the retina can trigger further steps, including microglia activation [57,58]. In parallel, hyperglycemia induces microglia activation via reactive oxygen species (ROS) [59]. Generation of ROS induces nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) nuclear translocation and extracellular signal–regulated kinase (ERK) phosphorylation in microglia [60,61,62,63] and hence production of cytokines, such as tumor necrosis factor (TNF)α, interleukins (IL)-1β, IL-6, IL-8, vascular cell adhesion molecule (VCAM-1) and intracellular adhesion molecule 1 (ICAM-1) [64,65,66].
Hyperglycemia can also influence pericyte loss in early stages of DR, a process which is accompanied by ATP release [67]. In this context, the purinergic P2X7 receptor gained interest, which is involved in regulation of the lumen diameter through ATP signaling [68]. Blocking the purinergic P2X7 receptor in high glucose conditions reduced retinal cell death and the inflammatory response [69].
3.2. Leukostasis in Diabetic Retinopathy
Early in diabetes, endothelial cells start to increase the expression of ICAM-1 and P-selectin [70]. Two possible positive activators of ICAM-1 are the microglia-derived cytokines TNFα and IL-1β, suggesting that inflammation might start prior to leukocytosis. ICAM-1 (CD54) is a ligand of the β2 integrin lymphocyte function-associated antigen-1, which is part of a family of leukocyte integrins that are characterized by their β- (CD18) and α-chains (CD11a). ICAM-1 is expressed on the leukocyte surface and enhanced expression of ICAM-1 increases the adherence of leukocytes to the vascular membrane. Binding to CD11/CD18 on the surface of endothelial cells activates leukocytes, such as neutrophils and monocytes and results in accumulation of these immune cells on the luminal vascular surface [71]. This accumulation benefits occlusions, non-perfusion of the retinal vessels and dysfunction of the BRB [72]. Knock-down of adhesion molecules could prevent leukostasis in animal models [73,74]. Leukostasis, among others, is responsible for disruption of the BRB, death of endothelial cells [75] and is an early feature of DR in rodents, emerging within weeks of the onset of hyperglycemia; it has also been described in patients [72,73,75].
Additionally, by binding to β2 integrin lymphocyte function-associated antigen-1, ICAM-1 activates a key adhesion pathway, leading to upregulation of inflammatory cytokines [76,77,78] and promotes an inflammatory cascade in the retina [75]. Animal studies found that retinal leukostasis was decreased in diabetic mice deficient for TNFα. In these mice, a reduction of vascular leakage three and six months after onset of diabetes and a reduction of neurodegeneration after three months of diabetes was detected [79]. However, the deficiency of TNFα did not reduce leukostasis at earlier time points (four and six weeks of diabetes), suggesting that earlier time points were not dependent on TNFα signaling [79]. The temporal relationship between leukostasis and inflammation needs to be further investigated to determine the exact mechanisms.
Beside the increase in inflammation, leukostasis also leads to non-perfusion areas in the retina causing tissue death and neurodegeneration. However, dying neurons could contribute to vascular closure and thus, also in this relation, the temporal relationship still needs to be studied.
4. Microglia
Microglia, the macrophages of the CNS, are derived from hematopoietic stem cells [80,81]. Amoeboid, mesodermal myeloid progenitors enter the retina during development and differentiate into ramified parenchymal microglia in the adult retina [80,82,83,84]. These glia represent 5–12% of the cells in the CNS and can be identified by their immunoreactivity for CD45 (pan-leukocyte), MHC class I, and MHC class II [85,86]. Microglia are a population of self-renewing, relatively long-lived, innate immune cells [87,88].
For a long time, resident ramified microglia were described as quiescent cells, waiting for their activation, but today it is known that ramified microglia are involved in multiple processes and contribute to tissue and neuronal homeostasis [89,90,91]. Microglia have protective and corrective properties, for example, microglia are in close contact with neurons, transiently contacting synapses with their processes to monitor their functional state [92] and synapse stability, ensuring healthy vision [93]. The processes of microglia are constantly in motion, extending, retracting and continuously scanning their microenvironment [94]. Additionally, neurons express microglia ligand proteins such as fractalkine on their surface, which can be specifically recognized by microglia surface receptors, such as fractalkine receptor. Thus, on one hand these interactions contribute to controlling microglia function and on the other hand, microglia themselves can sense neuronal changes and promptly react to very subtle changes in their microenvironment [94,95].
The activation of microglia is determined by extracellular signals, including neuronal damage, chronic neurodegeneration, dying cells, extracellular liposaccharides and nucleic acids, which are recognized by a broad range of receptors [82,96], such as toll-like receptors (TLR) and receptors of advanced glycation end products. These enable microglia to detect pathogens via signaling of the pro-inflammatory nuclear transcription factor NFκB [97]. Translocation of NFκB is followed by production of cytokines and other inflammatory mediators.
Upon activation, ramified microglia undergo a series of stereotypic, morphological, phenotypic, and functional changes [98]. During activation, microglia start to proliferate and change their morphology from ramified state, with long and thin processes, to amoeboid state with larger cell bodies and thicker and shorter processes [86,95,99,100,101]. Additionally, immunoreactivity and migratory properties of microglia are enhanced [95,100,101]. Thus, expression levels of pro- and anti-inflammatory mediators (interleukins, cytokines, chemokines, proteases, nitric oxide and ROS) are increased as is the phagocytic activity of microglia [102,103].
In vitro studies showed that microglia and macrophages can become both pro- (M1) and anti-inflammatory, but pro-healing (M2) [104,105]. M1 microglia are induced by Th-1 cytokines, interferon γ or lipopolysaccharide and express high levels of IL-12, IL-23, TNFα, IL-1β and IL-6 [106]. These cells are neurotoxic. M2 microglia are induced by Th-2 cytokines, such as IL-4, IL-10 and IL-13 and express high levels of IL-10 [106]. These cells are mainly phagocytic and their response is neuroprotective. Phagocytic microglia are responsible for clearance of cell debris and necrotic or apoptotic cells. However, in vivo, microglia had an intermediates phenotype [104,107], suggesting that the distinction between M1 and M2 microglia is hazy and that microglia can adapt their phenotype to meet demands [3,108].
Microglia activation is a highly regulated process; the amount is determined by the affected tissue and the extent of dysfunction, damage or infection [109,110]. Normally, activation of microglia has protective properties, but under certain conditions could also result in damage to the CNS. Dysregulation of microglia activation may result in severe complications, including vascular breakdown, glia dysfunction and neuronal death [111]. Furthermore, dysregulation was associated with many diseases as well as neurodegeneration [112,113], suggesting that activation could become maladaptive in certain conditions.
Involvement of microglia was described in most CNS disorders and progression and healing of these diseases highly depends on microglia activation [114]. Changes in glia already occurred in initial stages of the disease.
4.1. Microglia in the Retina
As part of the CNS, retinal microglia adopt the role of tissue macrophages. In the adult retina, ramified microglia are mainly located in the inner retinal layers, such as the nerve fiber, the ganglion cell and the inner and outer plexiform layers [87,115,116], whereas they were barely found in the inner nuclear and totally absent in the outer nuclear layer [86,99].
In the healthy retina, microglia are required for normal retinal growth, the immune system, neurogenesis, synaptic pruning, controlling development, formation of blood vessels, aging and retinal function via interaction with neurons, glia and endothelial cells and secretion of growth factors and cytokines as well as neuroprotective and anti-inflammatory mediators [117,118,119,120,121,122,123]. In pathogenesis of the retina, microglia play a role in infection, trauma and retinal detachment.
4.2. Inflammation in Diabetic Retinopathy
The correlation between inflammation and DR was discovered when diabetic patients treated with salicylates for rheumatoid arthritis had fewer complications than untreated patients [124]. Since this discovery, increasing evidence showed that inflammation indeed plays an important role in DR; now, it is known that the diabetic retina always present a low chronic level of inflammation [125,126]. In diabetic patients, this low-level inflammation may be tolerated for years without any damage, however, mounting diabetic alternations may increase inflammation over time and it becomes more severe and chronic.
Inflammation is already associated with many other retinal diseases [127] and in DR, inflammation might be induced by leukocyte adhesion to the retinal vasculature and alteration of the BRB. A first step might be perivascular accumulation of activated microglial cells.
4.3. Microglia Activation in Diabetic Retinopathy
For a long time, microglia were underappreciated in most studies of DR. However, in recent years, numerous studies showed that microglia indeed play a significant role in DR. Microglia are altered by hyperglycemia, ischemia, hypoxia, dyslipidemia and endoplasmic reticulum stress, but the exact mode of microglia activation in DR is still unknown [125,126,128].
The activation of microglia in DR has also been documented in patients. DR is accompanied by an increase in cytokines, which further increase activation of microglia resulting in uncontrolled microglia activation, which is involved in neurotoxicity and tissue damage. Hyperglycemia induced TLR-2, TLR-4 and NFκB expression via an increase in ROS [129], indicating that oxidative stress plays a major role in microglia activation. Decreasing oxidation stress, e.g., via VP10/39 would be a promising therapeutic for DR [130]. The activation of NFκB leads to further production of cytokines and other inflammatory mediators [97]. NFκB was increased in activated microglia after hypoxia induction and was required for retinal angiogenesis [65]. Pathways of microglia activation in DR are summarized in Figure 2.
Hypoxia-inducible factor-1 (HIF-1) is the key player for the cellular response to reduced oxygen levels [131,132]. The HIF-1α subunit is stabilized in hypoxic conditions, binds to the HIF-1β subunit and thereby activates transcription of target genes, which among others are involved in proliferation, angiogenesis and cell survival [133,134,135,136]. Especially in highly active photoreceptors HIF-1 is of highest importance. It was reported to be constitutively active [137] and protects against retinal damage [138]. HO-1 is a target product of HIF-1α and activation of the Nrf2/HO-1 pathway to reduce oxidative stress could be an interesting approach to treat DR [139].
ERK phosphorylation is also involved in microglia activation [140] ERK can be activated by different signals and may have different consequences, e.g., ROS induced phosphorylation of ERK is important for TNFα expression [63] and vascular endothelial growth factor (VEGF) mediated activation of ERK is important for endothelial cells survival and proliferation [141,142]. LPS treatment in human retinal pigment epithelial cells activated ERK signaling, which is required for cytokine gene transcription [143]. Furthermore, ERK activation is important to induce expression of IL-6, MCP-1, and ICAM-1 [62]. High glucose levels in the retina were reported to increase phosphorylation of ERK [144] and also streptozotocine-induced diabetic rats showed an increase in proliferation and ERK expression in the retinal pigment epithelium [145].
Activation of microglia in the retina involves proliferation, migration and changes in their morphology. In streptozotocin-induced diabetic rats, the morphology of microglia changed from ramified to amoeboid [26,146]. The number of microglia increased in DR, suggesting that proliferation or immigration is enhanced [118]. Microglia, which are normally absent in the outer nuclear layer, migrated into the outer plexiform layer and the photoreceptor layer, while the amount of microglia in the ganglion cell layer decreased [26,86,99,147]. In another study, the density of microglia was not enhanced in diabetic rats, but the amount of activated microglia was increased [148]. Whether microglia activation in DR is neuroprotective or neurotoxic, is currently under discussion [149,150,151,152,153]. In the rodent retina, microglia activation started 1 month after diabetes induction and microglia invaded the inner plexiform layer after four months. After 14–16 months, microglia were found to migrate into the outer nuclear and photoreceptor layers [14,26,147].
In the human retina, microglial activation was present at distinct stages of DR. The number of microglia was increased and these cells migrated into the inner retinal layers and cluster around micro-aneurysms and intra-retinal hemorrhages. In DME, high numbers of microglia were found throughout the retina and in the sub-retinal space [118]. In retina of patients with NPDR, microglia migrated into the plexiform layers and increased in number, whereas in PDR, microglia significantly increased in number and clustered around ischemic areas [118,154].
5. Molecular Pathways of Inflammation in Diabetic Retinopathy
A complex chain of mechanisms, mediators and signaling cascades contribute to inflammation in DR. These include inflammatory cells, such as microglia and neutrophils, as well as inflammatory mediators, such as cytokines, chemokines, neurotoxins, growth factors and adhesion molecules [14,28,155,156,157,158,159]. The majority of the currently described mediators are summarized in Table 1.
The inflammatory process starts early in DR and can become chronic [28,158,159,160,161]. Chronic inflammation, characterized by prolonged duration and tissue destruction, induces a pro-inflammatory shift and enhances neuro-inflammation [162]. These characteristics apply to development and progression of DR [163,164,165]. Chronic inflammation has already been implicated with other, age-related retinal diseases such as age-related macular degeneration and age-induced changes in microglia [166].
Initially, upregulation of growth factors and cytokines may provide neurotrophic signals to maintain neuronal function and support retinal cell survival. However, over longer periods enhanced expression of chemokines and cytokines are maladaptive and cause vascular damage, DME and neovascularization [167].
5.1. Cytokines
Increased levels of cytokines, including IL-1β [28,168,169,170,171], IL-6 [172,173], IL-8 [173], VEGF [177] and TNFα [28,168,178,179] have been found in diabetic patients and rodent models of diabetes [198,199,200]. The levels of TNFα and IL-8 are further increased with progression of the disease; the highest amounts were measured in PDR [201,202,203]. In rodent diabetes models, the increase in IL-6 and TNFα is maintained up to 24 weeks of age [204]. Induction of diabetes in rats using streptozotocin increased retinal IL-6 and TNFα, whereas anti-inflammatory IL-10 was decreased, suggesting a pro-inflammatory and neurotoxic shift [146]. There was a local increase in IL-6 in the vitreous humor of diabetic patients, which was correlated with progression of DR [205].
5.2. VEGF
VEGF was identified based on its vascular effects, but was later also described as an important cytokine and signaling molecule for neurons. VEGF is neuroprotective in peripheral and central neurons [206,207] and is induced by hypoxia through the transcription factor HIF-1 [208]. It caused changes in tight junction organization and increases vascular permeability, via protein kinase C [44,209,210]. Deficiency of the VEGF-A gene in Müller cells reduced the effects of diabetes in the retina and decreased leukostasis, inflammation and vascular leakage, suggesting that Müller cells play an important role in retinal inflammation [211].
5.3. TNFα
Enhanced levels of TNFα and cyclo-oxygenase-2 are detected early in DR [180] and TNFα expression interferes with progression of DR. TNFα and IL-1β positively influenced ICAM-1, suggesting that TNFα could be involved in leukostasis. CD40, a member of the TNF receptor family, is normally upregulated in the rodent diabetic retina. Blocking CD40 reduced ICAM-1 upregulation, retinal leukostasis and degeneration capillary [212]. TNFα also exerts a crucial role in BRB breakdown [213] and neuronal death, by increasing caspase-3 expression [214]. Blocking TNFα receptor TNFR1, not only decreased vascular alternation, but also reduced neuronal cell death [214].
5.4. Chemokines
Enhanced levels of chemokines, such as CCL-2, CCL-4, CXCL-9 and CXCL-10, were measured in vitreous samples of patients with PDR [181]. Additionally, DR increases the levels of chemokine monocyte chemoattractant protein (MCP)-1 [28,173,182], which is involved in recruiting and activating microglia and leukocytes as well as in fibrosis and angiogenesis [215]. In streptozotocin-induced diabetic rodents, upregulation of MCP-1 started during the initial stages of DR and increased further with disease progression [216]. The main sources of MCP-1 are neurons and MCP-1 is involved in microglia activation by inducing TNFα release from microglia via the p38 and ERK pathway [63]. ERK1/2 and p38 phosphorylation was increased in microglia of diabetic rats, which was associated with harmful microglia activation [217,218].
5.5. Novel Molecular Targets in Diabetic Retinopathy
Over the last years, intensive research identified several new mediators influencing inflammation and neurodegeneration in DR. These new signaling pathways will be important to identify novel and better therapeutics.
Retinol-binding protein 4 (RBP4) expression was enhanced in patients with diabetes and its increase was correlated with retinal neuronal degeneration, early-onset of microglia activation and increased expression of pro-IL-18 and activated IL-18 [174,175,176]. Neurodegeneration in RBP4 overexpressing mice was shown to be independent of retinal microvascular pathology, suggesting an independent pathway in which microglia could influence neurodegeneration. RBP4 might alter inflammation via TLR4, c-Jun N-terminal kinases and p38 mitogen-activated protein kinase signaling pathways [176].
Another novel mediator of inflammation in DR could be nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOX)-1/4. Inhibition of NOX1/4 with GKT137831 not only reduced leukocyte adherence to the vasculature and vascular leakage, but also hypoxia-induced ROS levels and the pro-inflammatory phenotype (M1) of microglia and macroglia [188], suggesting that NOX inhibitors might be a promising therapeutic target in DR.
CX3CL1 was involved in activation of microglia in mouse models of type 1 diabetes [183]. CX3CL1 is a neuronal, membrane-bound chemokine, which could be proteolytically cleaved, became soluble and activated the CX3CR1 receptor in microglia. CX3CL1 deficiency in diabetic mice increased levels of IL-1β, negatively influenced the number of neurons in the ganglion cell layer and positively influenced the number of microglia cells [183]. In a model of retinitis pigmentosa, a progressive degenerating disease, deletion of CX3CR1 receptor caused an increase in microglial infiltration into the photoreceptor layer and accelerated photoreceptor apoptosis via increasing phagocytosis [219]. The role of CX3CR1 in DR was also analyzed in mice with streptozotocin-induced diabetes [220]. Here, CX3CR1 deletion expedited the onset of DR and increased apoptosis in the retina. In mouse models of mouse type 1 diabetes, loss of CX3CR1 signaling led to increased systemic inflammation and perivascular clustering of proliferating microglia with increased IL-1β expression [221]. Thus, CX3CR1 signaling bears a protective effect in the diabetic retina and might provide effective and successful means for treating DR.
