Non-Transfusion-Dependent Thalassemia: An Update on Complications and Management

 and
and
Abstract
:1. Introduction
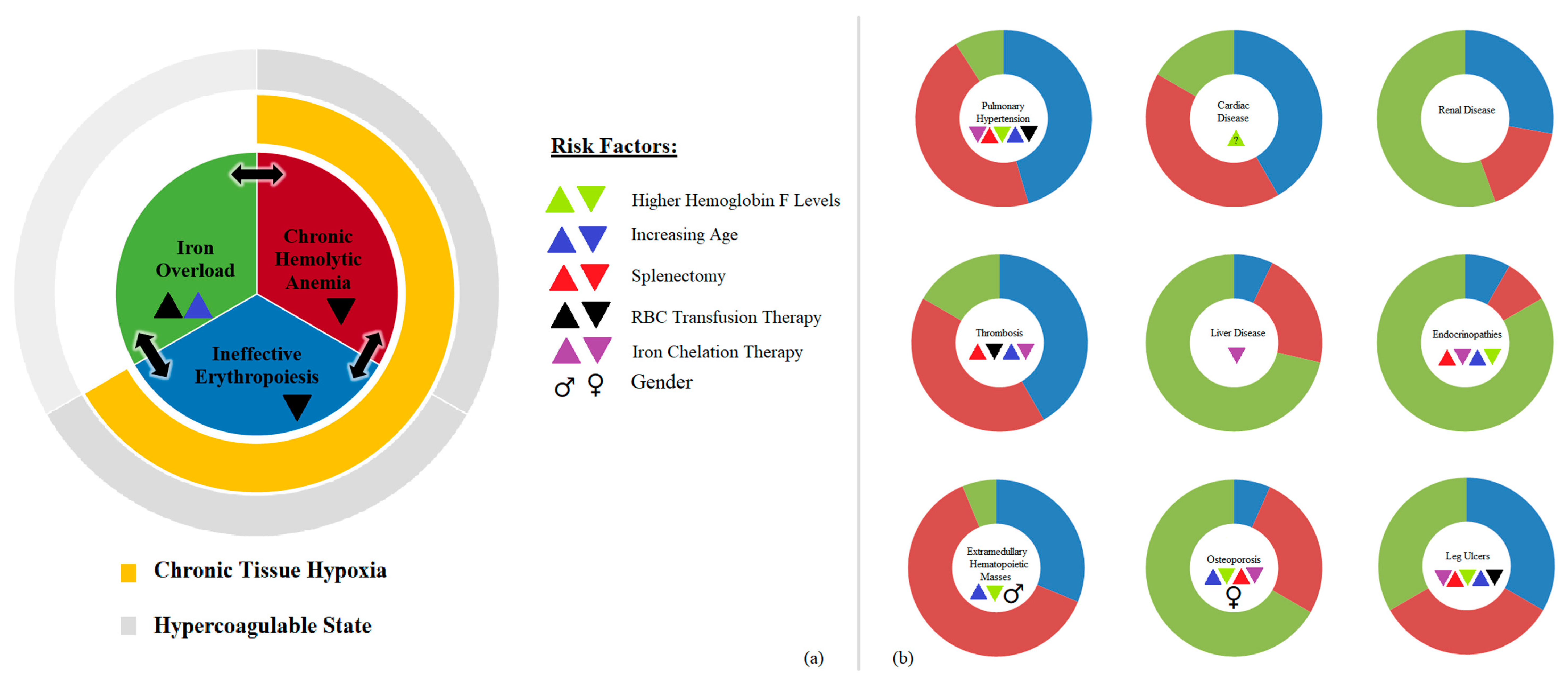
2. Pathophysiology
2.1. Ineffective Erythropoiesis
2.2. Chronic Hemolytic Anemia
2.3. Iron Overload
3. Morbidities in NTDT
3.1. Thrombosis
3.2. Cardiac Disease
3.3. Pulmonary Hypertension
3.4. Leg Ulcers
3.5. Hepatobiliary Complications
3.6. Extramedullary Hematopoiesis
3.7. Bone Disease
3.8. Endocrinopathies/Delayed Growth
3.9. Renal Disease
4. General Management
4.1. Transfusion Therapy
4.2. Splenectomy
4.3. Hydroxyurea Therapy
4.4. Iron Chelation
4.4.1. Deferasirox
4.4.2. Deferiprone
4.4.3. Deferoxamine
4.5. Hematopoietic Stem Cell Transplantation
5. Future Interventions
5.1. Improving Iron Dysregulation
5.2. Correcting Globin-Chain Imbalance
5.3. Improving Ineffective Erythropoiesis
6. Conclusions
Author Contributions
Conflicts of Interest
Abbreviations
| NTDT | Non-Transfusion Dependent Thalassemia |
| IOL | Iron Overload |
| LV | Left Ventricle |
| TDT | Transfusion Dependent Thalassemia |
| TMPRSS-6 | Transmembrane Protease Serine-6 |
| PHT | Pulmonary Hypertension |
| TI | Thalassemia Intermedia |
| CO | Cardiac Output |
| RBC | Red Blood Cell |
| PVR | Pulmonary Vascular Resistance |
| Hb | Hemoglobin |
| HCC | Hepatocellular Carcinoma |
| HbF | Fetal Hemoglobin |
| HCV | Hepatitis C Virus |
| HBV | Hepatitis B Virus |
| TIF | Thalassemia International Federation |
| LIC | Liver Iron Concentration |
| BMD | Bone Mineral Density |
| MRI | Magnetic Resonance Imaging |
| GI | Gastrointestinal |
| dw | Dry Weight |
| DNA | Deoxyribonucleic Acid |
| JAK2 | Janus Kinase-2 |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeats |
References
- Michlitsch, J.; Azimi, M.; Hoppe, C.; Walters, M.C.; Lubin, B.; Lorey, F.; Vichinsky, E. Newborn screening for hemoglobinopathies in california. Pediatr. Blood Cancer 2009, 52, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Weatherall, D.J.; Cappellini, M.D. Thalassaemia. Lancet 2017. [Google Scholar] [CrossRef]
- Weatherall, D.J.; Clegg, J.B. The Thalassaemia Syndromes; Blackwell Science Ltd.: Hoboken, NJ, USA, 2001. [Google Scholar]
- Steinberg, M.H.; Forget, B.G.; Higgs, D.R.; Weatherall, D.J. Disorders of Hemoglobin: Genetics, Pathophysiology, and Clinical Management; Cambridge University Press: New York, NY, USA, 2009. [Google Scholar]
- Saliba, A.N.; Taher, A.T. Morbidities in non-transfusion-dependent thalassemia. Ann. N. Y. Acad. Sci. 2016, 1368, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Izumi, H.; Morimoto, S.; Kuroda, M.; Kawamura, Y. Thalassemia intermedia complicated by hemochromatosis: Clinical and autopsy report of a case. Nihon Ketsueki Gakkai Zasshi 1969, 32, 336–352. [Google Scholar] [PubMed]
- Ben-Bassat, I.; Hertz, M.; Selzer, G.; Ramot, B. Extramedullary hematopoiesis with multiple tumor-simulating mediastinal masses in a patient with β-thalassemia intermedia. Isr. J. Med. Sci. 1977, 13, 1206–1210. [Google Scholar] [PubMed]
