Synthesized Heparan Sulfate Competitors Attenuate Pseudomonas aeruginosa Lung Infection

 and
and
Abstract
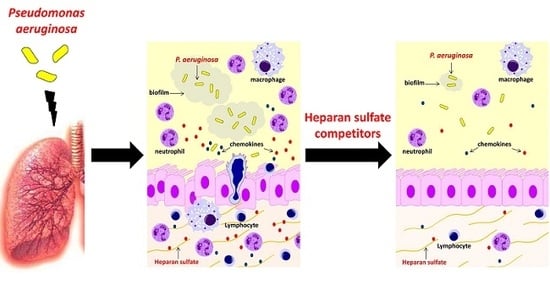
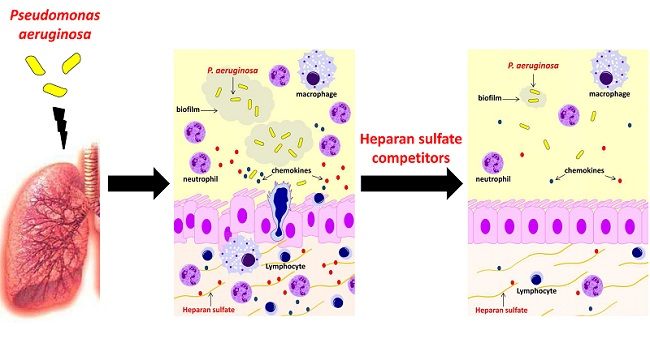
:
1. Introduction
2. Results
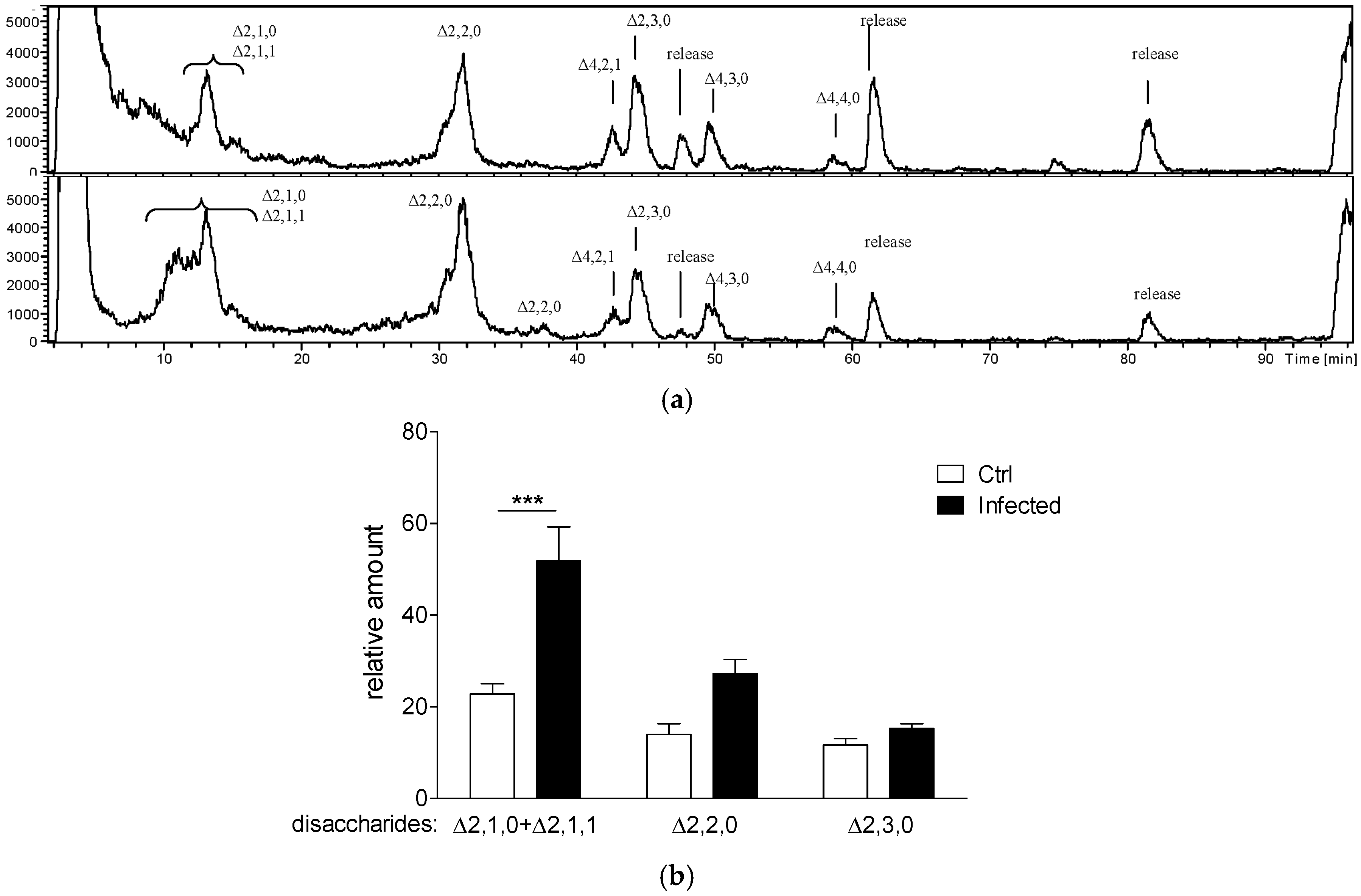