A study on bone marrow (BM)-derived pro-inflammatory monocytes and BM-derived reparative circulating angiogenic cells showed that altering acid sphingomyelinase-mediated sphingolipid signaling in BM-derived cells could normalize inflammation in DR [222]. Acid sphingomyelinase was significantly increased in retinal endothelial cells from diabetic patients. [223], suggesting that by decreasing sphingolipid signaling, inflammation in DR could be reduced.
Matrix metalloproteases (MMP) were also analyzed in the context of DR, because their levels were increased. Especially MMP-2 and MMP-9 levels were enhanced in patients and in animal models of diabetes [189,190,191,192]. MMPs play an acute role in inflammation and activation of chemokines. Elevated levels of glucose could induce MMP-2 expression [224]. Treating streptozotocin-diabetic rats with minocycline, an inhibitor of MMP-1 and MMP-9, together with a cyclooxygenase (COX) and tPA inhibitor prevented development of DR [193]. Furthermore, MMP-2 was sensitive to oxidative stress, upon increased superoxide levels, MMP-2 was activated and increased apoptosis of retinal capillary cells in diabetic rats [194]. Thus, inhibition of MMP-2 could be another promising therapeutic target in DR.
STAT3 signaling was previously connected to cytokine signaling in vascular inflammation. IL-6 induced STAT3 signaling and thereby increased retinal endothelial permeability and vascular leakage. This was accompanied by VEGF-induced reduction in ZO-1 and occluding, two tight junction proteins [195]. miR-146a reduced IL-6/STAT3/VEGF signaling in high glucose conditions and overexpression of this microRNA decreased apoptosis, suggesting that miR-146a is a potential target for reducing inflammation and degeneration in DR [196].
Furthermore, aberrant activation of Wnt signaling played a pathogenic role in DR [197]. Levels of β-catenin were increased in diabetic patients and animal models of diabetes [197]. In vitro, Wnt activation using β-catenin treatment in ARPE19 cells, a cell line derived from human retinal pigment epithelium, increased the expression of VEGF, NFκB and TNFα, as well as the generation of ROS [225]. Thus, blocking Wnt signaling could be a potential therapeutic target in treating DR by reducing inflammation. Indeed, several studies reported a reduction in pathology of DR after inhibition of Wnt signaling [226,227,228]. Inhibition of Wnt using DKK-1 reduced retinal inflammation in diabetic rats [229], while in patients with DR, serum levels of DKK-1 were reduced [230], indicating that there could be increased Wnt signaling in DR patients.
6. Neurodegeneration in Diabetic Retinopathy
The retina comprises four types of neurons: photoreceptors (rod and cones), bipolar cells, amacrine cells and ganglion cells, which are involved in photo transduction, modulation and signal transfer. The encoded visual signal is transduced to the brain through axons of the ganglion cells. Impairment of neurons in the retina may result in impairment of vision.
In DR, neuronal defects are among the earliest detectable changes, resulting in a retinal neuropathy [19]. Neuronal cell death occurs in cultured retinas [17], diabetic mice [231] and patients [232]. DR is always associated with impaired neuronal function. Thus, treatment must interfere not only with vascular alterations and inflammation, but also with neuronal defects. Loss of neurons in the retina starts even before development of clinical symptoms [233,234] and neuro-retinal function is weakened before vascular lesions [235,236,237,238,239].
Retinal ganglion neurons are the first to die in DR and thus, loss of their nerve fibers in the nerve fiber layer occurs [18,233,240,241,242,243]. A reduction in ganglion cells was described in both diabetic mice [19] and patients [18,244]. In animal models of diabetes retinal ganglion, cell loss started as early as 5 weeks after induction of hyperglycemia [245]. db/db diabetic mice developed neurodegeneration in the retina, starting at the age of eight weeks [104,204,246].
Neuronal cell death in DR is due to an increase in apoptosis [247,248]. The loss of neurons results in thinning of the inner retinal layers and the nerve fiber layer in diabetic mice [18,233,249,250,251]. In these mice, ganglion cell loss occurred but there was no difference in the density of pericyte or acellular capillaries, suggesting that neurodegeneration preceded the established clinical and morphometric vascular changes. Additionally, the remaining ganglion cell bodies began to swell and increased in size [243]. In patients with diabetes and no to minimal DR, there is significant, progressive loss of the nerve fiber layer, the ganglion cell layer and the inner plexiform layer [249].
Electroretinography (ERG) can be used to measure neuronal defects in DR patients. The oscillatory potential implicit time gives information about the electrophysiological communication between neuronal cells. ERG measurements detect local abnormalities or widespread pathology, even in very initial stages of the disease. In DR, patients show a reduction in electric activity [252].
7. The Influence of Microglia on Neurodegeneration in Diabetic Retinopathy
Activated microglia are strongly involved in neurodegeneration [28]. Production of neurotoxic factors, such as glutamate, oxidative stress, caspase-3, MMPs and nitrous oxide, which are all neurotoxic mediators, result in neuronal cell dysfunction as well as damage to pericytes and endothelial cells. An imbalance in retinal production of neuroprotective mediators and pro-inflammation cytokines was involved in the development of neurodegeneration in DR [185].
Glutamate is toxic to retinal ganglion neurons [204] and extracellular glutamate led to over-activation of ionotropic glutamate receptors, mainly α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) and N-methyl-d-aspartame (NMDA) receptors, which resulted in uncontrolled intracellular calcium responses and cell death [253,254]. Oxidative stress originates from aberrant production of mitochondria-derived ROS and super oxide induced by hyperglycemia [255]. Hyperglycemia-induced downregulation of neurotrophic mediators such as nerve growth factor (NGF), pigment epithelium-derived factor (PEDF), iron-responsive element-binding proteins (IRBP) and somatostatin, also contributed to neurodegeneration [184].
8. Microvascular Pathology and Defective BRB Integrity
Microvascular pathologies have been implicated in DR since the discovery of the disease. For a long time, many DR studies in both clinic and animal models focused on vascular dysfunction, including impaired endothelial cells, death of pericytes, thickening of retina capillary basement membrane and altered tight junctions [256,257]. Vascular changes are caused by leukostasis [258], microthrombosis [259] or invasion of Müller cells into the vascular lumen [260].
In the healthy retina, vascular endothelial cells and pericytes are responsible for nutrient supply, waste product removal and constitute the BRB. In DR, increased capillary permeability and capillary occlusion are the major pathologies to identify the complication in diabetic patients and categorize the state of disease progression.
DR progression is defined by a decrease in retinal perfusion and disruption of the BRB [261]. Changes in retinal blood vessel permeability reduced the quantity of occludin in retina endothelial cells, which caused disorganization of tight junction proteins and thickening of the vascular basal membrane [75,262,263]. Phospholipases A2 was shown to increase early in DR and was linked to BRB alterations [264]. Apoptosis was increased in pericytes and endothelial cells [265,266]. The loss of pericytes, caused by insulin response modifications, had severe consequences for the retinal vasculature such as unstable retinal perfusion, capillary hyper-perfusion, hematoretinian barrier abrogation, appearance of capillary dilation, micro-aneurysm formation, occlusion of capillaries, retinal ischemia, increased in vascular permeability and endothelial cell degeneration [267,268].
Leakage of the BRB allows serum proteins, such as circulating cytokines and chemokines, as well as high glucose levels and advanced glycation-end products into the retina parenchyma and thus additionally contribute to activation of microglia and immune cell infiltration into the retina. Thus, through disruption of the BRB, the retina is affected by both external and internal signals [63,269,270]. BRB disruption could also increase the production of neurotoxic glutamate and contribute to neurodegeneration [271].
9. Angiogenesis and Inflammation
Angiogenesis is the course of endothelial cell migration, proliferation, vessel formation and remodeling of the vascular system. The formation of new vessels from existing ones depends on degradation of the extracellular matrix. Angiogenesis is an extremely regulated process which vascular alteration in DR can disrupt resulting in uncontrolled sprouting.
In DR, neovascularization is caused by an imbalance of pro-angiogenic mediators and ischemia resulting in abnormal growth of new vessels, which interferes with the normal function of the retina, namely light transmission. The consequences are leaky vessels and an accumulation of fluids and proteins.
Angiogenesis and inflammation are not independent, but rather two interacting processes which share several mediators (e.g., VEGF) and signaling pathways. Thus, microglia might induce neovascularization by releasing pro-angiogenic mediators, including cytokines, growth factors and proteases [272,273]. Cytokines could directly or indirectly enhance vascular growth in endothelial cells [274,275,276]. Moreover, endothelial cells could produce pro-inflammatory molecules [277,278].
The major regulator and pro-angiogenic factor is VEGF, which was increased after hyperglycemia and hypoxia [279,280]. An imbalance in the expression of VEGF is responsible for the increased neovascularization in DR. Thus, altering VEGF expression is a major therapeutic target in DR. Angiopoietin-2 is also an important modulator of angiogenesis and a regulator of the BRB [187] which exhibited increased levels in patients with DME [186]. However, both mediators are also involved in inflammation and might act as pro-inflammatory mediators, further increasing microglia activation and expression of inflammatory cytokines [281,282].
10. Interaction of Microglia with Macroglia in the Retina
The retina contains two types of macroglia: Müller cells and astrocytes. Müller cells are important for the regulation of retinal metabolism and modulate neuronal and blood vessel function [46], whereas astrocytes provide nutritional and regulatory support.
In DR, Müller cells increased the expression of glial fibrillary acidic protein, experienced altered ability to convert glutamate into glutamine because they express less glutamine syntheses and their number also increased [147,283,284], suggesting, that Müller cells are strongly involved in neurodegeneration via glutamate signaling. Microglia could directly influence Müller cells, which respond to microglia activation on the molecular and functional levels. Via bidirectional signaling between the two cell types, the activation and migration of microglia was further increased. Müller cells increased the inflammatory response across the retinal layers by chemotaxis and adhesive cell contacts and thus increased the mobilization of migratory microglia [285].
In the healthy retina, astrocytes are only located in the nerve fiber layer and surround blood vessels as well as ganglion cells. In DR, astrocytes became activated, change their morphology, proliferate, migrate and secreted pro-inflammatory mediators such as IL-6, MCP-1 and VEGF [144,286]. The translocator protein, a biomarker for microglial and astrocyte gliosis in brain degeneration, was upregulated in retinal microglia during retinal inflammation and injury. In astrocytes, the endogenous ligand of translocator protein, diazepam-binding inhibitor was upregulated, suggesting an astrocyte-microglia interaction [287].
11. Treatment of Diabetic Retinopathy by Altering Microglia
Treatment of DR should be targeted at the first event of the complication. However, investigations about the exact temporal coherencies are still in progress. Cytokine upregulation, leukostasis and glia activation were among the first symptoms of DR and would be ideal targets for therapy to prevent downstream neurodegeneration.
11.1. Photocoagulation
Developed in the 1950s, photocoagulation was one of the first treatments for DR and is still standard of care. Via prolonged exposure to bright light, a reduction of neovascularization and DME was precipitated, together with microglia activation [288,289,290]. The microglia response was prolonged after light exposure and expression of antigens, such as CD11b, CD45, and F4/80, was induced.
Following photocoagulation, morphological changes from ramified to amoeboid microglia occurred and these microglia migrated into the outer nuclear layer and to the laser-induced injury [272,291,292]. Activation and infiltration into the outer nuclear layer started 6 h after light induction, but the number of amoeboid microglia declined 7 days after light exposure and the cells were then present mostly in the sub-retinal space [293]. Microglia activation peaked parallel to the apoptotic loss of cells 1 day after injury [293]. Expression of chemokines, pro-angiogenic mediators and inflammatory cytokines increased after laser-induced injury [272,293].
Song et al. (2017) showed that deletion of anaphylatoxin C5a receptor (C5aR), a receptor previously associated with age-related macular degeneration, reduced the light-induced migration of microglia. After exposure to light, C5aR mRNA was increased as was the number of Iba-1 positive microglia cells. Microglia cells expressed receptors for C5a, which promoted recruitment of microglia and macrophages [294].
11.2. VEGF Blocking
Blocking VEGF had inhibitory effects on vascularization, but was also suspected to have neurotoxic effects [295], however these effects are still under discussion. Beneficial effects of the treatment have been documented [296,297] and it seems that VEGF has a dual role in neuroprotection and neovascularization in hypoxic regions. Diverse isoforms of VEGF are currently used in the clinic. Furthermore, the PKCβ/HuR/VEGF pathway has been suggested as a potential pharmacological target for DR.
Inhibition of the receptors for VEGF, VEGF receptor 1 and/or 2, blocks retinal microglia migration and infiltration after laser-induced choroidal neovascularization [298]. Choroidal neovascularization is a serious complication of age-related macular degeneration. Zhou et al. (2017) showed that the number of M1 and M2 macrophages increased following neovascularization. M1 macrophages (CD80-positive cells) were most prominent in the choroid-retinal pigment epithelial complex, whereas the M2 macrophages (CD206-positive cells) were mostly increased in the retina itself [299]. Blocking VEGF with a neutralizing antibody significantly inhibited diabetes-related vascular leakage, leukostasis, expression of ICAM-1, abnormal localization and degeneration of the tight junction protein ZO-1, as well as the cell adhesion protein vascular endothelial cadherin [300]. Furthermore, expression of several cytokines and chemokines were decreased after blocking VEGF [300], suggesting that blocking VEGF might be a possible target for the reduction of microglia activation.
11.3. Steroid Therapy
Glucocorticoids and their pharmacological derivates are anti-inflammatory and anti-angiogenic mediators, which are already used for treatment of DME and PDR, but are only a second-choice treatment for patients with suboptimal response to anti-VEGF treatment [301,302]. Glucocorticoids such as the synthetic triamcinolone reduced the expression of VEGF, promoted the stability of the BRB, prevented photoreceptor degeneration, inhibited activation of microglia and Müller cells and reduced expression of TNFα and the activation of p38/SAPK signaling pathways [303,304,305]. Glucocorticoids had anti-inflammatory properties, including the inhibition of NFκB and induction of the phagocytic activity of monocytes and macrophages [306,307,308]. Glucocorticoid treatment might be beneficial for treatment of DR, by affecting both vasculature and neurons. However, the exact role of steroids in the treatment for DR and DME remains to be fully elucidated.
11.4. Direct Prevention of Microglia Activation
Blocking glial cell activation might be a strategy to prevent neurodegeneration in DR and inhibition of microglia activation has been recently investigated in clinical trials. There are two tetracycline antibiotics in the focus of research: minocycline and doxycycline.
Minocycline is a semisynthetic, second-generation tetracycline antibiotic with anti-inflammatory properties, which prevents microglia activation and is neuroprotective in mice [28,309,310]. Treatment with minocycline resulted in reduced production and release of inflammatory cytokines, such as TNFα and IL-1, reduction in the retinal vascular permeability, reduced disruption of tight junctions and reduced activation of caspase-3 [28,311,312]. However, retinal neuronal death was not reduced after minocycline treatment, which raised the question if minocycline alone could be a treatment in DR [311]. In patients with DME, minocycline improved visual function, central macular edema and vascular leakage, and reduced neuropathic pain in diabetic patients [313,314].
Doxycycline, also a semisynthetic, second-generation tetracycline was neuroprotective in mice by reducing cleaved caspase-3 levels and microglial activation [309,315]. In patients with severe NPDR or non-high-risk PDR, doxycycline treatment directly improved foveal sensitivity starting six months after treatment [316]. However, in patients with mild to moderate NPDR, there was no improvement in visual function, suggesting that doxycycline might have different effects at distinct stages of DR.
12. Conclusions and Perspectives
In the last years, it became evident that microglia play a key role in DR and chronic inflammation has now been linked to neurodegeneration in the retina. Dysregulation of microglia activation most probably is the major alteration, resulting in a shift from pro-survival to pro-neurotoxic. The temporal components need to be analyzed more closely to identify which alteration is the trigger, and which one is the cause. Thus, whether microglia activation is the cause or the result of neuronal alternations needs to be further investigated; however, the early increase in cytokines points to microglia activation as the catalyst for subsequent neurodegeneration. Furthermore, the mechanism behind microglia activation needs to be explored in-depth. It is still not known if leukostasis or hyperglycemia directly influences the activation of microglia. Focusing more on the role of the disruption of the BRB in microglia activation could also be important, because the leakage is followed by massive invasion of glia cells and further cytokine release. Numerous new molecular mediators were recently identified, suggesting new opportunities for potential therapeutics. Currently, the treatment of DR is insufficient and further studies are needed to evaluate the therapeutic potential of inhibiting microglia activation. Furthermore, treatment approaches involving multiple mechanisms, instead of only one target should be the center of research. When searching for a potential therapeutic, one should always keep in mind the multiple pathways involved and that the retina is a complex structure and interaction of all retinal cells needs to be considered.
Acknowledgments
We thank members of the Schmidt lab for all their support and helpful discussions and Cheryl Ernest for proofreading the manuscript. This work was supported by the German Research Foundation (DFG) grant 2159/2 to Mirko H. H. Schmidt.