- Taher, A.; Isma’eel, H.; Cappellini, M.D. Thalassemia intermedia: Revisited. Blood Cells Mol. Dis. 2006, 37, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Musallam, K.M.; Karimi, M.; El-Beshlawy, A.; Belhoul, K.; Daar, S.; Saned, M.S.; El-Chafic, A.H.; Fasulo, M.R.; Cappellini, M.D. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: The optimal care study. Blood 2010, 115, 1886–1892. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Taher, A.T.; Duca, L.; Cesaretti, C.; Halawi, R.; Cappellini, M.D. Levels of growth differentiation factor-15 are high and correlate with clinical severity in transfusion-independent patients with β-thalassemia intermedia. Blood Cells Mol. Dis. 2011, 47, 232–234. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.; Vichinsky, E.; Musallam, K.; Cappellini, M.D.; Viprakasit, V. Guidelines for the Management of Non Transfusion Dependent Thalassaemia (NTDT); Thalassemia International Federation: Nicosia, Cyprus, 2013. [Google Scholar]
- Aessopos, A.; Farmakis, D.; Karagiorga, M.; Voskaridou, E.; Loutradi, A.; Hatziliami, A.; Joussef, J.; Rombos, J.; Loukopoulos, D. Cardiac involvement in thalassemia intermedia: A multicenter study. Blood 2001, 97, 3411–3416. [Google Scholar] [CrossRef] [PubMed]
- Aessopos, A.; Tsironi, M.; Andreopoulos, A.; Farmakis, D. Heart disease in thalassemia intermedia. Hemoglobin 2009, 33 (Suppl. S1), S170–S176. [Google Scholar] [CrossRef] [PubMed]
- Rivella, S. The role of ineffective erythropoiesis in non-transfusion-dependent thalassemia. Blood Rev. 2012, 26 (Suppl. S1), S12–S15. [Google Scholar] [CrossRef]
- Melchiori, L.; Gardenghi, S.; Rivella, S. β-thalassemia: Hijaking ineffective erythropoiesis and iron overload. Adv. Hematol. 2010, 2010, 938640. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Musallam, K.M.; Saliba, A.N.; Graziadei, G.; Cappellini, M.D. Hemoglobin level and morbidity in non-transfusion-dependent thalassemia. Blood Cells Mol. Dis. 2015, 55, 108–109. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Poggiali, E.; Taher, A.T.; Musallam, K.M. Hypercoagulability in β-thalassemia: A status quo. Expert Rev. Hematol. 2012, 5, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C.; Nai, A. Ineffective erythropoiesis and regulation of iron status in iron loading anaemias. Br. J. Haematol. 2016, 172, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Musallam, K.M.; El-Beshlawy, A.; Karimi, M.; Daar, S.; Belhoul, K.; Saned, M.S.; Graziadei, G.; Cappellini, M.D. Age-related complications in treatment-naive patients with thalassaemia intermedia. Br. J. Haematol. 2010, 150, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Gardenghi, S.; Marongiu, M.F.; Ramos, P.; Guy, E.; Breda, L.; Chadburn, A.; Liu, Y.; Amariglio, N.; Rechavi, G.; Rachmilewitz, E.A.; et al. Ineffective erythropoiesis in β-thalassemia is characterized by increased iron absorption mediated by down-regulation of hepcidin and up-regulation of ferroportin. Blood 2007, 109, 5027–5035. [Google Scholar] [CrossRef] [PubMed]
- Tanno, T.; Porayette, P.; Sripichai, O.; Noh, S.J.; Byrnes, C.; Bhupatiraju, A.; Lee, Y.T.; Goodnough, J.B.; Harandi, O.; Ganz, T.; et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood 2009, 114, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Chauvet, C.; Viatte, L.; Danan, J.L.; Bigard, X.; Devaux, I.; Beaumont, C.; Kahn, A.; Vaulont, S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Investig. 2002, 110, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Nai, A.; Pagani, A.; Mandelli, G.; Lidonnici, M.R.; Silvestri, L.; Ferrari, G.; Camaschella, C. Deletion of tmprss6 attenuates the phenotype in a mouse model of β-thalassemia. Blood 2012, 119, 5021–5029. [Google Scholar] [CrossRef] [PubMed]