2.1. Evaluation of Specific HS Disaccharides and Their Levels in a Murine Model of Chronic P. aeruginosa Airways Infection
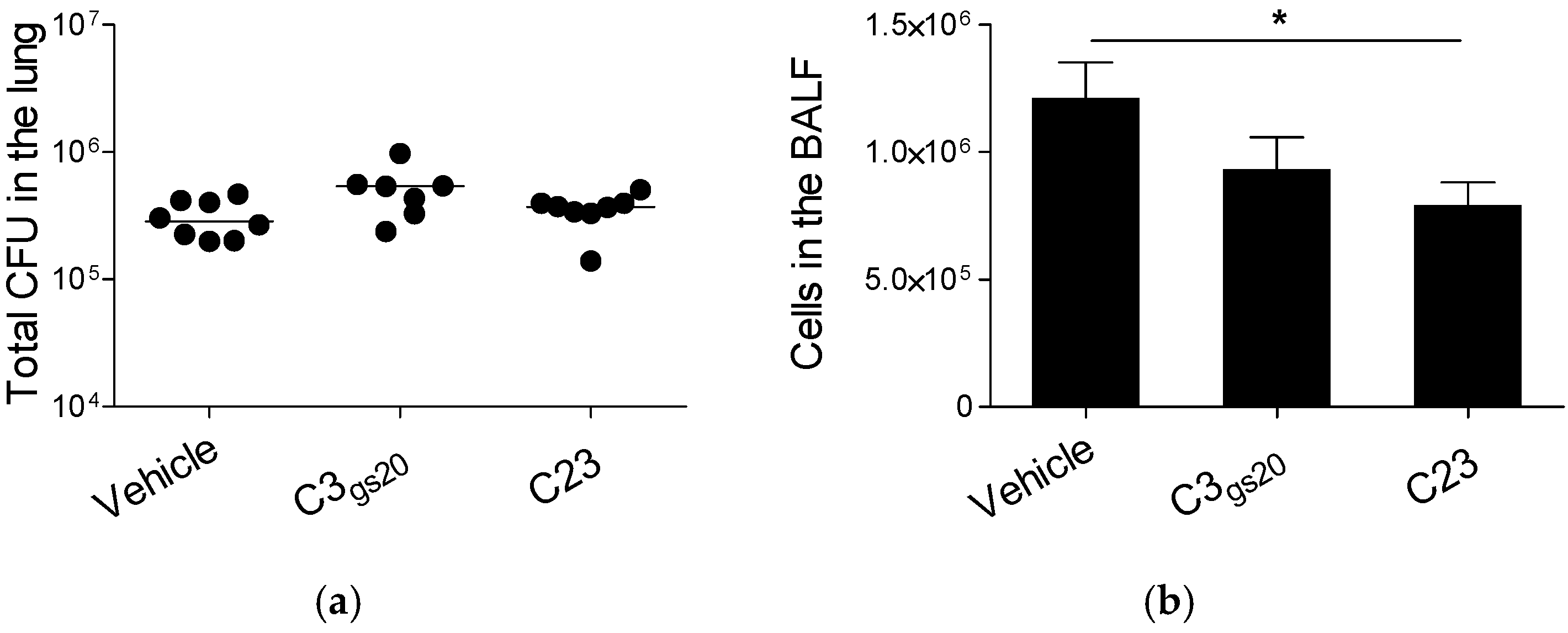
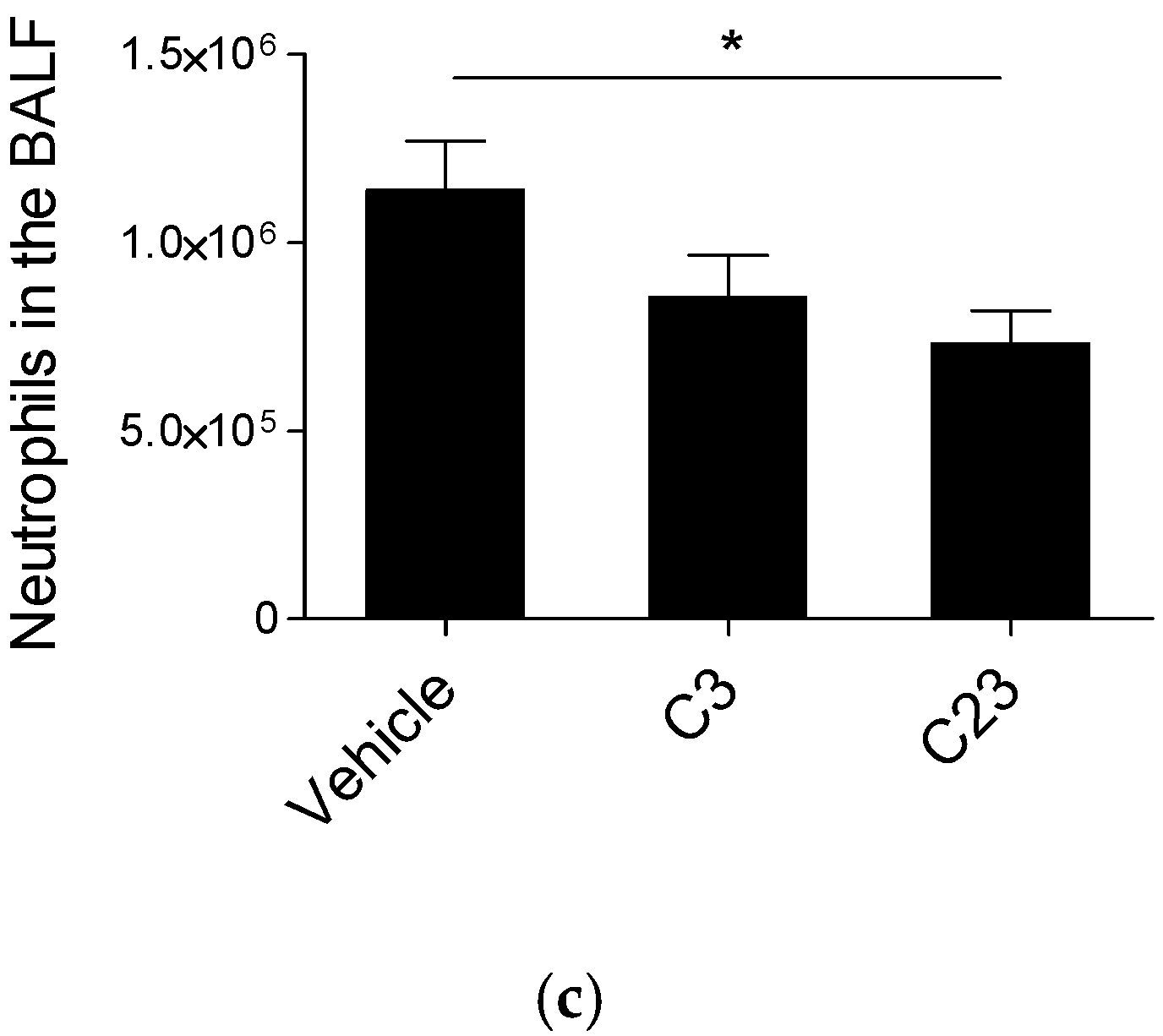
2.2. Structure and Efficacy of Synthesized HS Competitors in the