Author Contributions
Christine Altmann and Mirko H. H. Schmidt wrote and edited the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AMPA | α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate |
| C5aR | anaphylatoxin C5a receptor |
| BRB | blood-retina barrier |
| CCL | CC chemokine ligands |
| CNS | central nervous system |
| CX3CL | chemokine (C-X3-C motif) ligand |
| COX | cyclooxygenase |
| DME | diabetic macular edema |
| DR | diabetic retinopathy |
| ERG | electroretinogram |
| ERK | extracellular signal–regulated kinase |
| HIF | hypoxia-inducible factor |
| IL | interleukins |
| ICAM | intracellular adhesion molecule |
| IRBP | iron-responsive element-binding proteins |
| MMP | matrix metalloproteases |
| MCP | membrane cofactor protein |
| NMDA | N-methyl-d-aspartame |
| NOX | nicotinamide adenine dinucleotide phosphate (NADPH) oxidases |
| NGF | nerve growth factor |
| NO | nitric oxide |
| AGE | advanced glycation end products |
| LEP | leptin |
| NPDR | non-proliferating diabetic retinopathy |
| NF | nuclear factor |
| PEDF | pigment epithelium-derived factor |
| PDR | proliferating diabetic retinopathy |
| ROS | reactive oxygen species |
| RBP | retinol-binding protein |
| STAT | signal transducer and activator of transcription |
| TNF | tumor necrosis factor |
| VCAM | vascular cell adhesion molecule |
| VEGF | vascular endothelial growth factor |
| ZO | zonula occludens |
References
- Bourne, R.R.; Stevens, G.A.; White, R.A.; Smith, J.L.; Flaxman, S.R.; Price, H.; Jonas, J.B.; Keeffe, J.; Leasher, J.; Naidoo, K.; et al. Causes of Vision Loss Worldwide, 1990–2010: A Systematic Analysis. Lancet Glob. Health 2013, 1, e339–e349. [Google Scholar] [CrossRef]
- Fong, D.S.; Aiello, L.; Gardner, T.W.; King, G.L.; Blankenship, G.; Cavallerano, J.D.; Ferris, F.L., 3rd; Klein, R. Diabetic Retinopathy. Diabetes Care 2003, 26, S99–S102. [Google Scholar] [CrossRef] [PubMed]
- Banati, R.B.; Gehrmann, J.; Schubert, P.; Kreutzberg, G.W. Cytotoxicity of Microglia. Glia 1993, 7, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Fong, D.S.; Aiello, L.P.; Ferris, F.L., 3rd; Klein, R. Diabetic Retinopathy. Diabetes Care 2004, 27, 2540–2553. [Google Scholar] [CrossRef] [PubMed]
- Nentwich, M.M.; Ulbig, M.W. Diabetic Retinopathy—Ocular Complications of Diabetes Mellitus. World J. Diabetes 2015, 6, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic Retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef]
- Rathmann, W.; Giani, G. Global Prevalence of Diabetes: Estimates for the Year 2000 and Projections for 2030. Diabetes Care 2004, 27, 2568–2569. [Google Scholar] [CrossRef] [PubMed]
- Kowluru, R.A.; Chan, P.-S. Oxidative Stress and Diabetic Retinopathy. Exp. Diabetes Res. 2007, 2007, 43603. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, C.P.; Ferris, F.L., 3rd; Klein, R.E.; Lee, P.P.; Agardh, C.D.; Davis, M.; Dills, D.; Kampik, A.; Pararajasegaram, R.; Verdaguer, J.T. Proposed International Clinical Diabetic Retinopathy and Diabetic Macular Edema Disease Severity Scales. Ophthalmology 2003, 110, 1677–1682. [Google Scholar] [CrossRef]
- Arroba, A.I.; Valverde, A.M. Modulation of Microglia in the Retina: New Insights into Diabetic Retinopathy. Acta Diabetol. 2017, 54, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Stroud, S.; Mehta, A.; Rangasamy, S. New Treatments for Diabetic Retinopathy. Diabetes Obes. Metab. 2015, 17, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.W.; Lois, N.; Medina, R.J.; Adamson, P.; Curtis, T.M. Advances in Our Understanding of Diabetic Retinopathy. Clin. Sci. 2013, 125, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.A.; Lois, N.; Royle, P.; Clar, C.; Shyangdan, D.; Waugh, N. Current Treatments in Diabetic Macular Oedema: Systematic Review and Meta-Analysis. BMJ Open 2013, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Chen, H.; Su, S.B. Neuroinflammatory Responses in Diabetic Retinopathy. J. Neuroinflamm. 2015, 12, 141. [Google Scholar] [CrossRef] [PubMed]
- Aiello, L.P. Diabetic Retinopathy and Other Ocular Findings in the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study. Diabetes Care 2014, 37, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, F.; Cancarini, A.; dell’Omo, R.; Rezzola, S.; Romano, M.R.; Costagliola, C. Diabetic Retinopathy: Vascular and Inflammatory Disease. J. Diabetes Res. 2015, 2015, 582060. [Google Scholar] [CrossRef] [PubMed]
- Santiago, A.R.; Cristovao, A.J.; Santos, P.F.; Carvalho, C.M.; Ambrosio, A.F. High Glucose Induces Caspase-Independent Cell Death in Retinal Neural Cells. Neurobiol. Dis. 2007, 25, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.J.; Lieth, E.; Khin, S.A.; Antonetti, D.A.; Buchanan, A.G.; Gardner, T.W. Neural Apoptosis in the Retina During Experimental and Human Diabetes. Early Onset and Effect of Insulin. J. Clin. Investig. 1998, 102, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.J. A New View of Diabetic Retinopathy: A Neurodegenerative Disease of the Eye. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2003, 27, 283–290. [Google Scholar] [CrossRef]
- Ramirez, A.I.; de Hoz, R.; Salobrar-Garcia, E.; Salazar, J.J.; Rojas, B.; Ajoy, D.; Lopez-Cuenca, I.; Rojas, P.; Trivino, A.; Ramirez, J.M. The Role of Microglia in Retinal Neurodegeneration: Alzheimer’s Disease, Parkinson, and Glaucoma. Front. Aging Neurosci. 2017, 9, 214. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.M.; Yang, S.; Huang, S.S.; Tang, B.S.; Guo, J.F. Microglial Activation in the Pathogenesis of Huntington's Disease. Front. Aging Neurosci. 2017, 9, 193. [Google Scholar] [CrossRef] [PubMed]
- Lieth, E.; Gardner, T.W.; Barber, A.J.; Antonetti, D.A. Retinal Neurodegeneration: Early Pathology in Diabetes. Clin. Exp. Ophthalmol. 2000, 28, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Jackson, G.R.; Barber, A.J. Visual Dysfunction Associated with Diabetic Retinopathy. Curr. Diabetes Rep. 2010, 10, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Tyrberg, M.; Lindblad, U.; Melander, A.; Lovestam-Adrian, M.; Ponjavic, V.; Andreasson, S. Electrophysiological Studies in Newly Onset Type 2 Diabetes without Visible Vascular Retinopathy. Doc. Ophthalmol. 2011, 123, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Juen, S.; Kieselbach, G.F. Electrophysiological Changes in Juvenile Diabetics without Retinopathy. Arch. Ophthalmol. 1990, 108, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.X.; Ng, Y.K.; Ling, E.A. Neuronal and Microglial Response in the Retina of Streptozotocin-Induced Diabetic Rats. Vis. Neurosci. 2000, 17, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.; Hirose, G. Origin of the Retina from Both Sides of the Embryonic Brain: A Contribution to the Problem of Crossing at the Optic Chiasma. Science 1978, 202, 637–639. [Google Scholar] [CrossRef] [PubMed]
- Krady, J.K.; Basu, A.; Allen, C.M.; Xu, Y.; LaNoue, K.F.; Gardner, T.W.; Levison, S.W. Minocycline Reduces Proinflammatory Cytokine Expression, Microglial Activation, and Caspase-3 Activation in a Rodent Model of Diabetic Retinopathy. Diabetes 2005, 54, 1559–1565. [Google Scholar] [CrossRef] [PubMed]
- Pournaras, C.J. Retinal Oxygen Distribution. Its Role in the Physiopathology of Vasoproliferative Microangiopathies. Retina 1995, 15, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, J.; Braun, R.D.; Dunn, R., Jr.; Linsenmeier, R.A. Oxygen Distribution in the Macaque Retina. Investig. Ophthalmol. Vis. Sci. 1993, 34, 516–521. [Google Scholar]
- Reichenbach, A.; Stolzenburg, J.U.; Eberhardt, W.; Chao, T.I.; Dettmer, D.; Hertz, L. What Do Retinal Muller (Glial) Cells Do for Their Neuronal ‘Small Siblings’? J. Chem. Neuroanat. 1993, 6, 201–213. [Google Scholar] [CrossRef]
- Ames, A., 3rd. Energy Requirements of Cns Cells as Related to Their Function and to Their Vulnerability to Ischemia: A Commentary Based on Studies on Retina. Can. J. Physiol. Pharmacol. 1992, 70, S158–S164. [Google Scholar] [CrossRef] [PubMed]
- Kolko, M.; Vosborg, F.; Henriksen, U.L.; Hasan-Olive, M.M.; Diget, E.H.; Vohra, R.; Gurubaran, I.R.S.; Gjedde, A.; Mariga, S.T.; Skytt, D.M.; et al. Lactate Transport and Receptor Actions in Retina: Potential Roles in Retinal Function and Disease. Neurochem. Res. 2016, 41, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.B.; Lindsay, K.J.; Du, J. Glucose, Lactate, and Shuttling of Metabolites in Vertebrate Retinas. J. Neurosci. Res. 2015, 93, 1079–1092. [Google Scholar] [CrossRef] [PubMed]
- Winkler, B.S.; Arnold, M.J.; Brassell, M.A.; Sliter, D.R. Glucose Dependence of Glycolysis, Hexose Monophosphate Shunt Activity, Energy Status, and the Polyol Pathway in Retinas Isolated from Normal (Nondiabetic) Rats. Investig. Ophthalmol. Vis. Sci. 1997, 38, 62–71. [Google Scholar]
- Poitry-Yamate, C.L.; Poitry, S.; Tsacopoulos, M. Lactate Released by Muller Glial Cells Is Metabolized by Photoreceptors from Mammalian Retina. J. Neurosci. 1995, 15, 5179–5191. [Google Scholar] [PubMed]
- Philp, N.J.; Yoon, H.; Grollman, E.F. Monocarboxylate Transporter Mct1 Is Located in the Apical Membrane and Mct3 in the Basal Membrane of Rat Rpe. Am. J. Physiol. 1998, 274, R1824–R1828. [Google Scholar] [CrossRef] [PubMed]
- Castillo, X.; Rosafio, K.; Wyss, M.T.; Drandarov, K.; Buck, A.; Pellerin, L.; Weber, B.; Hirt, L. A Probable Dual Mode of Action for Both l- and d-Lactate Neuroprotection in Cerebral Ischemia. J. Cereb. Blood Flow Metab. 2015, 35, 1561–1569. [Google Scholar] [CrossRef] [PubMed]
- Horn, T.; Klein, J. Neuroprotective Effects of Lactate in Brain Ischemia: Dependence on Anesthetic Drugs. Neurochem. Int. 2013, 62, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Bouzat, P.; Sala, N.; Suys, T.; Zerlauth, J.B.; Marques-Vidal, P.; Feihl, F.; Bloch, J.; Messerer, M.; Levivier, M.; Meuli, R.; et al. Cerebral Metabolic Effects of Exogenous Lactate Supplementation on the Injured Human Brain. Intensive Care Med. 2014, 40, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Ichai, C.; Armando, G.; Orban, J.C.; Berthier, F.; Rami, L.; Samat-Long, C.; Grimaud, D.; Leverve, X. Sodium Lactate Versus Mannitol in the Treatment of Intracranial Hypertensive Episodes in Severe Traumatic Brain-Injured Patients. Intensive Care Med. 2009, 35, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia Metabolically Support Axons and Contribute to Neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Ballabh, P.; Braun, A.; Nedergaard, M. The Blood-Brain Barrier: An Overview: Structure, Regulation, and Clinical Implications. Neurobiol. Dis. 2004, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, D.A.; Lieth, E.; Barber, A.J.; Gardner, T.W. Molecular Mechanisms of Vascular Permeability in Diabetic Retinopathy. Semin. Ophthalmol. 1999, 14, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Gardner, T.W.; Lieth, E.; Khin, S.A.; Barber, A.J.; Bonsall, D.J.; Lesher, T.; Rice, K.; Brennan, W.A., Jr. Astrocytes Increase Barrier Properties and ZO-1 Expression in Retinal Vascular Endothelial Cells. Investig. Ophthalmol. Vis. Sci. 1997, 38, 2423–2427. [Google Scholar]
- Abbott, N.J.; Revest, P.A.; Romero, I.A. Astrocyte-Endothelial Interaction: Physiology and Pathology. Neuropathol. Appl. Neurobiol. 1992, 18, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.M.; Genuth, S.; Lachin, J.; Cleary, P.; Crofford, O.; Davis, M.; Rand, L.; Siebert, C. The Effect of Intensive Treatment of Diabetes on the Development and Progression of Long-Term Complications in Insulin-Dependent Diabetes Mellitus. N. Engl. J. Med. 1993, 329, 977–986. [Google Scholar] [PubMed]
- UK Prospective Diabetes Study (Ukpds) Group. Intensive Blood-Glucose Control with Sulphonylureas or Insulin Compared with Conventional Treatment and Risk of Complications in Patients with Type 2 Diabetes (Ukpds 33). Lancet 1998, 352, 837–853. [Google Scholar]
- Hudson, B.I.; Schmidt, A.M. Rage: A Novel Target for Drug Intervention in Diabetic Vascular Disease. Pharm. Res. 2004, 21, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Stitt, A.W. The Role of Advanced Glycation in the Pathogenesis of Diabetic Retinopathy. Exp. Mol. Pathol. 2003, 75, 95–108. [Google Scholar] [CrossRef]
- Barile, G.R.; Pachydaki, S.I.; Tari, S.R.; Lee, S.E.; Donmoyer, C.M.; Ma, W.; Rong, L.L.; Buciarelli, L.G.; Wendt, T.; Horig, H.; et al. The Rage Axis in Early Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2916–2924. [Google Scholar] [CrossRef] [PubMed]
- Aiello, L.P. The Potential Role of PKCβ in Diabetic Retinopathy and Macular Edema. Surv. Ophthalmol. 2002, 47, S263–S269. [Google Scholar] [CrossRef]
- Caldwell, R.B.; Bartoli, M.; Behzadian, M.A.; El-Remessy, A.E.; Al-Shabrawey, M.; Platt, D.H.; Liou, G.I.; Caldwell, R.W. Vascular Endothelial Growth Factor and Diabetic Retinopathy: Role of Oxidative Stress. Curr. Drug Targets 2005, 6, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Soufi, F.G.; Vardyani, M.; Sheervalilou, R.; Mohammadi, M.; Somi, M.H. Long-Term Treatment with Resveratrol Attenuates Oxidative Stress Pro-Inflammatory Mediators and Apoptosis in Streptozotocin-Nicotinamide-Induced Diabetic Rats. Gen. Physiol. Biophys. 2012, 31, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Rains, J.L.; Jain, S.K. Oxidative Stress, Insulin Signaling, and Diabetes. Free Radic. Biol. Med. 2011, 50, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Du, X.L.; Edelstein, D.; Rossetti, L.; Fantus, I.G.; Goldberg, H.; Ziyadeh, F.; Wu, J.; Brownlee, M. Hyperglycemia-Induced Mitochondrial Superoxide Overproduction Activates the Hexosamine Pathway and Induces Plasminogen Activator Inhibitor-1 Expression by Increasing Sp1 Glycosylation. Proc. Natl. Acad. Sci. USA 2000, 97, 12222–12226. [Google Scholar] [CrossRef] [PubMed]
- Milne, R.; Brownstein, S. Advanced Glycation End Products and Diabetic Retinopathy. Amino Acids 2013, 44, 1397–1407. [Google Scholar] [CrossRef] [PubMed]
- Zong, H.; Ward, M.; Stitt, A.W. Ages, Rage, and Diabetic Retinopathy. Curr. Diabetes Rep. 2011, 11, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Smith, M.A.; Miller, C.M.; Kern, T.S. Diabetes-Induced Nitrative Stress in the Retina, and Correction by Aminoguanidine. J. Neurochem. 2002, 80, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Stern, D.M.; Yan, S.D.; Yan, S.F.; Schmidt, A.M. Receptor for Advanced Glycation Endproducts (Rage) and the Complications of Diabetes. Ageing Res. Rev. 2002, 1, 1–15. [Google Scholar] [CrossRef]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.P.; et al. Normalizing Mitochondrial Superoxide Production Blocks Three Pathways of Hyperglycaemic Damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.K.; Lee, C.M.; Lee, D.S.; Na, G.; Lee, D.Y.; Choi, I.; Park, S.G.; Seo, S.K.; Yang, J.W.; Choi, J.S.; et al. The 15-Deoxy-Delta12,14-Prostaglandin J2 Inhibits Lpsstimulated Inflammation Via Enhancement of the Plateletactivating Factor Acetylhydrolase Activity in Human Retinal Pigment Epithelial Cells. Int. J. Mol. Med. 2014, 33, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Dong, N.; Chang, L.; Wang, B.; Chu, L. Retinal Neuronal Mcp-1 Induced by Ages Stimulates TNFα Expression in Rat Microglia Via P38, Erk, and Nf-κB Pathways. Mol. Vis. 2014, 20, 616–628. [Google Scholar] [PubMed]