- Tanno, T.; Bhanu, N.V.; Oneal, P.A.; Goh, S.H.; Staker, P.; Lee, Y.T.; Moroney, J.W.; Reed, C.H.; Luban, N.L.; Wang, R.H.; et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat. Med. 2007, 13, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Borgna Pignatti, C.; Carnelli, V.; Caruso, V.; Dore, F.; De Mattia, D.; Di Palma, A.; Di Gregorio, F.; Romeo, M.A.; Longhi, R.; Mangiagli, A.; et al. Thromboembolic events in β thalassemia major: An italian multicenter study. Acta Haematol. 1998, 99, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Robbiolo, L.; Bottasso, B.M.; Coppola, R.; Fiorelli, G.; Mannucci, A.P. Venous thromboembolism and hypercoagulability in splenectomized patients with thalassaemia intermedia. Br. J. Haematol. 2000, 111, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Musallam, K.M.; Karimi, M.; El-Beshlawy, A.; Belhoul, K.; Daar, S.; Saned, M.; Cesaretti, C.; Cappellini, M.D. Splenectomy and thrombosis: The case of thalassemia intermedia. J. Thromb. Haemost. 2010, 8, 2152–2158. [Google Scholar] [CrossRef] [PubMed]
- Sonakul, D.; Fucharoen, S. Pulmonary thromboembolism in thalassemic patients. Southeast Asian J. Trop. Med. Public Health 1992, 23 (Suppl. S2), 25–28. [Google Scholar] [PubMed]
- Musallam, K.M.; Beydoun, A.; Hourani, R.; Nasreddine, W.; Raad, R.; Koussa, S.; Taher, A.T. Brain magnetic resonance angiography in splenectomized adults with β-thalassemia intermedia. Eur. J. Haematol. 2011, 87, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Aessopos, A.; Farmakis, D.; Karagiorga, M.; Rombos, I.; Loucopoulos, D. Pseudoxanthoma elasticum lesions and cardiac complications as contributing factors for strokes in β-thalassemia patients. Stroke 1997, 28, 2421–2424. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Taher, A.T.; Karimi, M.; Rachmilewitz, E.A. Cerebral infarction in β-thalassemia intermedia: Breaking the silence. Thromb. Res. 2012, 130, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Nasreddine, W.; Beydoun, A.; Hourani, R.; Hankir, A.; Koussa, S.; Haidar, M.; Taher, A.T. Brain positron emission tomography in splenectomized adults with β-thalassemia intermedia: Uncovering yet another covert abnormality. Ann. Hematol. 2012, 91, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Amoozgar, H.; Zeighami, S.; Haghpanah, S.; Karimi, M. A comparison of heart function and arrhythmia in clinically asymptomatic patients with β thalassemia intermedia and β thalassemia major. Hematology 2017, 22, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Amoozgar, H.; Farhani, N.; Karimi, M. Early echocardiographic findings in β-thalassemia intermedia patients using standard and tissue doppler methods. Pediatr. Cardiol. 2011, 32, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Aessopos, A.; Farmakis, D.; Deftereos, S.; Tsironi, M.; Tassiopoulos, S.; Moyssakis, I.; Karagiorga, M. Thalassemia heart disease: A comparative evaluation of thalassemia major and thalassemia intermedia. Chest 2005, 127, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Roghi, A.; Cappellini, M.D.; Wood, J.C.; Musallam, K.M.; Patrizia, P.; Fasulo, M.R.; Cesaretti, C.; Taher, A.T. Absence of cardiac siderosis despite hepatic iron overload in italian patients with thalassemia intermedia: An MRI T2* study. Ann. Hematol. 2010, 89, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Origa, R.; Barella, S.; Argiolas, G.M.; Bina, P.; Agus, A.; Galanello, R. No evidence of cardiac iron in 20 never- or minimally-transfused patients with thalassemia intermedia. Haematologica 2008, 93, 1095–1096. [Google Scholar] [CrossRef] [PubMed]