- Palsamy, P.; Subramanian, S. Ameliorative Potential of Resveratrol on Proinflammatory Cytokines, Hyperglycemia Mediated Oxidative Stress, and Pancreatic β-Cell Dysfunction in Streptozotocin-Nicotinamide-Induced Diabetic Rats. J. Cell. Physiol. 2010, 224, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, A.; Yoshida, S.; Ishibashi, T.; Kuwano, M.; Inomata, H. Suppression of Retinal Neovascularization by the Nf-κB Inhibitor Pyrrolidine Dithiocarbamate in Mice. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1624–1629. [Google Scholar]
- Wang, A.L.; Yu, A.C.; He, Q.H.; Zhu, X.; Tso, M.O. Ages Mediated Expression and Secretion of TNFα in Rat Retinal Microglia. Exp. Eye Res. 2007, 84, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, M.; Kern, T.S.; Lorenzi, M. Accelerated Death of Retinal Microvascular Cells in Human and Experimental Diabetic Retinopathy. J. Clin. Investig. 1996, 97, 2883–2890. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, H.; Sugiyama, T.; Wu, D.M.; Kobayashi, M.; Yamanishi, S.; Katsumura, K.; Puro, D.G. Atp: A Vasoactive Signal in the Pericyte-Containing Microvasculature of the Rat Retina. J. Physiol. 2003, 551, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Platania, C.B.M.; Giurdanella, G.; di Paola, L.; Leggio, G.M.; Drago, F.; Salomone, S.; Bucolo, C. P2X7 Receptor Antagonism: Implications in Diabetic Retinopathy. Biochem. Pharmacol. 2017, 138, 130–139. [Google Scholar] [CrossRef] [PubMed]
- McLeod, D.S.; Lefer, D.J.; Merges, C.; Lutty, G.A. Enhanced Expression of Intracellular Adhesion Molecule-1 and P-Selectin in the Diabetic Human Retina and Choroid. Am. J. Pathol. 1995, 147, 642–653. [Google Scholar] [PubMed]
- Valle, A.; Giamporcaro, G.M.; Scavini, M.; Stabilini, A.; Grogan, P.; Bianconi, E.; Sebastiani, G.; Masini, M.; Maugeri, N.; Porretti, L.; et al. Reduction of Circulating Neutrophils Precedes and Accompanies Type 1 Diabetes. Diabetes 2013, 62, 2072–2077. [Google Scholar] [CrossRef] [PubMed]
- Chibber, R.; Ben-Mahmud, B.M.; Chibber, S.; Kohner, E.M. Leukocytes in Diabetic Retinopathy. Curr. Diabetes Rev. 2007, 3, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Joussen, A.M.; Poulaki, V.; Le, M.L.; Koizumi, K.; Esser, C.; Janicki, H.; Schraermeyer, U.; Kociok, N.; Fauser, S.; Kirchhof, B.; et al. A Central Role for Inflammation in the Pathogenesis of Diabetic Retinopathy. FASEB J. 2004, 18, 1450–1452. [Google Scholar] [CrossRef] [PubMed]
- Leal, E.C.; Manivannan, A.; Hosoya, K.; Terasaki, T.; Cunha-Vaz, J.; Ambrosio, A.F.; Forrester, J.V. Inducible Nitric Oxide Synthase Isoform Is a Key Mediator of Leukostasis and Blood-Retinal Barrier Breakdown in Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5257–5265. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Khosrof, S.; Bursell, S.E.; Rohan, R.; Murata, T.; Clermont, A.C.; Aiello, L.P.; Ogura, Y.; Adamis, A.P. Prevention of Leukostasis and Vascular Leakage in Streptozotocin-Induced Diabetic Retinopathy Via Intercellular Adhesion Molecule-1 Inhibition. Proc. Natl. Acad. Sci. USA 1999, 96, 10836–10841. [Google Scholar] [CrossRef] [PubMed]
- Long, E.O. Intercellular Adhesion Molecule 1 (Icam-1): Getting a Grip on Leukocyte Adhesion. J. Immunol. 2011, 186. [Google Scholar] [CrossRef] [PubMed]
- Rothlein, R.; Dustin, M.L.; Marlin, S.D.; Springer, T.A. A Human Intercellular Adhesion Molecule (Icam-1) Distinct from Lfa-1. J. Immunol. 1986, 137, 1270–1274. [Google Scholar] [PubMed]
- Dustin, M.L.; Rothlein, R.; Bhan, A.K.; Dinarello, C.A.; Springer, T.A. Induction by Il 1 and Interferon-γ: Tissue Distribution, Biochemistry, and Function of a Natural Adherence Molecule (Icam-1). J. Immunol. 1986, 137, 245–254. [Google Scholar] [PubMed]
- Huang, H.; Gandhi, J.K.; Zhong, X.; Wei, Y.; Gong, J.; Duh, E.J.; Vinores, S.A. TNFα Is Required for Late Brb Breakdown in Diabetic Retinopathy, and Its Inhibition Prevents Leukostasis and Protects Vessels and Neurons from Apoptosis. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1336–1344. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.Y.; Kohsaka, S.; Rezaie, P. The Origin and Cell Lineage of Microglia: New Concepts. Brain Res. Rev. 2007, 53, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Saijo, K.; Glass, C.K. Microglial Cell Origin and Phenotypes in Health and Disease. Nat. Rev. Immunol. 2011, 11, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Polazzi, E.; Monti, B. Microglia and Neuroprotection: From in Vitro Studies to Therapeutic Applications. Prog. Neurobiol. 2010, 92, 293–315. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, E.; Thanos, S. Microglia-Targeted Pharmacotherapy in Retinal Neurodegenerative Diseases. Curr. Drug Targets 2004, 5, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Gehrmann, J.; Banati, R.B.; Kreutzberg, G.W. Microglia in the Immune Surveillance of the Brain: Human Microglia Constitutively Express Hla-Dr Molecules. J. Neuroimmunol. 1993, 48, 189–198. [Google Scholar] [CrossRef]
- Provis, J.M.; Diaz, C.M.; Penfold, P.L. Microglia in Human Retina: A Heterogeneous Population with Distinct Ontogenies. Perspect. Dev. Neurobiol. 1996, 3, 213–222. [Google Scholar] [PubMed]
- Santiago, A.R.; Baptista, F.I.; Santos, P.F.; Cristóvão, G.; Ambrósio, A.F.; Cunha, R.A.; Gomes, C.A. Role of Microglia Adenosine A2A Receptors in Retinal and Brain Neurodegenerative Diseases. Mediat. Inflamm. 2014, 2014, 465694. [Google Scholar] [CrossRef] [PubMed]
- Ajami, B.; Bennett, J.L.; Krieger, C.; Tetzlaff, W.; Rossi, F.M. Local Self-Renewal Can Sustain CNS Microglia Maintenance and Function Throughout Adult Life. Nat. Neurosci. 2007, 10, 1538–1543. [Google Scholar] [CrossRef] [PubMed]
- Derecki, N.C.; Cronk, J.C.; Kipnis, J. The Role of Microglia in Brain Maintenance: Implications for Rett Syndrome. Trends Immunol. 2013, 34, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Teeling, J. Microglia and Macrophages of the Central Nervous System: The Contribution of Microglia Priming and Systemic Inflammation to Chronic Neurodegeneration. Semin. Immunopathol. 2013, 35, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Kierdorf, K.; Prinz, M. Microglia in Steady State. J. Clin. Investig. 2017, 127, 3201–3209. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting Microglia Directly Monitor the Functional State of Synapses in Vivo and Determine the Fate of Ischemic Terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhao, L.; Zhang, J.; Fariss, R.N.; Ma, W.; Kretschmer, F.; Wang, M.; Qian, H.H.; Badea, T.C.; Diamond, J.S.; et al. Requirement for Microglia for the Maintenance of Synaptic Function and Integrity in the Mature Retina. J. Neurosci. 2016, 36, 2827–2842. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. Atp Mediates Rapid Microglial Response to Local Brain Injury in Vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Dheen, S.T.; Kaur, C.; Ling, E.A. Microglial Activation and Its Implications in the Brain Diseases. Curr. Med. Chem. 2007, 14, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Li, C.H.; Liu, Y.L. Toll-Like Receptor (TLR)-2/4 Expression in Retinal Ganglion Cells in a High-Glucose Environment and Its Implications. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Graeber, M.B.; Kreutzberg, G.W. Peripheral Nerve Lesion Produces Increased Levels of Major Histocompatibility Complex Antigens in the Central Nervous System. J. Neuroimmunol. 1989, 21, 117–123. [Google Scholar] [CrossRef]
- Chen, L.; Yang, P.; Kijlstra, A. Distribution, Markers, and Functions of Retinal Microglia. Ocul. Immunol. Inflamm. 2002, 10, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Kreutzberg, G.W. Microglia: A Sensor for Pathological Events in the Cns. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Tambuyzer, B.R.; Ponsaerts, P.; Nouwen, E.J. Microglia: Gatekeepers of Central Nervous System Immunology. J. Leukoc. Biol. 2009, 85, 352–370. [Google Scholar] [CrossRef] [PubMed]
- Langmann, T. Microglia Activation in Retinal Degeneration. J. Leukoc. Biol. 2007, 81, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J.; Kreutzberg, G.W. Response of Endogenous Glial Cells to Motor Neuron Degeneration Induced by Toxic Ricin. J. Comp. Neurol. 1988, 268, 248–263. [Google Scholar] [CrossRef] [PubMed]
- Arroba, A.I.; Alcalde-Estevez, E.; Garcia-Ramirez, M.; Cazzoni, D.; de la Villa, P.; Sanchez-Fernandez, E.M.; Mellet, C.O.; Fernandez, J.M.G.; Hernandez, C.; Simo, R.; et al. Modulation of Microglia Polarization Dynamics During Diabetic Retinopathy in db/db Mice. Biochim. Biophys. Acta 2016, 1862, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Yi, H.; Xu, L.; Zhang, Z. Fluoxetine and S-Citalopram Inhibit M1 Activation and Promote M2 Activation of Microglia in Vitro. Neuroscience 2015, 294, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative Activation of Macrophages: An Immunologic Functional Perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef] [PubMed]
- Lampron, A.; Elali, A.; Rivest, S. Innate Immunity in the Cns: Redefining the Relationship between the CNS and Its Environment. Neuron 2013, 78, 214–232. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Li, W.; Rodriguez, M.L. Role of Microglia in CNS Inflammation. FEBS Lett. 2011, 585, 3798–3805. [Google Scholar] [CrossRef] [PubMed]
- Raivich, G.; Bohatschek, M.; Kloss, C.U.; Werner, A.; Jones, L.L.; Kreutzberg, G.W. Neuroglial Activation Repertoire in the Injured Brain: Graded Response, Molecular Mechanisms and Cues to Physiological Function. Brain Res. Brain Res. Rev. 1999, 30, 77–105. [Google Scholar] [CrossRef]
- Colton, C.; Wilcock, D.M. Assessing Activation States in Microglia. CNS Neurol. Disord. Drug Targets 2010, 9, 174–191. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.P.; Sun, H.L.; Wu, L.M.; Guo, X.J.; Dou, H.L.; Tso, M.O.; Zhao, L.; Li, S.M. Baicalein Reduces Inflammatory Process in a Rodent Model of Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Frick, L.; Pittenger, C. Microglial Dysregulation in Ocd, Tourette Syndrome, and Pandas. J. Immunol. Res. 2016, 2016, 8606057. [Google Scholar] [CrossRef] [PubMed]
- Von Bernhardi, R.; Bernhardi, L.E.; Eugenín, J. Microglial Cell Dysregulation in Brain Aging and Neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.W.; Beggs, S. Sublime Microglia: Expanding Roles for the Guardians of the CNS. Cell 2014, 158, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.A.; Perry, V.H.; Gordon, S. Immunohistochemical Localization of a Macrophage-Specific Antigen in Developing Mouse Retina: Phagocytosis of Dying Neurons and Differentiation of Microglial Cells to Form a Regular Array in the Plexiform Layers. J. Cell Biol. 1983, 97, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.M.; Martin-Oliva, D.; Ferrer-Martin, R.M.; Tassi, M.; Calvente, R.; Sierra, A.; Carrasco, M.C.; Marin-Teva, J.L.; Navascues, J.; Cuadros, M.A. Microglial Response to Light-Induced Photoreceptor Degeneration in the Mouse Retina. J. Comp. Neurol. 2010, 518, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Cui, J.; Li, L.; Hitchcock, P.F.; Li, Y. The Role of Microglia in the Neurogenesis of Zebrafish Retina. Biochem. Biophys. Res. Commun. 2012, 421, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.Y.; Green, W.R.; Tso, M.O. Microglial Activation in Human Diabetic Retinopathy. Arch. Ophthalmol. 2008, 126, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Jiao, H.; Natoli, R.; Valter, K.; Provis, J.M.; Rutar, M. Spatiotemporal Cadence of Macrophage Polarisation in a Model of Light-Induced Retinal Degeneration. PLoS ONE 2015, 10, e0143952. [Google Scholar] [CrossRef] [PubMed]
- Arroba, A.I.; Alvarez-Lindo, N.; van Rooijen, N.; de la Rosa, E.J. Microglia-Mediated Igf-I Neuroprotection in the Rd10 Mouse Model of Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9124–9130. [Google Scholar] [CrossRef] [PubMed]
- Bilimoria, P.M.; Stevens, B. Microglia Function during Brain Development: New Insights from Animal Models. Brain Res. 2015, 1617, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New Roles for the Synaptic Stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Checchin, D.; Sennlaub, F.; Levavasseur, E.; Leduc, M.; Chemtob, S. Potential Role of Microglia in Retinal Blood Vessel Formation. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3595–3602. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.D.; Field, R.A. Diabetic Retinopathy and Rheumatoid Arthritis. Lancet 1964, 2, 17–18. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.; Al-Shabrawey, M.; Caldwell, R.W.; Caldwell, R.B. Inflammation and Diabetic Retinal Microvascular Complications. J. Cardiovasc. Dis. Res. 2011, 2, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Kern, T.S. Inflammation in Diabetic Retinopathy. Prog. Retin. Eye Res. 2011, 30, 343–358. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, Stress, and Diabetes. J. Clin. Investig. 2005, 115, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, S.; Perego, C.; Pischiutta, F.; Zanier, E.R.; de Simoni, M.G. The Ischemic Environment Drives Microglia and Macrophage Function. Front. Neurol. 2015, 6, 81. [Google Scholar] [CrossRef] [PubMed]
- Rajamani, U.; Jialal, I. Hyperglycemia Induces Toll-Like Receptor-2 and -4 Expression and Activity in Human Microvascular Retinal Endothelial Cells: Implications for Diabetic Retinopathy. J. Diabetes Res. 2014, 2014, 790902. [Google Scholar] [CrossRef] [PubMed]
- Pittala, V.; Fidilio, A.; Lazzara, F.; Platania, C.B.M.; Salerno, L.; Foresti, R.; Drago, F.; Bucolo, C. Effects of Novel Nitric Oxide-Releasing Molecules against Oxidative Stress on Retinal Pigmented Epithelial Cells. Oxid. Med. Cell. Longev. 2017, 2017, 1420892. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.E.; Gu, J.; Schau, M.; Bunn, H.F. Regulation of Hypoxia-Inducible Factor 1α Is Mediated by an O2-Dependent Degradation Domain Via the Ubiquitin-Proteasome Pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7987–7992. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.H.; Rue, E.A.; Semenza, G.L. Hypoxia-Inducible Factor 1 Is a Basic-Helix-Loop-Helix-Pas Heterodimer Regulated by Cellular O2 Tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Vadlapatla, R.K.; Vadlapudi, A.D.; Mitra, A.K. Hypoxia-Inducible Factor-1 (HIF-1): A Potential Target for Intervention in Ocular Neovascular Diseases. Curr. Drug Targets 2013, 14, 919–935. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.A.; Giaccia, A.J. Hypoxia, Gene Expression, and Metastasis. Cancer Metastasis Rev. 2007, 26, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Lukashev, D.; Ohta, A.; Sitkovsky, M. Hypoxia-Dependent Anti-Inflammatory Pathways in Protection of Cancerous Tissues. Cancer Metastasis Rev. 2007, 26, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.; Graham, C.H. Hypoxia-Driven Selection of the Metastatic Phenotype. Cancer Metastasis Rev. 2007, 26, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.M.; Groot, A.J.; van der Groep, P.; Sersansie, R.; Vooijs, M.; van Diest, P.J.; van Noorden, C.J.F.; Schlingemann, R.O.; Klaassen, I. Active Hif-1 in the Normal Human Retina. J. Histochem. Cytochem. 2010, 58, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Grimm, C.; Wenzel, A.; Groszer, M.; Mayser, H.; Seeliger, M.; Samardzija, M.; Bauer, C.; Gassmann, M.; Reme, C.E. HIF-1-Induced Erythropoietin in the Hypoxic Retina Protects against Light-Induced Retinal Degeneration. Nat. Med. 2002, 8, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Foresti, R.; Bucolo, C.; Platania, C.M.; Drago, F.; Dubois-Rande, J.L.; Motterlini, R. Nrf2 Activators Modulate Oxidative Stress Responses and Bioenergetic Profiles of Human Retinal Epithelial Cells Cultured in Normal or High Glucose Conditions. Pharmacol. Res. 2015, 99, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Cho, C.L.; Liang, C.L.; Chen, S.D.; Liliang, P.C.; Wang, S.Y.; Chen, H.J. Inhibition of the MEK/ERK Pathway Reduces Microglial Activation and Interleukin-1-β Expression in Spinal Cord Ischemia/Reperfusion Injury in Rats. J. Thorac. Cardiovasc. Surg. 2007, 133, 934–941. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, M.E.; Wang, W.; Chen, X.; Caberoy, N.B.; Guo, F.; Shen, C.; Ji, Y.; Tian, H.; Wang, H.; Chen, R.; et al. Secretogranin III as a Disease-Associated Ligand for Antiangiogenic Therapy of Diabetic Retinopathy. J. Exp. Med. 2017, 214, 1029–1047. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Zhang, X.M.; Chen, B.Y.; Yang, X.J. Vegf Activates Divergent Intracellular Signaling Components to Regulate Retinal Progenitor Cell Proliferation and Neuronal Differentiation. Development 2006, 133, 2201–2210. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.B.; Monick, M.M.; Hunninghake, G.W. Both Erk and P38 Kinases Are Necessary for Cytokine Gene Transcription. Am. J. Respir. Cell Mol. Biol. 1999, 20, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.S.; Huang, Q.; Gurel, Z.; Sorenson, C.M.; Sheibani, N. High Glucose Alters Retinal Astrocytes Phenotype through Increased Production of Inflammatory Cytokines and Oxidative Stress. PLoS ONE 2014, 9, e103148. [Google Scholar] [CrossRef] [PubMed]
- Al-Hussaini, H.; Kilarkaje, N. Effects of Diabetes on Retinal Pigment Epithelial Cell Proliferation and Mitogen-Activated Protein Kinase Signaling in Dark Agouti Rats. Exp. Toxicol. Pathol. 2015, 67, 117–124. [Google Scholar] [CrossRef] [PubMed]
- McVicar, C.M.; Hamilton, R.; Colhoun, L.M.; Gardiner, T.A.; Brines, M.; Cerami, A.; Stitt, A.W. Intervention with an Erythropoietin-Derived Peptide Protects against Neuroglial and Vascular Degeneration During Diabetic Retinopathy. Diabetes 2011, 60, 2995–3005. [Google Scholar] [CrossRef] [PubMed]
- Rungger-Brandle, E.; Dosso, A.A.; Leuenberger, P.M. Glial Reactivity, an Early Feature of Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1971–1980. [Google Scholar]
- Chen, X.; Zhou, H.; Gong, Y.; Wei, S.; Zhang, M. Early Spatiotemporal Characterization of Microglial Activation in the Retinas of Rats with Streptozotocin-Induced Diabetes. Graefe’s Arch. Clin. Exp. Ophthalmol. 2015, 253, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; Todd, K.G. Differential Regulation of Trophic and Proinflammatory Microglial Effectors Is Dependent on Severity of Neuronal Injury. Glia 2008, 56, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Roque, R.S.; Imperial, C.J.; Caldwell, R.B. Microglial Cells Invade the Outer Retina as Photoreceptors Degenerate in Royal College of Surgeons Rats. Investig. Ophthalmol. Vis. Sci. 1996, 37, 196–203. [Google Scholar]
- Hughes, E.H.; Schlichtenbrede, F.C.; Murphy, C.C.; Sarra, G.M.; Luthert, P.J.; Ali, R.R.; Dick, A.D. Generation of Activated Sialoadhesin-Positive Microglia During Retinal Degeneration. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2229–2234. [Google Scholar] [CrossRef]
- Zeng, H.Y.; Zhu, X.A.; Zhang, C.; Yang, L.P.; Wu, L.M.; Tso, M.O. Identification of Sequential Events and Factors Associated with Microglial Activation, Migration, and Cytotoxicity in Retinal Degeneration in Rd Mice. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2992–2999. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.F.; Streilein, J.W. Light-Induced Migration of Retinal Microglia into the Subretinal Space. Investig. Ophthalmol. Vis. Sci. 2001, 42, 3301–3310. [Google Scholar]
- Karlstetter, M.; Scholz, R.; Rutar, M.; Wong, W.T.; Provis, J.M.; Langmann, T. Retinal Microglia: Just Bystander or Target for Therapy? Prog. Retin. Eye Res. 2015, 45, 30–57. [Google Scholar] [CrossRef] [PubMed]
- Ascaso, F.J.; Huerva, V.; Grzybowski, A. The Role of Inflammation in the Pathogenesis of Macular Edema Secondary to Retinal Vascular Diseases. Mediat. Inflamm. 2014, 2014, 432685. [Google Scholar] [CrossRef] [PubMed]
- Khalfaoui, T.; Lizard, G.; Ouertani-Meddeb, A. Adhesion Molecules (ICAM-1 and VCAM-1) and Diabetic Retinopathy in Type 2 Diabetes. J. Mol. Histol. 2008, 39, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Grant, M.B.; Afzal, A.; Spoerri, P.; Pan, H.; Shaw, L.C.; Mames, R.N. The Role of Growth Factors in the Pathogenesis of Diabetic Retinopathy. Expert Opin. Investig. Drugs 2004, 13, 1275–1293. [Google Scholar] [CrossRef] [PubMed]
- Schroder, S.; Palinski, W.; Schmid-Schonbein, G.W. Activated Monocytes and Granulocytes, Capillary Nonperfusion, and Neovascularization in Diabetic Retinopathy. Am. J. Pathol. 1991, 139, 81–100. [Google Scholar] [PubMed]
- Adamis, A.P. Is Diabetic Retinopathy an Inflammatory Disease? Br. J. Ophthalmol. 2002, 86, 363–365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gerhardinger, C.; Lorenzi, M. Early Complement Activation and Decreased Levels of Glycosylphosphatidylinositol-Anchored Complement Inhibitors in Human and Experimental Diabetic Retinopathy. Diabetes 2002, 51, 3499–3504. [Google Scholar] [CrossRef] [PubMed]
- Joussen, A.M.; Poulaki, V.; Mitsiades, N.; Cai, W.Y.; Suzuma, I.; Pak, J.; Ju, S.T.; Rook, S.L.; Esser, P.; Mitsiades, C.S.; et al. Suppression of Fas-Fasl-Induced Endothelial Cell Apoptosis Prevents Diabetic Blood-Retinal Barrier Breakdown in a Model of Streptozotocin-Induced Diabetes. FASEB J. 2003, 17, 76–78. [Google Scholar] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-Mediated Neurotoxicity: Uncovering the Molecular Mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Kern, T.S. Contributions of Inflammatory Processes to the Development of the Early Stages of Diabetic Retinopathy. Exp. Diabetes Res. 2007, 2007, 95103. [Google Scholar] [CrossRef] [PubMed]
- Kaul, K.; Hodgkinson, A.; Tarr, J.M.; Kohner, E.M.; Chibber, R. Is Inflammation a Common Retinal-Renal-Nerve Pathogenic Link in Diabetes? Curr. Diabetes Rev. 2010, 6, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Kojima, S.; Yamada, T.; Tamai, M. Quantitative Analysis of Interleukin-6 in Vitreous from Patients with Proliferative Vitreoretinal Diseases. Jpn. J. Ophthalmol. 2001, 45, 40–45. [Google Scholar] [CrossRef]
- Ma, W.; Wong, W.T. Aging Changes in Retinal Microglia and Their Relevance to Age-Related Retinal Disease. Adv. Exp. Med. Biol. 2016, 854, 73–78. [Google Scholar] [PubMed]
- Zhang, L.; Nair, A.; Krady, K.; Corpe, C.; Bonneau, R.H.; Simpson, I.A.; Vannucci, S.J. Estrogen Stimulates Microglia and Brain Recovery from Hypoxia-Ischemia in Normoglycemic but Not Diabetic Female Mice. J. Clin. Investig. 2004, 113, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, S.; Xia, X. Role of Intravitreal Inflammatory Cytokines and Angiogenic Factors in Proliferative Diabetic Retinopathy. Curr. Eye Res. 2012, 37, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Carmo, A.; Cunha-Vaz, J.G.; Carvalho, A.P.; Lopes, M.C. l-Arginine Transport in Retinas from Streptozotocin Diabetic Rats: Correlation with the Level of Il-1β and No Synthase Activity. Vis. Res. 1999, 39, 3817–3823. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Odenbach, S. Role of Interleukin-1β in the Pathogenesis of Diabetic Retinopathy. Br. J. Ophthalmol. 2004, 88, 1343–1347. [Google Scholar] [CrossRef] [PubMed]
- Gerhardinger, C.; Costa, M.B.; Coulombe, M.C.; Toth, I.; Hoehn, T.; Grosu, P. Expression of Acute-Phase Response Proteins in Retinal Muller Cells in Diabetes. Investig. Ophthalmol. Vis. Sci. 2005, 46, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, C.; Agardh, C.D.; Agardh, E. Profile of Intraocular Tumour Necrosis Factor-α and Interleukin-6 in Diabetic Subjects with Different Degrees of Diabetic Retinopathy. Acta Ophthalmol. 2013, 91, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Sohn, H.J.; Han, D.H.; Kim, I.T.; Oh, I.K.; Kim, K.H.; Lee, D.Y.; Nam, D.H. Changes in Aqueous Concentrations of Various Cytokines after Intravitreal Triamcinolone Versus Bevacizumab for Diabetic Macular Edema. Am. J. Ophthalmol. 2011, 152, 686–694. [Google Scholar] [CrossRef] [PubMed]
- Van Hoek, M.; Dehghan, A.; Zillikens, M.C.; Hofman, A.; Witteman, J.C.; Sijbrands, E.J. An RBP4 Promoter Polymorphism Increases Risk of Type 2 Diabetes. Diabetologia 2008, 51, 1423–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munkhtulga, L.; Nagashima, S.; Nakayama, K.; Utsumi, N.; Yanagisawa, Y.; Gotoh, T.; Omi, T.; Kumada, M.; Zolzaya, K.; Lkhagvasuren, T.; et al. Regulatory SNP in the RBP4 Gene Modified the Expression in Adipocytes and Associated with Bmi. Obesity 2010, 18, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Otalora, L.; Martin, A.A.; Moiseyev, G.; Vanlandingham, P.; Wang, Q.; Farjo, R.; Yeganeh, A.; Quiambao, A.; Farjo, K.M. Transgenic Mice Overexpressing Serum Retinol-Binding Protein Develop Progressive Retinal Degeneration through a Retinoid-Independent Mechanism. Mol. Cell. Biol. 2015, 35, 2771–2789. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Boulton, M. The Pathogenesis of Diabetic Retinopathy: Old Concepts and New Questions. Eye (Lond.) 2002, 16, 242–260. [Google Scholar] [CrossRef] [PubMed]
- Joussen, A.M.; Poulaki, V.; Mitsiades, N.; Kirchhof, B.; Koizumi, K.; Dohmen, S.; Adamis, A.P. Nonsteroidal Anti-Inflammatory Drugs Prevent Early Diabetic Retinopathy via TNFα Suppression. FASEB J. 2002, 16, 438–440. [Google Scholar] [PubMed]
- Behl, Y.; Krothapalli, P.; Desta, T.; DiPiazza, A.; Roy, S.; Graves, D.T. Diabetes-Enhanced Tumor Necrosis Factor-α Production Promotes Apoptosis and the Loss of Retinal Microvascular Cells in Type 1 and Type 2 Models of Diabetic Retinopathy. Am. J. Pathol. 2008, 172, 1411–1418. [Google Scholar] [CrossRef] [PubMed]
- Joussen, A.M.; Murata, T.; Tsujikawa, A.; Kirchhof, B.; Bursell, S.E.; Adamis, A.P. Leukocyte-Mediated Endothelial Cell Injury and Death in the Diabetic Retina. Am. J. Pathol. 2001, 158, 147–152. [Google Scholar] [CrossRef]
- Nawaz, M.I.; van Raemdonck, K.; Mohammad, G.; Kangave, D.; van Damme, J.; El-Asrar, A.M.A.; Struyf, S. Autocrine CCL2, CXCL4, CXCL9 and CXCL10 Signal in Retinal Endothelial Cells and Are Enhanced in Diabetic Retinopathy. Exp. Eye Res. 2013, 109, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, C.; Segura, R.M.; Fonollosa, A.; Carrasco, E.; Francisco, G.; Simo, R. Interleukin-8, Monocyte Chemoattractant Protein-1 and IL-10 in the Vitreous Fluid of Patients with Proliferative Diabetic Retinopathy. Diabet. Med. 2005, 22, 719–722. [Google Scholar] [CrossRef] [PubMed]
- Cardona, S.M.; Mendiola, A.S.; Yang, Y.C.; Adkins, S.L.; Torres, V.; Cardona, A.E. Disruption of Fractalkine Signaling Leads to Microglial Activation and Neuronal Damage in the Diabetic Retina. ASN Neuro 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Simo, R.; Hernandez, C. Novel Approaches for Treating Diabetic Retinopathy Based on Recent Pathogenic Evidence. Prog. Retin. Eye Res. 2015, 48, 160–180. [Google Scholar] [CrossRef] [PubMed]
- Simo, R.; Sundstrom, J.M.; Antonetti, D.A. Ocular Anti-VEGF Therapy for Diabetic Retinopathy: The Role of Vegf in the Pathogenesis of Diabetic Retinopathy. Diabetes Care 2014, 37, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.I.; Hykin, P.G.; Gregor, Z.J.; Boulton, M.; Cree, I.A. Angiopoietin Concentrations in Diabetic Retinopathy. Br. J. Ophthalmol. 2005, 89, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, S.; Srinivasan, R.; Maestas, J.; McGuire, P.G.; Das, A. A Potential Role for Angiopoietin 2 in the Regulation of the Blood-Retinal Barrier in Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3784–3791. [Google Scholar] [CrossRef] [PubMed]
- Deliyanti, D.; Wilkinson-Berka, J.L. Inhibition of Nox1/4 with Gkt137831: A Potential Novel Treatment to Attenuate Neuroglial Cell Inflammation in the Retina. J. Neuroinflamm. 2015, 12, 136. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, G.; Kowluru, R.A. Novel Role of Mitochondrial Matrix Metalloproteinase-2 in the Development of Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3832–3841. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, G.; Kowluru, R.A. Matrix Metalloproteinase-2 in the Development of Diabetic Retinopathy and Mitochondrial Dysfunction. Lab. Investig. 2010, 90, 1365–1372. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, G.; Kowluru, R.A. Diabetic Retinopathy and Signaling Mechanism for Activation of Matrix Metalloproteinase-9. J. Cell. Physiol. 2012, 227, 1052–1061. [Google Scholar] [CrossRef] [PubMed]
- Giebel, S.J.; Menicucci, G.; McGuire, P.G.; Das, A. Matrix Metalloproteinases in Early Diabetic Retinopathy and Their Role in Alteration of the Blood-Retinal Barrier. Lab. Investig. 2005, 85, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, L.K.; Addepalli, V. Attenuation of Diabetic Retinopathy by Enhanced Inhibition of MMP-2 and MMP-9 Using Aspirin and Minocycline in Streptozotocin-Diabetic Rats. Am. J. Transl. Res. 2010, 2, 181–189. [Google Scholar] [PubMed]
- Kowluru, R.A.; Kanwar, M. Oxidative Stress and the Development of Diabetic Retinopathy: Contributory Role of Matrix Metalloproteinase-2. Free Radic. Biol. Med. 2009, 46, 1677–1685. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.H.; Park, S.W.; Kim, K.J.; Bae, J.S.; Lee, E.H.; Paek, S.H.; Kim, S.U.; Ye, S.; Kim, J.H.; Cho, C.H. Endothelial STAT3 Activation Increases Vascular Leakage through Downregulating Tight Junction Proteins: Implications for Diabetic Retinopathy. J. Cell. Physiol. 2017, 232, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
- Ye, E.A.; Steinle, J.J. miR-146a Suppresses STAT3/VEGF Pathways and Reduces Apoptosis through IL-6 Signaling in Primary Human Retinal Microvascular Endothelial Cells in High Glucose Conditions. Vis. Res. 2017, 139, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, Y.; Zhou, T.; Zhou, K.K.; Mott, R.; Wu, M.; Boulton, M.; Lyons, T.J.; Gao, G.; Ma, J.X. Activation of the Wnt Pathway Plays a Pathogenic Role in Diabetic Retinopathy in Humans and Animal Models. Am. J. Pathol. 2009, 175, 2676–2685. [Google Scholar] [CrossRef] [PubMed]
- Abu el Asrar, A.M.; Maimone, D.; Morse, P.H.; Gregory, S.; Reder, A.T. Cytokines in the Vitreous of Patients with Proliferative Diabetic Retinopathy. Am. J. Ophthalmol. 1992, 114, 731–736. [Google Scholar] [CrossRef]
- Patel, J.I.; Saleh, G.M.; Hykin, P.G.; Gregor, Z.J.; Cree, I.A. Concentration of Haemodynamic and Inflammatory Related Cytokines in Diabetic Retinopathy. Eye 2008, 22, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Yuuki, T.; Kanda, T.; Kimura, Y.; Kotajima, N.; Tamura, J.; Kobayashi, I.; Kishi, S. Inflammatory Cytokines in Vitreous Fluid and Serum of Patients with Diabetic Vitreoretinopathy. J. Diabetes Complicat. 2001, 15, 257–259. [Google Scholar] [CrossRef]