- Aessopos, A.; Samarkos, M.; Voskaridou, E.; Papaioannou, D.; Tsironi, M.; Kavouklis, E.; Vaiopoulos, G.; Stamatelos, G.; Loukopoulos, D. Arterial calcifications in β-thalassemia. Angiology 1998, 49, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Soergel, K.H.; Sommers, S.C. Idiopathic pulmonary hemosiderosis and related syndromes. Am. J. Med. 1962, 32, 499–511. [Google Scholar] [CrossRef]
- Du, Z.D.; Roguin, N.; Milgram, E.; Saab, K.; Koren, A. Pulmonary hypertension in patients with thalassemia major. Am. Heart J. 1997, 134, 532–537. [Google Scholar] [CrossRef]
- Rashidi Ghader, F.; Vahidshahi, K. Evaluation of the prevalence of pulmonary hypertension in thalassemia intermedia. Res. J. Biol. Sci. 2008, 3, 794–797. [Google Scholar]
- Derchi, G.; Galanello, R.; Bina, P.; Cappellini, M.D.; Piga, A.; Lai, M.E.; Quarta, A.; Casu, G.; Perrotta, S.; Pinto, V.; et al. Prevalence and risk factors for pulmonary arterial hypertension in a large group of β-thalassemia patients using right heart catheterization: A webthal study. Circulation 2014, 129, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.; Musallam, K.M.; Cappellini, M.D.; Daar, S.; El-Beshlawy, A.; Belhoul, K.; Saned, M.S.; Temraz, S.; Koussa, S.; Taher, A.T. Risk factors for pulmonary hypertension in patients with β thalassemia intermedia. Eur. J. Intern. Med. 2011, 22, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Gladwin, M.T.; Sachdev, V.; Jison, M.L.; Shizukuda, Y.; Plehn, J.F.; Minter, K.; Brown, B.; Coles, W.A.; Nichols, J.S.; Ernst, I.; et al. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N. Engl. J. Med. 2004, 350, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Haddad, A.; Tyan, P.; Radwan, A.; Mallat, N.; Taher, A. Β-thalassemia intermedia: A bird’s-eye view. Turk. J. Haematol. 2014, 31, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Gimmon, Z.; Wexler, M.R.; Rachmilewitz, E.A. Juvenile leg ulceration in β-thalassemia major and intermedia. Plast. Reconstr. Surg. 1982, 69, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, N.F.; Muraca, G.M.; O’Donnell, A.; Premawardhena, A.; Fisher, C.; Weatherall, D.J. Studies in haemoglobin E β-thalassaemia. Br. J. Haematol. 2008, 141, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Levin, C.; Koren, A. Healing of refractory leg ulcer in a patient with thalassemia intermedia and hypercoagulability after 14 years of unresponsive therapy. Isr. Med. Assoc. 2011, 13, 316–318. [Google Scholar]
- Matta, B.N.; Abbas, O.; Maakaron, J.E.; Koussa, S.; Daderian, R.H.; Taher, A.T. Leg ulcers in patients with β-thalassaemia intermedia: A single centre’s experience. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Sankaran, V.G.; Cappellini, M.D.; Duca, L.; Nathan, D.G.; Taher, A.T. Fetal hemoglobin levels and morbidity in untransfused patients with β-thalassemia intermedia. Blood 2012, 119, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Moukhadder, H.M.; Halawi, R.; Cappellini, M.D.; Taher, A.T. Hepatocellular carcinoma as an emerging morbidity in the thalassemia syndromes: A comprehensive review. Cancer 2017, 123, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V. Iron, hemochromatosis, and hepatocellular carcinoma. Gastroenterology 2004, 127, S79–S86. [Google Scholar] [CrossRef] [PubMed]