- Doganay, S.; Evereklioglu, C.; Er, H.; Turkoz, Y.; Sevinc, A.; Mehmet, N.; Savli, H. Comparison of Serum No, TNFα, IL-1β, SIL-2r, IL-6 and IL-8 Levels with Grades of Retinopathy in Patients with Diabetes Mellitus. Eye 2002, 16, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Schram, M.T.; Chaturvedi, N.; Schalkwijk, C.G.; Fuller, J.H.; Stehouwer, C.D. Markers of Inflammation Are Cross-Sectionally Associated with Microvascular Complications and Cardiovascular Disease in Type 1 Diabetes—The Eurodiab Prospective Complications Study. Diabetologia 2005, 48, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Demircan, N.; Safran, B.G.; Soylu, M.; Ozcan, A.A.; Sizmaz, S. Determination of Vitreous Interleukin-1 (IL-1) and Tumour Necrosis Factor (TNF) Levels in Proliferative Diabetic Retinopathy. Eye 2006, 20, 1366–1369. [Google Scholar] [CrossRef] [PubMed]
- Bogdanov, P.; Corraliza, L.; Villena, J.A.; Carvalho, A.R.; Garcia-Arumi, J.; Ramos, D.; Ruberte, J.; Simo, R.; Hernandez, C. The db/db Mouse: A Useful Model for the Study of Diabetic Retinal Neurodegeneration. PLoS ONE 2014, 9, e97302. [Google Scholar] [CrossRef] [PubMed]
- Funatsu, H.; Yamashita, H.; Noma, H.; Mimura, T.; Yamashita, T.; Hori, S. Increased Levels of Vascular Endothelial Growth Factor and Interleukin-6 in the Aqueous Humor of Diabetics with Macular Edema. Am. J. Ophthalmol. 2002, 133, 70–77. [Google Scholar] [CrossRef]
- Jin, K.L.; Mao, X.O.; Greenberg, D.A. Vascular Endothelial Growth Factor: Direct Neuroprotective Effect in in Vitro Ischemia. Proc. Natl. Acad. Sci. USA 2000, 97, 10242–10247. [Google Scholar] [CrossRef] [PubMed]
- Sondell, M.; Lundborg, G.; Kanje, M. Vascular Endothelial Growth Factor Has Neurotrophic Activity and Stimulates Axonal Outgrowth, Enhancing Cell Survival and Schwann Cell Proliferation in the Peripheral Nervous System. J. Neurosci. 1999, 19, 5731–5740. [Google Scholar] [PubMed]
- Goldberg, M.A.; Schneider, T.J. Similarities between the Oxygen-Sensing Mechanisms Regulating the Expression of Vascular Endothelial Growth Factor and Erythropoietin. J. Biol. Chem. 1994, 269, 4355–4359. [Google Scholar] [PubMed]
- Aiello, L.P.; Bursell, S.E.; Clermont, A.; Duh, E.; Ishii, H.; Takagi, C.; Mori, F.; Ciulla, T.A.; Ways, K.; Jirousek, M.; et al. Vascular Endothelial Growth Factor-Induced Retinal Permeability Is Mediated by Protein Kinase C in Vivo and Suppressed by an Orally Effective β-Isoform-Selective Inhibitor. Diabetes 1997, 46, 1473–1480. [Google Scholar] [CrossRef] [PubMed]
- Aiello, L.P.; Wong, J.S. Role of Vascular Endothelial Growth Factor in Diabetic Vascular Complications. Kidney Int. Suppl. 2000, 77, S113–S119. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, X.; Elliott, M.H.; Zhu, M.; Le, Y.Z. Muller Cell-Derived VEGF Is Essential for Diabetes-Induced Retinal Inflammation and Vascular Leakage. Diabetes 2010, 59, 2297–2305. [Google Scholar] [CrossRef] [PubMed]
- Portillo, J.A.; Greene, J.A.; Okenka, G.; Miao, Y.; Sheibani, N.; Kern, T.S.; Subauste, C.S. Cd40 Promotes the Development of Early Diabetic Retinopathy in Mice. Diabetologia 2014, 57, 2222–2231. [Google Scholar] [CrossRef] [PubMed]
- Aveleira, C.A.; Lin, C.M.; Abcouwer, S.F.; Ambrosio, A.F.; Antonetti, D.A. TNFα Signals through Pkczeta/Nf-κb to Alter the Tight Junction Complex and Increase Retinal Endothelial Cell Permeability. Diabetes 2010, 59, 2872–2882. [Google Scholar] [CrossRef] [PubMed]
- Costa, G.N.; Vindeirinho, J.; Cavadas, C.; Ambrosio, A.F.; Santos, P.F. Contribution of TNF Receptor 1 to Retinal Neural Cell Death Induced by Elevated Glucose. Mol. Cell. Neurosci. 2012, 50, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Gharaee-Kermani, M.; Denholm, E.M.; Phan, S.H. Costimulation of Fibroblast Collagen and Transforming Growth Factor β1 Gene Expression by Monocyte Chemoattractant Protein-1 Via Specific Receptors. J. Biol. Chem. 1996, 271, 17779–17784. [Google Scholar] [CrossRef] [PubMed]
- Dong, N.; Li, X.; Xiao, L.; Yu, W.; Wang, B.; Chu, L. Upregulation of Retinal Neuronal MCP-1 in the Rodent Model of Diabetic Retinopathy and Its Function in Vitro. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7567–7575. [Google Scholar] [CrossRef] [PubMed]
- Koistinaho, M.; Koistinaho, J. Role of P38 and P44/42 Mitogen-Activated Protein Kinases in Microglia. Glia 2002, 40, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.S.; El-Remessy, A.B.; Matragoon, S.; Zhang, W.; Patel, Y.; Khan, S.; Al-Gayyar, M.M.; El-Shishtawy, M.M.; Liou, G.I. Retinal Microglial Activation and Inflammation Induced by Amadori-Glycated Albumin in a Rat Model of Diabetes. Diabetes 2011, 60, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Zabel, M.K.; Zhao, L.; Zhang, Y.; Gonzalez, S.R.; Ma, W.; Wang, X.; Fariss, R.N.; Wong, W.T. Microglial Phagocytosis and Activation Underlying Photoreceptor Degeneration Is Regulated by CX3CL1-CX3CR1 Signaling in a Mouse Model of Retinitis Pigmentosa. Glia 2016, 64, 1479–1491. [Google Scholar] [CrossRef] [PubMed]
- Beli, E.; Dominguez, J.M., 2nd; Hu, P.; Thinschmidt, J.S.; Caballero, S.; Calzi, S.L.; Luo, D.; Shanmugam, S.; Salazar, T.E.; Duan, Y.; et al. CX3CR1 Deficiency Accelerates the Development of Retinopathy in a Rodent Model of Type 1 Diabetes. J. Mol. Med. 2016, 94, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Mendiola, A.S.; Garza, R.; Cardona, S.M.; Mythen, S.A.; Lira, S.A.; Akassoglou, K.; Cardona, A.E. Fractalkine Signaling Attenuates Perivascular Clustering of Microglia and Fibrinogen Leakage During Systemic Inflammation in Mouse Models of Diabetic Retinopathy. Front. Cell. Neurosci. 2016, 10, 303. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, H.; Navitskaya, S.; O’Reilly, S.; Gallimore, J.; Mize, H.; Beli, E.; Wang, Q.; Kady, N.; Huang, C.; Blanchard, G.J.; et al. Role of Acid Sphingomyelinase in Shifting the Balance between Proinflammatory and Reparative Bone Marrow Cells in Diabetic Retinopathy. Stem Cells 2016, 34, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Kady, N.; Yan, Y.; Salazar, T.; Wang, Q.; Chakravarthy, H.; Huang, C.; Beli, E.; Navitskaya, S.; Grant, M.; Busik, J. Increase in Acid Sphingomyelinase Level in Human Retinal Endothelial Cells and Cd34+ Circulating Angiogenic Cells Isolated from Diabetic Individuals Is Associated with Dysfunctional Retinal Vasculature and Vascular Repair Process in Diabetes. J. Clin. Lipidol. 2017, 11, 694–703. [Google Scholar] [CrossRef] [PubMed]
- Noda, K.; Ishida, S.; Inoue, M.; Obata, K.; Oguchi, Y.; Okada, Y.; Ikeda, E. Production and Activation of Matrix Metalloproteinase-2 in Proliferative Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2163–2170. [Google Scholar] [CrossRef]
- Zhou, T.; Hu, Y.; Chen, Y.; Zhou, K.K.; Zhang, B.; Gao, G.; Ma, J.X. The Pathogenic Role of the Canonical Wnt Pathway in Age-Related Macular Degeneration. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4371–4379. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Lee, K.; Zhang, B.; Zhou, T.; He, X.; Gao, G.; Murray, A.R.; Ma, J.X. Identification of a Novel Inhibitor of the Canonical Wnt Pathway. Mol. Cell. Biol. 2011, 31, 3038–3051. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Hu, Y.; Ding, L.; Chen, Y.; Takahashi, Y.; Mott, R.; Ma, J.X. Therapeutic Potential of a Monoclonal Antibody Blocking the Wnt Pathway in Diabetic Retinopathy. Diabetes 2012, 61, 2948–2957. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cheng, R.; Lee, K.; Tyagi, P.; Ding, L.; Kompella, U.B.; Chen, J.; Xu, X.; Ma, J.X. Nanoparticle-Mediated Expression of a Wnt Pathway Inhibitor Ameliorates Ocular Neovascularization. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Qiu, F.; He, J.; Zhou, Y.; Bai, X.; Wu, G.; Wang, X.; Liu, Z.; Chen, Y.; Ma, J.X.; Liu, Z. Plasma and Vitreous Fluid Levels of Dickkopf-1 in Patients with Diabetic Retinopathy. Eye 2014, 28, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Jiang, J.; Wang, S.; Xia, X. Dkk1 Inhibits Proliferation and Migration in Human Retinal Pigment Epithelial Cells Via the Wnt/Β-Catenin Signaling Pathway. Exp. Ther. Med. 2016, 12, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Hammes, H.P.; Federoff, H.J.; Brownlee, M. Nerve Growth Factor Prevents Both Neuroretinal Programmed Cell Death and Capillary Pathology in Experimental Diabetes. Mol. Med. 1995, 1, 527–534. [Google Scholar] [PubMed]
- Kerrigan, L.A.; Zack, D.J.; Quigley, H.A.; Smith, S.D.; Pease, M.E. Tunel-Positive Ganglion Cells in Human Primary Open-Angle Glaucoma. Arch. Ophthalmol. 1997, 115, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, H.W.; Verbraak, F.D.; Kok, P.H.; Stehouwer, M.; Garvin, M.K.; Sonka, M.; DeVries, J.H.; Schlingemann, R.O.; Abramoff, M.D. Early Neurodegeneration in the Retina of Type 2 Diabetic Patients. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2715–2719. [Google Scholar] [CrossRef] [PubMed]
- Simo, R.; Hernandez, C. Neurodegeneration in the Diabetic Eye: New Insights and Therapeutic Perspectives. Trends Endocrinol. Metab. 2014, 25, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Parisi, V.; Uccioli, L. Visual Electrophysiological Responses in Persons with Type 1 Diabetes. Diabetes Metab. Res. Rev. 2001, 17, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Ghirlanda, G.; di Leo, M.A.; Caputo, S.; Cercone, S.; Greco, A.V. From Functional to Microvascular Abnormalities in Early Diabetic Retinopathy. Diabetes Metab. Rev. 1997, 13, 15–35. [Google Scholar] [CrossRef]
- Bresnick, G.H. Diabetic Retinopathy Viewed as a Neurosensory Disorder. Arch. Ophthalmol. 1986, 104, 989–990. [Google Scholar] [CrossRef] [PubMed]
- Greenstein, V.C.; Shapiro, A.; Zaidi, Q.; Hood, D.C. Psychophysical Evidence for Post-Receptoral Sensitivity Loss in Diabetics. Investig. Ophthalmol. Vis. Sci. 1992, 33, 2781–2790. [Google Scholar]
- Bearse, M.A., Jr.; Han, Y.; Schneck, M.E.; Barez, S.; Jacobsen, C.; Adams, A.J. Local Multifocal Oscillatory Potential Abnormalities in Diabetes and Early Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2004, 45, 3259–3265. [Google Scholar] [CrossRef] [PubMed]
- Wolter, J.R. Diabetic Retinopathy. Am. J. Ophthalmol. 1961, 51, 1123–1141. [Google Scholar] [CrossRef]
- Bloodworth, J.M., Jr. Diabetic Retinopathy. Diabetes 1962, 11, 1–22. [Google Scholar] [PubMed]
- Araszkiewicz, A.; Zozulinska-Ziolkiewicz, D.; Meller, M.; Bernardczyk-Meller, J.; Pilacinski, S.; Rogowicz-Frontczak, A.; Naskret, D.; Wierusz-Wysocka, B. Neurodegeneration of the Retina in Type 1 Diabetic Patients. Polskie Archiwum Medycyny Wewnętrznej 2012, 122, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Kern, T.S.; Barber, A.J. Retinal Ganglion Cells in Diabetes. J. Physiol. 2008, 586, 4401–4408. [Google Scholar] [CrossRef] [PubMed]
- Gillies, M.C.; Su, T.; Stayt, J.; Simpson, J.M.; Naidoo, D.; Salonikas, C. Effect of High Glucose on Permeability of Retinal Capillary Endothelium in Vitro. Investig. Ophthalmol. Vis. Sci. 1997, 38, 635–642. [Google Scholar]
- Kusari, J.; Zhou, S.; Padillo, E.; Clarke, K.G.; Gil, D.W. Effect of Memantine on Neuroretinal Function and Retinal Vascular Changes of Streptozotocin-Induced Diabetic Rats. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5152–5159. [Google Scholar] [CrossRef] [PubMed]
- Robinson, R.; Barathi, V.A.; Chaurasia, S.S.; Wong, T.Y.; Kern, T.S. Update on Animal Models of Diabetic Retinopathy: From Molecular Approaches to Mice and Higher Mammals. Dis. Model. Mech. 2012, 5, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, E.; Hernandez, C.; de Torres, I.; Farres, J.; Simo, R. Lowered Cortistatin Expression Is an Early Event in the Human Diabetic Retina and Is Associated with Apoptosis and Glial Activation. Mol. Vis. 2008, 14, 1496–1502. [Google Scholar] [PubMed]
- Garcia-Ramirez, M.; Hernandez, C.; Villarroel, M.; Canals, F.; Alonso, M.A.; Fortuny, R.; Masmiquel, L.; Navarro, A.; Garcia-Arumi, J.; Simo, R. Interphotoreceptor Retinoid-Binding Protein (IRBP) Is Downregulated at Early Stages of Diabetic Retinopathy. Diabetologia 2009, 52, 2633–2641. [Google Scholar] [CrossRef] [PubMed]
- Sohn, E.H.; van Dijk, H.W.; Jiao, C.; Kok, P.H.; Jeong, W.; Demirkaya, N.; Garmager, A.; Wit, F.; Kucukevcilioglu, M.; van Velthoven, M.E.; et al. Retinal Neurodegeneration May Precede Microvascular Changes Characteristic of Diabetic Retinopathy in Diabetes Mellitus. Proc. Natl. Acad. Sci. USA 2016, 113, E2655–E2664. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Ma, J.; Li, Y.; Zhang, Z. Association between Retinal Neuronal Degeneration and Visual Function Impairment in Type 2 Diabetic Patients without Diabetic Retinopathy. Sci. China Life Sci. 2015, 58, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Lopes de Faria, J.M.; Russ, H.; Costa, V.P. Retinal Nerve Fibre Layer Loss in Patients with Type 1 Diabetes Mellitus without Retinopathy. Br. J. Ophthalmol. 2002, 86, 725–728. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, S.E. Erg in Juvenile Diabetics: A Prognostic Study. In Symposium on the Treatment of Diabetic Retinopathy; Goldberg, M., Fine, S.L., Eds.; Department of Health, Education and Welfare: Arlington, VA, USA, 1969; pp. 681–689. [Google Scholar]
- Santiago, A.R.; Gaspar, J.M.; Baptista, F.I.; Cristovao, A.J.; Santos, P.F.; Kamphuis, W.; Ambrosio, A.F. Diabetes Changes the Levels of Ionotropic Glutamate Receptors in the Rat Retina. Mol. Vis. 2009, 15, 1620–1630. [Google Scholar] [PubMed]
- Ng, Y.K.; Zeng, X.X.; Ling, E.A. Expression of Glutamate Receptors and Calcium-Binding Proteins in the Retina of Streptozotocin-Induced Diabetic Rats. Brain Res. 2004, 1018, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. The Pathobiology of Diabetic Complications: A Unifying Mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Tonkiss, J.; Roy, S. Aging Increases Retinal Vascular Lesions Characteristic of Early Diabetic Retinopathy. Biogerontology 2010, 11, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yanoff, M.; Liu, X.; Ye, X. Retinal Capillary Pericyte Apoptosis in Early Human Diabetic Retinopathy. Chin. Med. J. 1997, 110, 659–663. [Google Scholar] [PubMed]
- Barouch, F.C.; Miyamoto, K.; Allport, J.R.; Fujita, K.; Bursell, S.E.; Aiello, L.P.; Luscinskas, F.W.; Adamis, A.P. Integrin-Mediated Neutrophil Adhesion and Retinal Leukostasis in Diabetes. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1153–1158. [Google Scholar]
- Oosthuyse, B.; Moons, L.; Storkebaum, E.; Beck, H.; Nuyens, D.; Brusselmans, K.; van Dorpe, J.; Hellings, P.; Gorselink, M.; Heymans, S.; et al. Deletion of the Hypoxia-Response Element in the Vascular Endothelial Growth Factor Promoter Causes Motor Neuron Degeneration. Nat. Genet. 2001, 28, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Bek, T. Glial Cell Involvement in Vascular Occlusion of Diabetic Retinopathy. Acta Ophthalmol. Scand. 1997, 75, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Barot, M.; Gokulgandhi, M.R.; Patel, S.; Mitra, A.K. Microvascular Complications and Diabetic Retinopathy: Recent Advances and Future Implications. Future Med. Chem. 2013, 5, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Antonetti, D.A.; Barber, A.J.; Khin, S.; Lieth, E.; Tarbell, J.M.; Gardner, T.W. Vascular Permeability in Experimental Diabetes Is Associated with Reduced Endothelial Occludin Content: Vascular Endothelial Growth Factor Decreases Occludin in Retinal Endothelial Cells. Penn State Retina Research Group. Diabetes 1998, 47, 1953–1959. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.J.; Antonetti, D.A.; Gardner, T.W. Altered Expression of Retinal Occludin and Glial Fibrillary Acidic Protein in Experimental Diabetes. The Penn State Retina Research Group. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3561–3568. [Google Scholar]
- Lupo, G.; Motta, C.; Giurdanella, G.; Anfuso, C.D.; Alberghina, M.; Drago, F.; Salomone, S.; Bucolo, C. Role of Phospholipases A2 in Diabetic Retinopathy: In Vitro and in Vivo Studies. Biochem. Pharmacol. 2013, 86, 1603–1613. [Google Scholar] [CrossRef] [PubMed]
- Alder, V.A.; Su, E.N.; Yu, D.Y.; Cringle, S.J.; Yu, P.K. Diabetic Retinopathy: Early Functional Changes. Clin. Exp. Pharmacol. Physiol. 1997, 24, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Curtis, T.M.; Hamilton, R.; Yong, P.H.; McVicar, C.M.; Berner, A.; Pringle, R.; Uchida, K.; Nagai, R.; Brockbank, S.; Stitt, A.W. Müller Glial Dysfunction During Diabetic Retinopathy in Rats Is Linked to Accumulation of Advanced Glycation End-Products and Advanced Lipoxidation End-Products. Diabetologia 2011, 54, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, N.; Grigorian, R.A.; Tutela, A.; Zarbin, M.A. Diabetic Macular Edema: Pathogenesis and Treatment. Surv. Ophthalmol. 2009, 54, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Durham, J.T.; Herman, I.M. Microvascular Modifications in Diabetic Retinopathy. Curr. Diabetes Rep. 2011, 11, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Legacy, J.; Hanea, S.; Theoret, J.; Smith, P.D. Granulocyte Macrophage Colony-Stimulating Factor Promotes Regeneration of Retinal Ganglion Cells in Vitro through a Mammalian Target of Rapamycin-Dependent Mechanism. J. Neurosci. Res. 2013, 91, 771–779. [Google Scholar] [CrossRef] [PubMed]
- Sappington, R.M.; Chan, M.; Calkins, D.J. Interleukin-6 Protects Retinal Ganglion Cells from Pressure-Induced Death. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2932–2942. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.; Foulds, W.S.; Ling, E.-A. Hypoxia-Ischemia and Retinal Ganglion Cell Damage. Clin. Ophthalmol. 2008, 2, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Heiduschka, P.; Alex, A.F.; Niekamper, D.; Eter, N. Behaviour of CD11B-Positive Cells in an Animal Model of Laser-Induced Choroidal Neovascularisation. Ophthalmologica 2017, 237, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, A.; Seandel, M.; Kupriyanova, T.A.; Partridge, J.J.; Madsen, M.A.; Hahn-Dantona, E.A.; Quigley, J.P.; Deryugina, E.I. Proangiogenic Role of Neutrophil-Like Inflammatory Heterophils during Neovascularization Induced by Growth Factors and Human Tumor Cells. Blood 2006, 107, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. Il-1 Is Required for Tumor Invasiveness and Angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef] [PubMed]
- Naldini, A.; Leali, D.; Pucci, A.; Morena, E.; Carraro, F.; Nico, B.; Ribatti, D.; Presta, M. Cutting Edge: Il-1β Mediates the Proangiogenic Activity of Osteopontin-Activated Human Monocytes. J. Immunol. 2006, 177, 4267–4270. [Google Scholar] [CrossRef] [PubMed]
- Leali, D.; Dell’Era, P.; Stabile, H.; Sennino, B.; Chambers, A.F.; Naldini, A.; Sozzani, S.; Nico, B.; Ribatti, D.; Presta, M. Osteopontin (ETA-1) and Fibroblast Growth Factor-2 Cross-Talk in Angiogenesis. J. Immunol. 2003, 171, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Speyer, C.L.; Ward, P.A. Role of Endothelial Chemokines and Their Receptors During Inflammation. J. Investig. Surg. 2011, 24, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Sprague, A.H.; Khalil, R.A. Inflammatory Cytokines in Vascular Dysfunction and Vascular Disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Dor, Y.; Porat, R.; Keshet, E. Vascular Endothelial Growth Factor and Vascular Adjustments to Perturbations in Oxygen Homeostasis. Am. J. Physiol. Cell Physiol. 2001, 280, C1367–C1374. [Google Scholar] [CrossRef] [PubMed]
- Boyer, D.S.; Hopkins, J.J.; Sorof, J.; Ehrlich, J.S. Anti-Vascular Endothelial Growth Factor Therapy for Diabetic Macular Edema. Ther. Adv. Endocrinol. Metab. 2013, 4, 151–169. [Google Scholar] [CrossRef] [PubMed]
- Aplin, A.C.; Gelati, M.; Fogel, E.; Carnevale, E.; Nicosia, R.F. Angiopoietin-1 and Vascular Endothelial Growth Factor Induce Expression of Inflammatory Cytokines before Angiogenesis. Physiol. Genom. 2006, 27, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Angelo, L.S.; Kurzrock, R. Vascular Endothelial Growth Factor and Its Relationship to Inflammatory Mediators. Clin. Cancer Res. 2007, 13, 2825–2830. [Google Scholar] [CrossRef] [PubMed]
- Lieth, E.; LaNoue, K.F.; Antonetti, D.A.; Ratz, M. Diabetes Reduces Glutamate Oxidation and Glutamine Synthesis in the Retina. The Penn State Retina Research Group. Exp. Eye Res. 2000, 70, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, M.; Gerhardinger, C.; Lorenzi, M. Muller Cell Changes in Human Diabetic Retinopathy. Diabetes 1998, 47, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Ma, W.; Zhao, L.; Fariss, R.N.; Wong, W.T. Adaptive Muller Cell Responses to Microglial Activation Mediate Neuroprotection and Coordinate Inflammation in the Retina. J. Neuroinflamm. 2011, 8, 173. [Google Scholar] [CrossRef] [PubMed]
- Nagayach, A.; Patro, N.; Patro, I. Astrocytic and Microglial Response in Experimentally Induced Diabetic Rat Brain. Metab. Brain Dis. 2014, 29, 747–761. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, X.; Zhao, L.; Ma, W.; Rodriguez, I.R.; Fariss, R.N.; Wong, W.T. Macroglia-Microglia Interactions Via Tspo Signaling Regulates Microglial Activation in the Mouse Retina. J. Neurosci. 2014, 34, 3793–3806. [Google Scholar] [CrossRef] [PubMed]
- Reme, C.E.; Rol, P.; Grothmann, K.; Kaase, H.; Terman, M. Bright Light Therapy in Focus: Lamp Emission Spectra and Ocular Safety. Technol. Health Care 1996, 4, 403–413. [Google Scholar] [PubMed]
- Rozanowska, M.; Sarna, T. Light-Induced Damage to the Retina: Role of Rhodopsin Chromophore Revisited. Photochem. Photobiol. 2005, 81, 1305–1330. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, A.; Grimm, C.; Samardzija, M.; Reme, C.E. Molecular Mechanisms of Light-Induced Photoreceptor Apoptosis and Neuroprotection for Retinal Degeneration. Prog. Retin. Eye Res. 2005, 24, 275–306. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Liang, K.J.; Fariss, R.N.; Wong, W.T. Ex Vivo Dynamic Imaging of Retinal Microglia Using Time-Lapse Confocal Microscopy. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4169–4176. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Garcia, S.; Reichhart, N.; Hernandez-Matas, C.; Zabulis, X.; Kociok, N.; Brockmann, C.; Joussen, A.M.; Strauss, O. In Vivo Analysis of the Time and Spatial Activation Pattern of Microglia in the Retina Following Laser-Induced Choroidal Neovascularization. Exp. Eye Res. 2015, 139, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shen, J.K.; Lam, T.T.; Zeng, H.Y.; Chiang, S.K.; Yang, F.; Tso, M.O. Activation of Microglia and Chemokines in Light-Induced Retinal Degeneration. Mol. Vis. 2005, 11, 887–895. [Google Scholar] [PubMed]
- Song, D.; Sulewski, M.E., Jr.; Wang, C.; Song, J.; Bhuyan, R.; Sterling, J.; Clark, E.; Song, W.C.; Dunaief, J.L. Complement C5a Receptor Knockout Has Diminished Light-Induced Microglia/Macrophage Retinal Migration. Mol. Vis. 2017, 23, 210–218. [Google Scholar] [PubMed]
- Roodhart, J.M.; Langenberg, M.H.; Witteveen, E.; Voest, E.E. The Molecular Basis of Class Side Effects Due to Treatment with Inhibitors of the VEGF/VEGFR Pathway. Curr. Clin. Pharmacol. 2008, 3, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Zapata, M.J.; Marti-Carvajal, A.J.; Sola, I.; Pijoan, J.I.; Buil-Calvo, J.A.; Cordero, J.A.; Evans, J.R. Anti-Vascular Endothelial Growth Factor for Proliferative Diabetic Retinopathy. Cochrane Database Syst. Rev. 2014, CD008721. [Google Scholar] [CrossRef] [PubMed]
- Arevalo, J.F. Intravitreal Bevacizumab as Anti-Vascular Endothelial Growth Factor in the Management of Complications of Proliferative Diabetic Retinopathy. Med. Hypothesis Discov. Innov. Ophthalmol. 2013, 2, 20–24. [Google Scholar] [PubMed]
- Huang, H.; Parlier, R.; Shen, J.K.; Lutty, G.A.; Vinores, S.A. VEGF Receptor Blockade Markedly Reduces Retinal Microglia/Macrophage Infiltration into Laser-Induced CNV. PLoS ONE 2013, 8, e71808. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yoshida, S.; Kubo, Y.; Yoshimura, T.; Kobayashi, Y.; Nakama, T.; Yamaguchi, M.; Ishikawa, K.; Oshima, Y.; Ishibashi, T. Different Distributions of M1 and M2 Macrophages in a Mouse Model of Laser-Induced Choroidal Neovascularization. Mol. Med. Rep. 2017, 15, 3949–3956. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Wang, H.; Liu, Y.; Li, W.; Kim, D.; Huang, H. Blockade of Vascular Endothelial Growth Factor Receptor 1 Prevents Inflammation and Vascular Leakage in Diabetic Retinopathy. J. Ophthalmol. 2015, 2015, 605946. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.S.; Sun, J.K.; Aiello, L.P. Role of Steroids in the Management of Diabetic Macular EDEMA and Proliferative Diabetic Retinopathy. Semin. Ophthalmol. 2009, 24, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Regillo, C.D.; Callanan, D.G.; Do, D.V.; Fine, H.F.; Holekamp, N.M.; Kuppermann, B.D.; Singer, M.A.; Singh, R.P. Use of Corticosteroids in the Treatment of Patients with Diabetic Macular Edema Who Have a Suboptimal Response to Anti-Vegf: Recommendations of an Expert Panel. Ophthalmic Surg. Lasers Imaging Retina 2017, 48, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, S.; Gomi, F.; Oshima, Y.; Tohyama, M.; Tano, Y. Vascular Endothelial Growth Factor Reduced and Connective Tissue Growth Factor Induced by Triamcinolone in Arpe19 Cells under Oxidative Stress. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1062–1068. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Yang, L.; Li, H.; Gong, H.; Cheng, L.; Zheng, H.; Zhang, L.M.; Lan, Y. Downregulation of VEGF mRNA Expression by Triamcinolone Acetonide Acetate-Loaded Chitosan Derivative Nanoparticles in Human Retinal Pigment Epithelial Cells. Int. J. Nanomed. 2012, 7, 4649–4660. [Google Scholar]
- Shen, W.; Lee, S.R.; Araujo, J.; Chung, S.H.; Zhu, L.; Gillies, M.C. Effect of Glucocorticoids on Neuronal and Vascular Pathology in a Transgenic Model of Selective Muller Cell Ablation. Glia 2014, 62, 1110–1124. [Google Scholar] [CrossRef] [PubMed]
- McColl, A.; Bournazos, S.; Franz, S.; Perretti, M.; Morgan, B.P.; Haslett, C.; Dransfield, I. Glucocorticoids Induce Protein S-Dependent Phagocytosis of Apoptotic Neutrophils by Human Macrophages. J. Immunol. 2009, 183, 2167–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heasman, S.J.; Giles, K.M.; Ward, C.; Rossi, A.G.; Haslett, C.; Dransfield, I. Glucocorticoid-Mediated Regulation of Granulocyte Apoptosis and Macrophage Phagocytosis of Apoptotic Cells: Implications for the Resolution of Inflammation. J. Endocrinol. 2003, 178, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Glezer, I.; Simard, A.R.; Rivest, S. Neuroprotective Role of the Innate Immune System by Microglia. Neuroscience 2007, 147, 867–883. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; Todd, K.G. Hypoxia-Activated Microglial Mediators of Neuronal Survival Are Differentially Regulated by Tetracyclines. Glia 2006, 53, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Yrjanheikki, J.; Tikka, T.; Keinanen, R.; Goldsteins, G.; Chan, P.H.; Koistinaho, J. A Tetracycline Derivative, Minocycline, Reduces Inflammation and Protects against Focal Cerebral Ischemia with a Wide Therapeutic Window. Proc. Natl. Acad. Sci. USA 1999, 96, 13496–13500. [Google Scholar] [CrossRef] [PubMed]
- Abcouwer, S.F.; Lin, C.M.; Shanmugam, S.; Muthusamy, A.; Barber, A.J.; Antonetti, D.A. Minocycline Prevents Retinal Inflammation and Vascular Permeability Following Ischemia-Reperfusion Injury. J. Neuroinflamm. 2013, 10, 149. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.L.; Yu, A.C.; Lau, L.T.; Lee, C.; Le, M.W.; Zhu, X.; Tso, M.O. Minocycline Inhibits LPS-Induced Retinal Microglia Activation. Neurochem. Int. 2005, 47, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Cukras, C.A.; Petrou, P.; Chew, E.Y.; Meyerle, C.B.; Wong, W.T. Oral Minocycline for the Treatment of Diabetic Macular Edema (DME): Results of a Phase I/II Clinical Study. Investig. Ophthalol. Vis. Sci. 2012, 53, 3865–3874. [Google Scholar] [CrossRef] [PubMed]
- Douglas, Y.; Bhatwadekar, A.D.; Shaw, L.C.; Carnegie, D.; Caballero, S.; Li, Q.; Calzi, S.L.; Raizada, M.K.; Stitt, A.W.; Grant, M.B. Bone Marrow-CNS Connections: Implications in the Pathogenesis of Diabetic Retinopathy. Prog. Retin. Eye Res. 2012, 31, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Jantzie, L.L.; Cheung, P.Y.; Todd, K.G. Doxycycline Reduces Cleaved Caspase-3 and Microglial Activation in an Animal Model of Neonatal Hypoxia-Ischemia. J. Cereb. Blood Flow Metab. 2005, 25, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Scott, I.U.; Jackson, G.R.; Quillen, D.A.; Larsen, M.; Klein, R.; Liao, J.; Holfort, S.; Munch, I.C.; Gardner, T.W. Effect of Doxycycline vs. Placebo on Retinal Function and Diabetic Retinopathy Progression in Patients with Severe Nonproliferative or Non-High-Risk Proliferative Diabetic Retinopathy: A Randomized Clinical Trial. JAMA Ophthalmol. 2014, 132, 535–543. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The mouse retina is a model system to analyzing angiogenesis. The mouse retina is a robust tool to analyze in vivo angiogenesis. The avascular retinal vasculature develops gradually as vessels start to grow into the tissue. Starting at postnatal day 0 (P0), blood vessels branch out in a single plane until approximately P8, which allows monitoring of the vessel growth in an intact system including endothelial cells, pericytes, neurons, microglia, astrocytes and Müller cells. Genotype-specific alteration can be analyzed, which may provide insight into molecular and cellular mechanisms of the regulation of angiogenesis. (A) Overview of the retina vasculature stained with the endothelial cell marker platelet endothelial cell adhesion molecule (PECAM; CD31) at P5. The retinal leaves show the branching of the multiple retinal vasculatures. The vessels were stained with (B) PECAM and C ERG to mark endothelial cells and their nuclei, respectively. Scale bar in (A) 500 µm, in (B,C) 250 µm.
Figure 1.
The mouse retina is a model system to analyzing angiogenesis. The mouse retina is a robust tool to analyze in vivo angiogenesis. The avascular retinal vasculature develops gradually as vessels start to grow into the tissue. Starting at postnatal day 0 (P0), blood vessels branch out in a single plane until approximately P8, which allows monitoring of the vessel growth in an intact system including endothelial cells, pericytes, neurons, microglia, astrocytes and Müller cells. Genotype-specific alteration can be analyzed, which may provide insight into molecular and cellular mechanisms of the regulation of angiogenesis. (A) Overview of the retina vasculature stained with the endothelial cell marker platelet endothelial cell adhesion molecule (PECAM; CD31) at P5. The retinal leaves show the branching of the multiple retinal vasculatures. The vessels were stained with (B) PECAM and C ERG to mark endothelial cells and their nuclei, respectively. Scale bar in (A) 500 µm, in (B,C) 250 µm.