- Kew, M.C. Hepatic iron overload and hepatocellular carcinoma. Cancer Lett. 2009, 286, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Borgna-Pignatti, C.; Garani, M.C.; Forni, G.L.; Cappellini, M.D.; Cassinerio, E.; Fidone, C.; Spadola, V.; Maggio, A.; Restivo Pantalone, G.; Piga, A.; et al. Hepatocellular carcinoma in thalassaemia: An update of the italian registry. Br. J. Haematol. 2014, 167, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Puliyel, M.; Sposto, R.; Berdoukas, V.A.; Hofstra, T.C.; Nord, A.; Carson, S.; Wood, J.; Coates, T.D. Ferritin trends do not predict changes in total body iron in patients with transfusional iron overload. Am. J. Hematol. 2014, 89, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.; El Rassi, F.; Isma’eel, H.; Koussa, S.; Inati, A.; Cappellini, M.D. Correlation of liver iron concentration determined by R2 magnetic resonance imaging with serum ferritin in patients with thalassemia intermedia. Haematologica 2008, 93, 1584–1586. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Taher, A.T.; Rachmilewitz, E.A. Β-thalassemia intermedia: A clinical perspective. Cold Spring Harb. Perspect. Med. 2012, 2, a013482. [Google Scholar] [CrossRef] [PubMed]
- Haidar, R.; Mhaidli, H.; Taher, A.T. Paraspinal extramedullary hematopoiesis in patients with thalassemia intermedia. Eur. Spine J. 2010, 19, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Dore, F.; Cianciulli, P.; Rovasio, S.; Oggiano, L.; Bonfigli, S.; Murineddu, M.; Pardini, S.; Simonetti, G.; Gualdi, G.; Papa, G.; et al. Incidence and clinical study of ectopic erythropoiesis in adult patients with thalassemia intermedia. Ann. Ital. Med. Int. 1992, 7, 137–140. [Google Scholar] [PubMed]
- Cappellini, M.D.; Musallam, K.M.; Taher, A.T. Insight onto the pathophysiology and clinical complications of thalassemia intermedia. Hemoglobin 2009, 33 (Suppl. S1), S145–S159. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Taher, A.T.; Cappellini, M.D.; Sankaran, V.G. Clinical experience with fetal hemoglobin induction therapy in patients with β-thalassemia. Blood 2013, 121, 2199–2212. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Cappellini, M.D.; Wood, J.C.; Motta, I.; Graziadei, G.; Tamim, H.; Taher, A.T. Elevated liver iron concentration is a marker of increased morbidity in patients with β thalassemia intermedia. Haematologica 2011, 96, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Musallam, K.M.; Cappellini, M.D.; Weatherall, D.J. Optimal management of β thalassaemia intermedia. Br. J. Haematol. 2011, 152, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Forni, G.L.; Perrotta, S.; Giusti, A.; Quarta, G.; Pitrolo, L.; Cappellini, M.D.; D’Ascola, D.G.; Borgna Pignatti, C.; Rigano, P.; Filosa, A.; et al. Neridronate improves bone mineral density and reduces back pain in β-thalassaemia patients with osteoporosis: Results from a phase 2, randomized, parallel-arm, open-label study. Br. J. Haematol 2012, 158, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, M.G.; Macklin, E.A.; Trachtenberg, F.L.; Fung, E.B.; Cheung, A.M.; Vichinsky, E.; Olivieri, N.; Kirby, M.; Kwiatkowski, J.L.; Cunningham, M.; et al. Differences in the prevalence of growth, endocrine and vitamin d abnormalities among the various thalassaemia syndromes in north america. Br. J. Haematol. 2009, 146, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Baldini, M.; Marcon, A.; Cassin, R.; Ulivieri, F.M.; Spinelli, D.; Cappellini, M.D.; Graziadei, G. Β-thalassaemia intermedia: Evaluation of endocrine and bone complications. BioMed Res. Int. 2014, 2014, 174581. [Google Scholar] [CrossRef] [PubMed]
- Inati, A.; Noureldine, M.A.; Mansour, A.; Abbas, H.A. Endocrine and bone complications in β-thalassemia intermedia: Current understanding and treatment. BioMed Res. Int. 2015, 2015, 813098. [Google Scholar] [CrossRef] [PubMed]
- Nassar, A.H.; Usta, I.M.; Taher, A.M. Β-thalassemia intermedia and pregnancy: Should we anticoagulate? J. Thromb. Haemost. 2006, 4, 1413–1414. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M. Chronic hypoxia and tubulointerstitial injury: A final common pathway to end-stage renal failure. J. Am. Soc. Nephrol. 2006, 17, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.M.; Lawler, E.V.; Mackenzie, H.S. The hyperfiltration theory: A paradigm shift in nephrology. Kidney Int. 1996, 49, 1774–1777. [Google Scholar] [CrossRef] [PubMed]
- Mallat, N.S.; Musallam, K.M.; Mallat, S.G.; Ziyadeh, F.N.; Koussa, S.; Taher, A.T. End stage renal disease in six patients with β-thalassemia intermedia. Blood Cells Mol. Dis. 2013, 51, 146–148. [Google Scholar] [CrossRef] [PubMed]
- Derchi, G.; Forni, G.L.; Formisano, F.; Cappellini, M.D.; Galanello, R.; D’Ascola, G.; Bina, P.; Magnano, C.; Lamagna, M. Efficacy and safety of sildenafil in the treatment of severe pulmonary hypertension in patients with hemoglobinopathies. Haematologica 2005, 90, 452–458. [Google Scholar] [PubMed]
- Taher, A.; Isma’eel, H.; Mehio, G.; Bignamini, D.; Kattamis, A.; Rachmilewitz, E.A.; Cappellini, M.D. Prevalence of thromboembolic events among 8860 patients with thalassaemia major and intermedia in the mediterranean area and iran. Thromb. Haemost. 2006, 96, 488–491. [Google Scholar] [PubMed]
- O’Driscoll, A.; Mackie, I.J.; Porter, J.B.; Machin, S.J. Low plasma heparin cofactor II levels in thalassaemia syndromes are corrected by chronic blood transfusion. Br. J. Haematol. 1995, 90, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Cappellini, M.D.; Taher, A.T. Evaluation of the 5mg/g liver iron concentration threshold and its association with morbidity in patients with β-thalassemia intermedia. Blood Cells Mol. Dis. 2013, 51, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Musallam, K.M.; Cappellini, M.D.; Daar, S.; Karimi, M.; El-Beshlawy, A.; Graziadei, G.; Magestro, M.; Wulff, J.; Pietri, G.; Taher, A.T. Serum ferritin level and morbidity risk in transfusion-independent patients with β-thalassemia intermedia: The orient study. Haematologica 2014, 99, e218–e221. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Porter, J.B.; Viprakasit, V.; Kattamis, A.; Chuncharunee, S.; Sutcharitchan, P.; Siritanaratkul, N.; Galanello, R.; Karakas, Z.; Lawniczek, T.; et al. Deferasirox effectively reduces iron overload in non-transfusion-dependent thalassemia (ntdt) patients: 1-year extension results from the thalassa study. Ann. Hematol. 2013, 92, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Musallam, K.M.; Viprakasit, V.; Porter, J.B.; Cappellini, M.D. Iron chelation therapy for non-transfusion-dependent thalassemia (NTDT): A status quo. Blood Cells Mol. Dis. 2014, 52, 88–90. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Cohen, A.; Piga, A.; Bejaoui, M.; Perrotta, S.; Agaoglu, L.; Aydinok, Y.; Kattamis, A.; Kilinc, Y.; Porter, J.; et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with β-thalassemia. Blood 2006, 107, 3455–3462. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Porter, J.; El-Beshlawy, A.; Li, C.K.; Seymour, J.F.; Elalfy, M.; Gattermann, N.; Giraudier, S.; Lee, J.W.; Chan, L.L.; et al. Tailoring iron chelation by iron intake and serum ferritin: The prospective EPIC study of deferasirox in 1744 patients with transfusion-dependent anemias. Haematologica 2010, 95, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Anastasi, S.; Lisi, R.; Abbate, G.; Caruso, V.; Giovannini, M.; De Sanctis, V. Absence of teratogenicity of deferasirox treatment during pregnancy in a thalassaemic patient. Pediatr. Endocrinol. Rev. 2011, 8 (Suppl. S2), 345–347. [Google Scholar] [PubMed]
- Rombos, Y.; Tzanetea, R.; Konstantopoulos, K.; Simitzis, S.; Zervas, C.; Kyriaki, P.; Kavouklis, M.; Aessopos, A.; Sakellaropoulos, N.; Karagiorga, M.; et al. Chelation therapy in patients with thalassemia using the orally active iron chelator deferiprone (L1). Haematologica 2000, 85, 115–117. [Google Scholar] [PubMed]
- Pootrakul, P.; Sirankapracha, P.; Sankote, J.; Kachintorn, U.; Maungsub, W.; Sriphen, K.; Thakernpol, K.; Atisuk, K.; Fucharoen, S.; Chantraluksri, U.; et al. Clinical trial of deferiprone iron chelation therapy in β-thalassaemia/haemoglobin e patients in thailand. Br. J. Haematol. 2003, 122, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Calvaruso, G.; Vitrano, A.; Di Maggio, R.; Lai, E.; Colletta, G.; Quota, A.; Gerardi, C.; Rigoli, L.C.; Sacco, M.; Pitrolo, L.; et al. Deferiprone versus deferoxamine in thalassemia intermedia: Results from a 5-year long-term italian multicenter randomized clinical trial. Am. J. Hematol. 2015, 90, 634–638. [Google Scholar] [CrossRef] [PubMed]
- Cossu, P.; Toccafondi, C.; Vardeu, F.; Sanna, G.; Frau, F.; Lobrano, R.; Cornacchia, G.; Nucaro, A.; Bertolino, F.; Loi, A.; et al. Iron overload and desferrioxamine chelation therapy in β-thalassemia intermedia. Eur. J. Pediatr. 1981, 137, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Pennell, D.J.; Berdoukas, V.; Karagiorga, M.; Ladis, V.; Piga, A.; Aessopos, A.; Gotsis, E.D.; Tanner, M.A.; Smith, G.C.; Westwood, M.A.; et al. Randomized controlled trial of deferiprone or deferoxamine in β-thalassemia major patients with asymptomatic myocardial siderosis. Blood 2006, 107, 3738–3744. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.; Martin, M.; Mizanin, J.; Konkle, D.F.; Schwartz, E. Vision and hearing during deferoxamine therapy. J. Pediatr. 1990, 117, 326–330. [Google Scholar] [CrossRef]
- Baronciani, D.; Angelucci, E.; Potschger, U.; Gaziev, J.; Yesilipek, A.; Zecca, M.; Orofino, M.G.; Giardini, C.; Al-Ahmari, A.; Marktel, S.; et al. Hemopoietic stem cell transplantation in thalassemia: A report from the european society for blood and bone marrow transplantation hemoglobinopathy registry, 2000–2010. Bone Marrow Transpl. 2016, 51, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Rai, P.; Malik, P. Gene therapy for hemoglobin disorders—A mini-review. J. Rare Dis. Res. Treat. 2016, 1, 25–31. [Google Scholar] [PubMed]