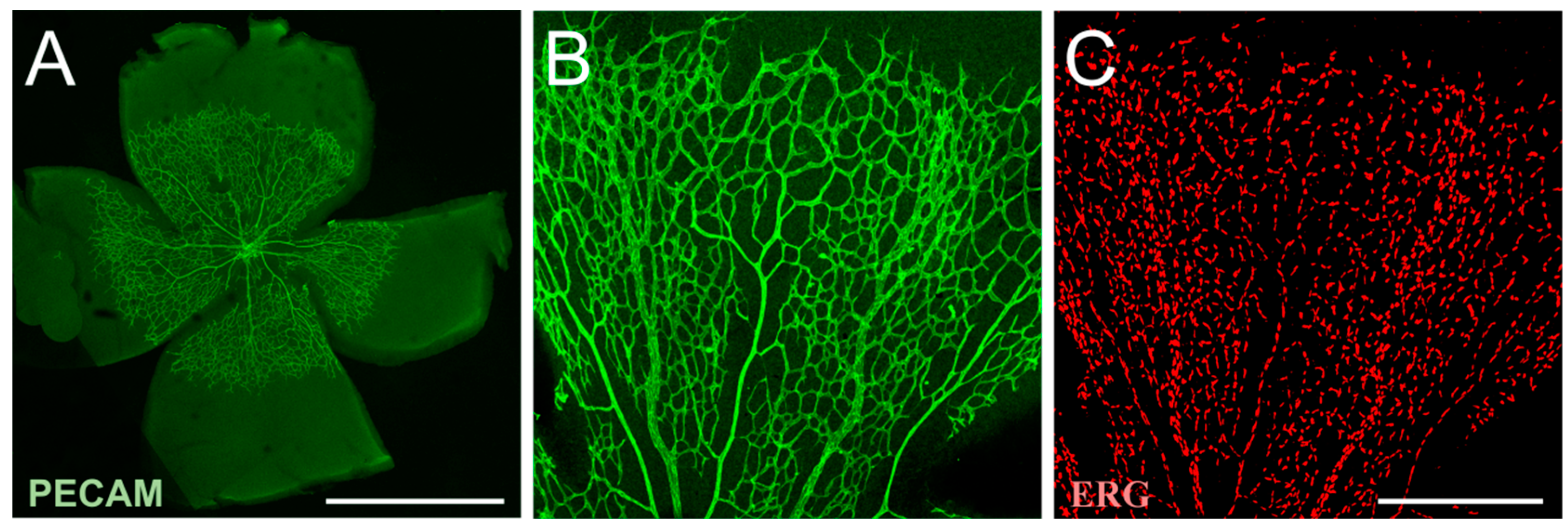
Figure 2.
Possible mechanisms underlying microglial activation and subsequent inflammatory responses in diabetic retinopathy. Ramified microglia are influenced by hyperglycemia, alterations in ischemia, vessel leakage and upregulation of various mediators. Microglia activation means morphological changes, proliferation and migration. This activation results in an inflammatory response, including downregulation (down arrow) of cytokine IL-10 and growth factors as well as upregulation (up arrow) of various cytokines, chemokines and neurotoxins.
Figure 2.
Possible mechanisms underlying microglial activation and subsequent inflammatory responses in diabetic retinopathy. Ramified microglia are influenced by hyperglycemia, alterations in ischemia, vessel leakage and upregulation of various mediators. Microglia activation means morphological changes, proliferation and migration. This activation results in an inflammatory response, including downregulation (down arrow) of cytokine IL-10 and growth factors as well as upregulation (up arrow) of various cytokines, chemokines and neurotoxins.
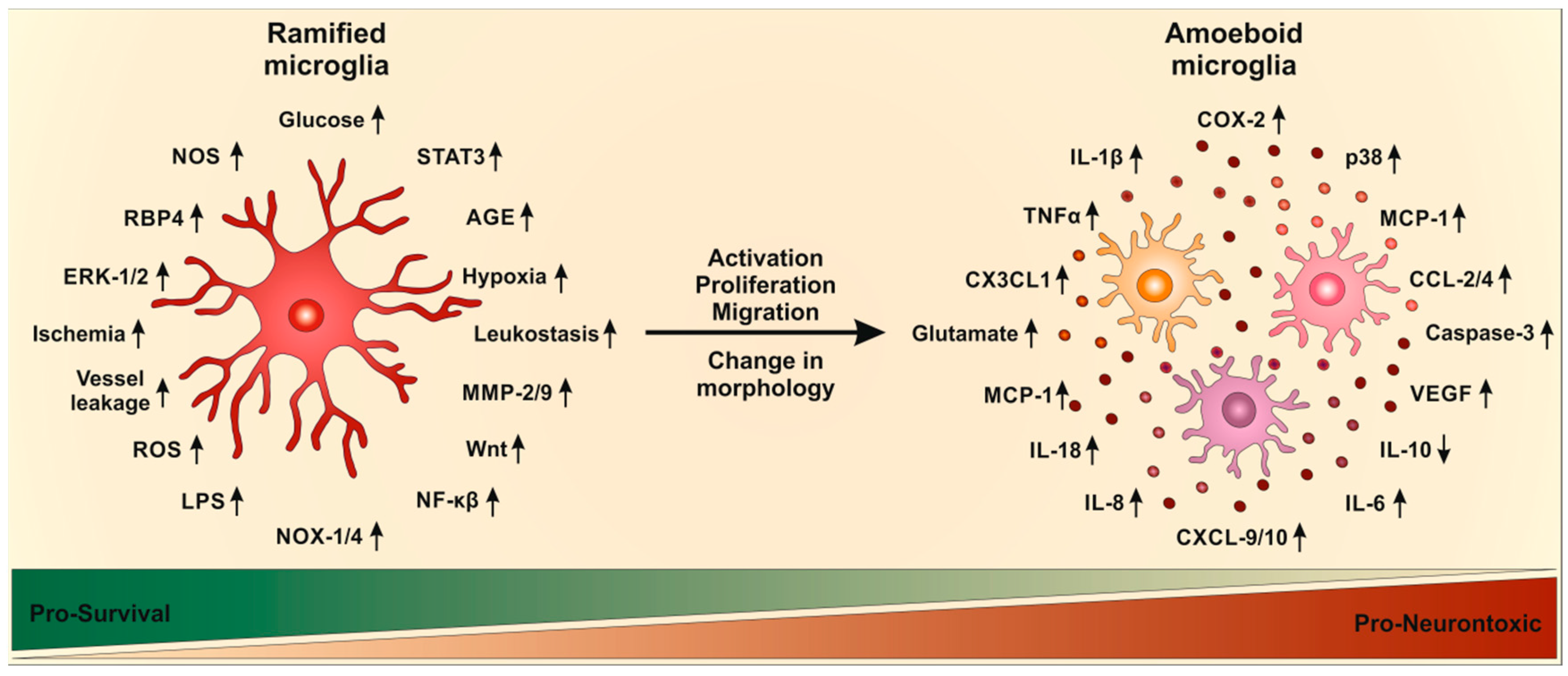

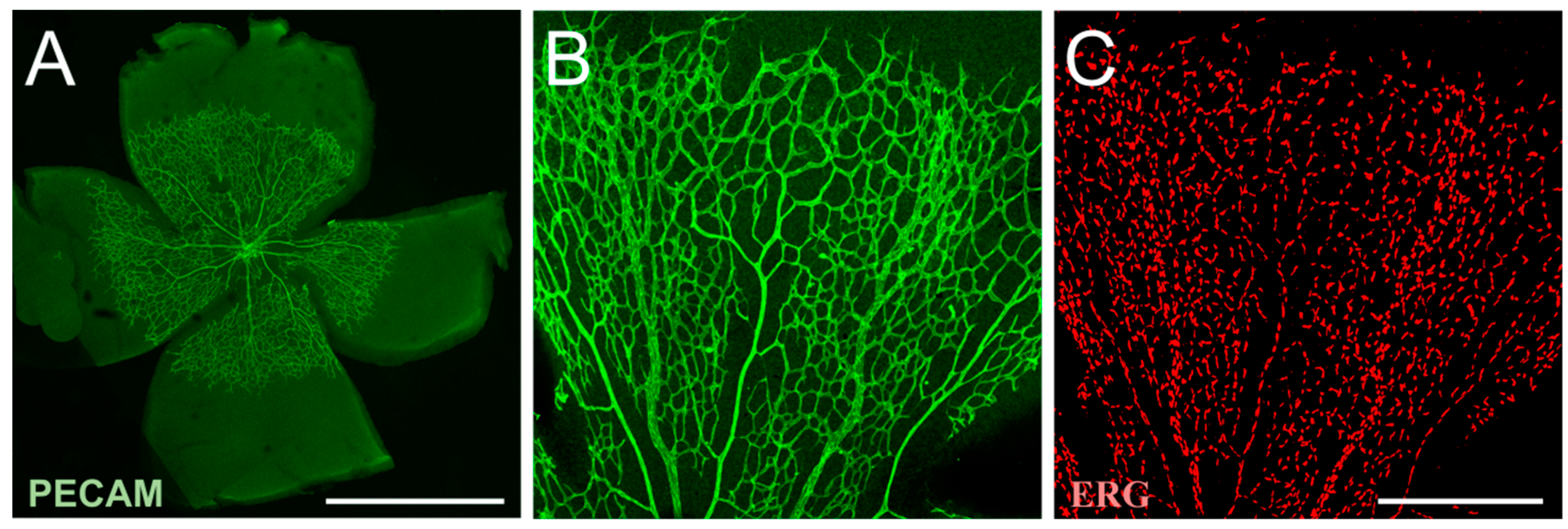{kind=link}
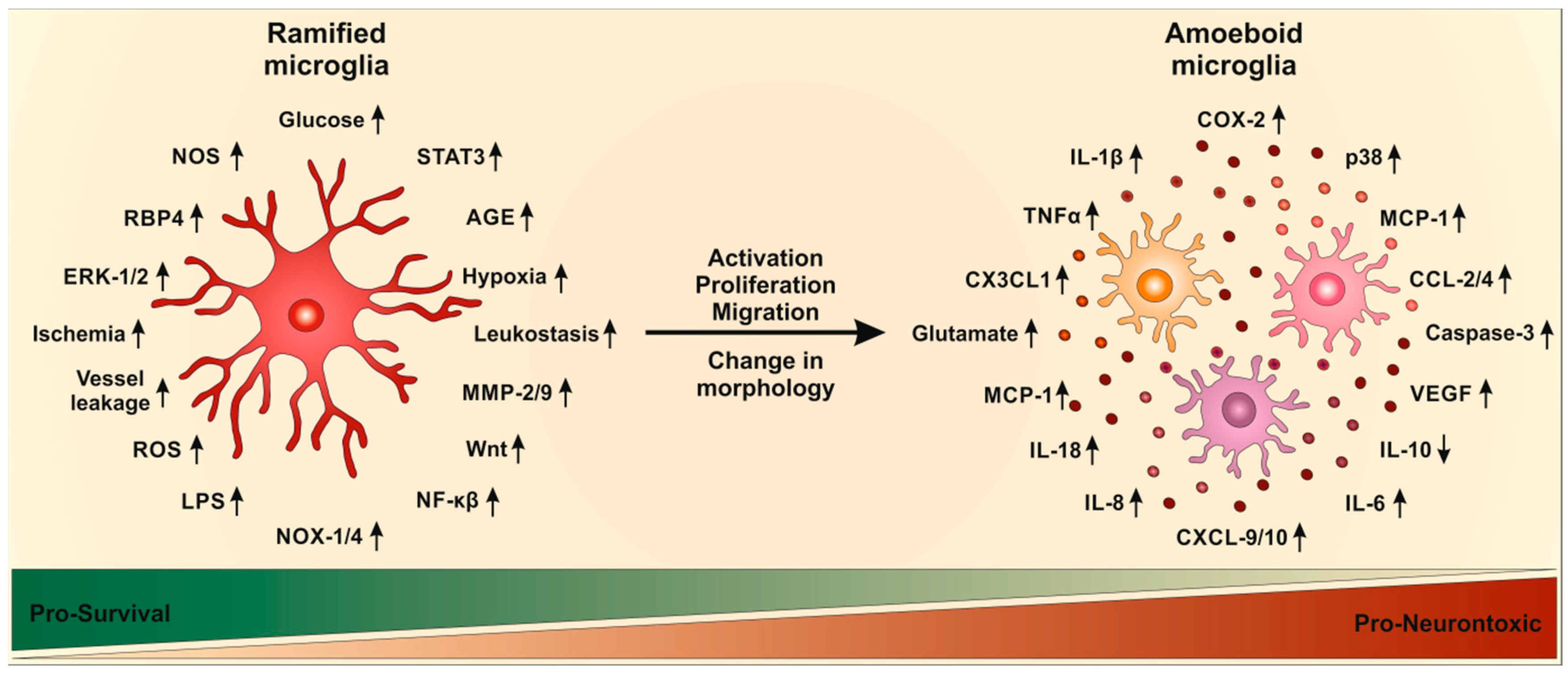{kind=link}
Table 1.
Mediators in diabetic retinopathy, including cytokines, chemokines, growth factors, adhesion molecules, neurotoxins and others. These mediators are either upregulated (up arrow) or downregulated (down arrow) in diabetic retinopathy.
Table 1.
Mediators in diabetic retinopathy, including cytokines, chemokines, growth factors, adhesion molecules, neurotoxins and others. These mediators are either upregulated (up arrow) or downregulated (down arrow) in diabetic retinopathy.
| Mediators | Regulation | Relevance | References |
|---|---|---|---|
| Cytokines | |||
| IL-1β | ↑ | Immuno-stimulation Increased ICAM-1 | [28,168,169,170,171] |
| IL-6 | ↑ | Immuno-stimulation | [172,173] |
| IL-8 | ↑ | Immuno-stimulation | [173] |
| IL-18 | ↑ | Immuno-stimulation | [174,175,176] |
| VEGF | ↑ | Immuno-stimulation Angio-stimulation Neuroprotective | [177] |
| TNFα | ↑ | Immuno-stimulation Increased ICAM-1 Increased leukostasis | [28,168,178,179,180] |
| COX-2 | ↑ | Immuno-stimulation | [180] |
| Chemokines | |||
| CCL-2 | ↑ | Immuno-stimulation | [181] |
| CCL-4 | ↑ | Immuno-stimulation | [181] |
| CXCL-9 | ↑ | Immuno-stimulation | [181] |
| CXCL-10 | ↑ | Immuno-stimulation | [181] |
| MCP-1 | ↑ | Immuno-stimulation Increased fibrosis Angio-stimulation | [28,173,182] |
| CX3CL1 | ↑ | Immuno-stimulation Neuroprotective | [183] |
| Growth factors | |||
| NGF | ↓ | Cellular toxicity | [184] |
| PEDF | ↓ | Cellular toxicity | [184] |
| IRBP | ↓ | Cellular toxicity | [184] |
| Somatostatin | ↓ | Cellular toxicity | [184] |
| Adhesion molecules | |||
| ICAM-1 | ↑ | Increased leukostasis | [64,65,66] |
| VCAM-1 | ↑ | Increased leukostasis | [64,65,66] |
| Neurotoxins | |||
| ROS | ↑ | Cellular toxicity | [185] |
| NO | ↑ | Cellular toxicity | [185] |
| Glutamate | ↑ | Cellular toxicity | [185] |
| Caspase-3 | ↑ | Cellular toxicity | [185] |
| Others | |||
| LPS | ↑ | Immuno-stimulation Cellular toxicity | [106] |
| AGE | ↑ | Immuno-stimulation Cellular toxicity | [49,50,51,52,53,54,55,56] |
| LEP | ↑ | Immuno-stimulation Cellular toxicity | [49,50,51,52,53,54,55,56] |
| Angiopoietin-2 | ↑ | Angio-stimulation | [186,187] |
| RBP4 | ↑ | Immuno-stimulation Neurotoxic Increased vascular leakage | [174,175,176] |
| NOX-1/4 | ↑ | Immuno-stimulation Increased leukostasis Increased ROS Increased vascular leakage | [188] |
| MMP-2/9 | ↑ | Immuno-stimulation Increased chemokines Neurotoxic | [189,190,191,192,193,194] |
| STAT3 | ↑ | Immuno-stimulation Increased vascular leakage | [195,196] |
| Wnt | ↑ | Immuno-stimulation Increased ROS | [197] |
IL: interleukin; VEGF: vascular endothelial growth factor; TNF: tumor necrosis factor; COX: cyclooxygenase; CCL: chemokine (C-C motif) ligand; CXCL: chemokine (C-X-C motif) ligand; MCP: monocyte chemoattractant protein-1; CX3CL: chemokine (C-X3-C motif) ligand; NGF: nerve growth factor; PEDF: pigment epithelium-derived factor; IRBP: iron-responsive element-binding protein; ICAM: intercellular adhesion molecule; VCAM: vascular cell adhesion protein; ROS: reactive oxygen species; NO: nitric oxide; LPS: lipopolysaccharide; AGE: advanced glycation end products; LEP: leptin; RBP: retinol binding protein; NOX: nicotinamide adenine dinucleotide phosphate (NADPH) oxidase; MMP: matrix metalloproteinase; STAT: signal transducer and activator of transcription; Wnt: wingless.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Altmann, C.; Schmidt, M.H.H. The Role of Microglia in Diabetic Retinopathy: Inflammation, Microvasculature Defects and Neurodegeneration. Int. J. Mol. Sci. 2018, 19, 110. https://doi.org/10.3390/ijms19010110
AMA Style
Altmann C, Schmidt MHH. The Role of Microglia in Diabetic Retinopathy: Inflammation, Microvasculature Defects and Neurodegeneration. International Journal of Molecular Sciences. 2018; 19(1):110. https://doi.org/10.3390/ijms19010110
Chicago/Turabian StyleAltmann, Christine, and Mirko H. H. Schmidt. 2018. "The Role of Microglia in Diabetic Retinopathy: Inflammation, Microvasculature Defects and Neurodegeneration" International Journal of Molecular Sciences 19, no. 1: 110. https://doi.org/10.3390/ijms19010110
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.