- Ben Salah, N.; Bou-Fakhredin, R.; Mellouli, F.; Taher, A.T. Revisiting β thalassemia intermedia: Past, present, and future prospects. Hematology 2017, 22, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Casu, C.; Oikonomidou, P.R.; Chen, H.; Nandi, V.; Ginzburg, Y.; Prasad, P.; Fleming, R.E.; Shah, Y.M.; Valore, E.V.; Nemeth, E.; et al. Minihepcidin peptides as disease modifiers in mice affected by β-thalassemia and polycythemia vera. Blood 2016, 128, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Casu, C.; Aghajan, M.; Oikonomidou, P.R.; Guo, S.; Monia, B.P.; Rivella, S. Combination of tmprss6- aso and the iron chelator deferiprone improves erythropoiesis and reduces iron overload in a mouse model of β-thalassemia intermedia. Haematologica 2016, 101, e8–e11. [Google Scholar] [CrossRef] [PubMed]
- Bank, A.; Dorazio, R.; Leboulch, P. A phase I/II clinical trial of β-globin gene therapy for β-thalassemia. Ann. N. Y. Acad. Sci. 2005, 1054, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Raja, J.V.; Rachchh, M.A.; Gokani, R.H. Recent advances in gene therapy for thalassemia. J. Pharm. Bioallied Sci. 2012, 4, 194–201. [Google Scholar] [PubMed]
- Cox, D.B.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, V.G.; Weiss, M.J. Anemia: Progress in molecular mechanisms and therapies. Nat. Med. 2015, 21, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Aydinok, Y.; Karakas, Z.; Cassinerio, E.; Siritanaratkul, N.; Kattamis, A.; Maggio, A.; Hollaender, N.; Mahuzier, B.; Gadbaw, B.; Taher, A.T. Efficacy and safety of ruxolitinib in regularly transfused patients with thalassemia: Results from single-arm, multicenter, phase 2a truth study. Blood 2016, 128, 852. [Google Scholar]
- Attie, K.M.; Allison, M.J.; McClure, T.; Boyd, I.E.; Wilson, D.M.; Pearsall, A.E.; Sherman, M.L. A phase 1 study of ACE-536, a regulator of erythroid differentiation, in healthy volunteers. Am. J. Hematol. 2014, 89, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Piga, A.G.; Tartaglione, I.; Gamberini, R.; Voskaridou, E.; Melpignano, A.; Ricchi, P.; Caruso, V.; Pietrangelo, A.; Zhang, X.; Wilson, D.M. Luspatercept increases hemoglobin, decreases transfusion burden and improves iron overload in adults with β-thalassemia. Am. Soc. Hematol. 2016, 128, 851. [Google Scholar]


{kind=link}
| Homozygosity for mild forms of β+ thalassemia |
| Compound heterozygosity for β+/β0 thalassemia |
| Compound heterozygosity for β-thalassemia and another β chain variant (e.g., β-thal/hemoglobin HbE) |
| Coinheritance of homozygous β-thalassemia and hereditary persistence of fetal Hemoglobin [HPFH]) |
| Coinheritance of homozygous β+ thalassemia with α-thalassemia (e.g., β+/β+ with −α/−α, −−/αα, −α/αα, or −−/−α) |
| Coinheritance of heterozygous β-thalassemia and triplicated or quadruplicated α genes (eg, αα/ααα or αα/ααα) |
| Dominant forms of β-thalassemia |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sleiman, J.; Tarhini, A.; Bou-Fakhredin, R.; Saliba, A.N.; Cappellini, M.D.; Taher, A.T. Non-Transfusion-Dependent Thalassemia: An Update on Complications and Management. Int. J. Mol. Sci. 2018, 19, 182. https://doi.org/10.3390/ijms19010182
Sleiman J, Tarhini A, Bou-Fakhredin R, Saliba AN, Cappellini MD, Taher AT. Non-Transfusion-Dependent Thalassemia: An Update on Complications and Management. International Journal of Molecular Sciences. 2018; 19(1):182. https://doi.org/10.3390/ijms19010182
Chicago/Turabian StyleSleiman, Joseph, Ali Tarhini, Rayan Bou-Fakhredin, Antoine N. Saliba, Maria Domenica Cappellini, and Ali T. Taher. 2018. "Non-Transfusion-Dependent Thalassemia: An Update on Complications and Management" International Journal of Molecular Sciences 19, no. 1: 182. https://doi.org/10.3390/ijms19010182