Mouse Model of Acute P. aeruginosa Airways Infection
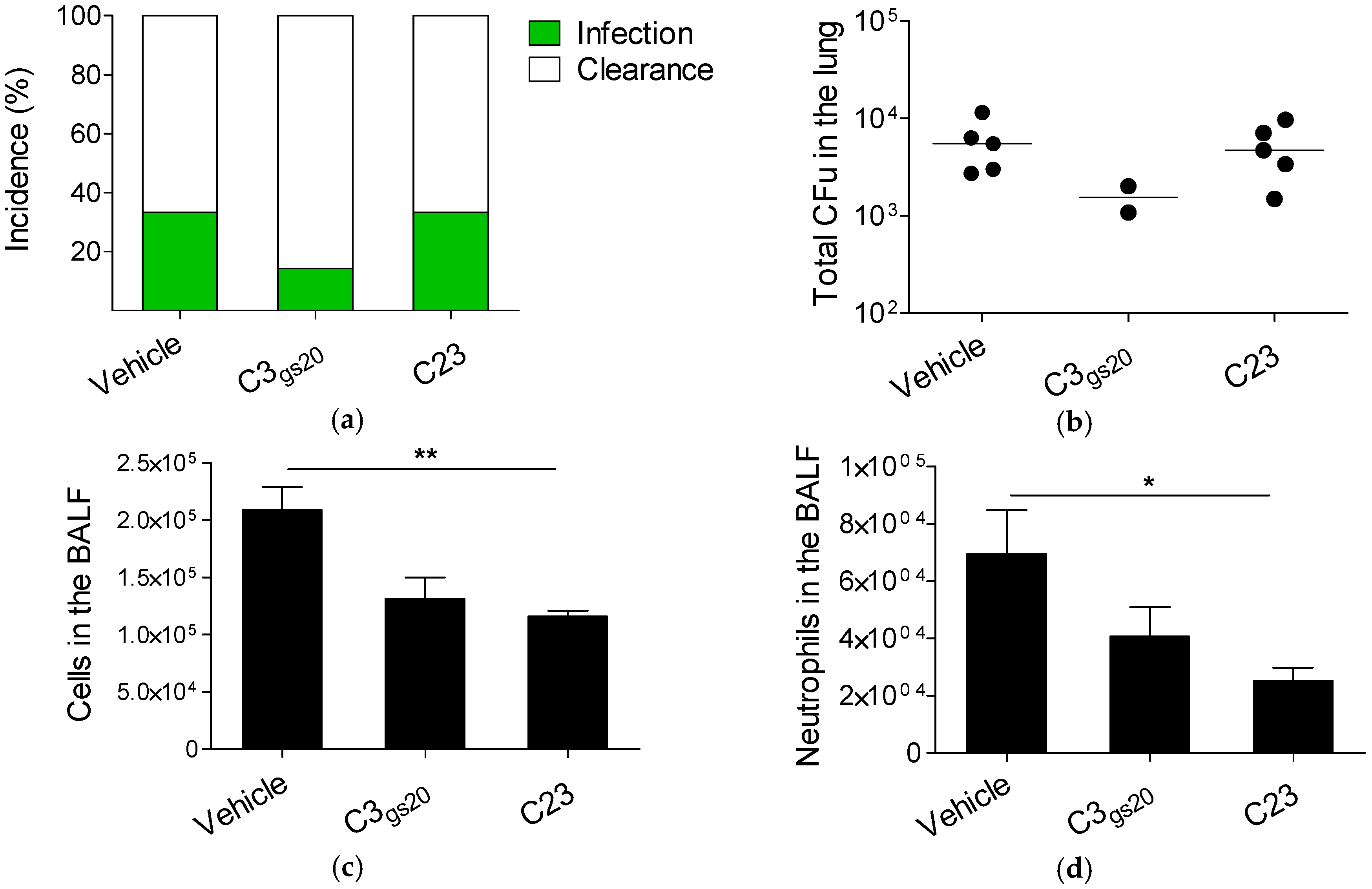
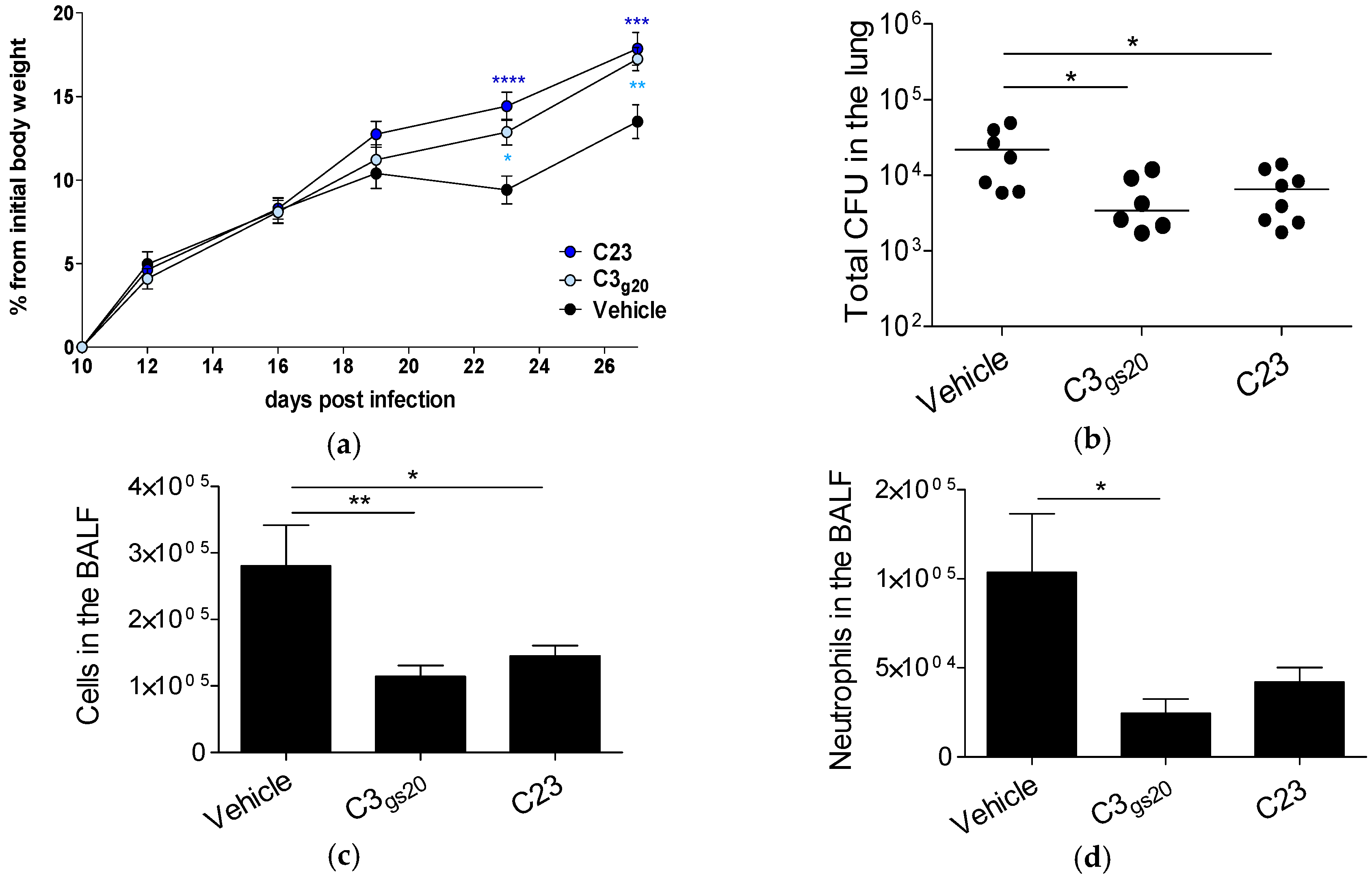
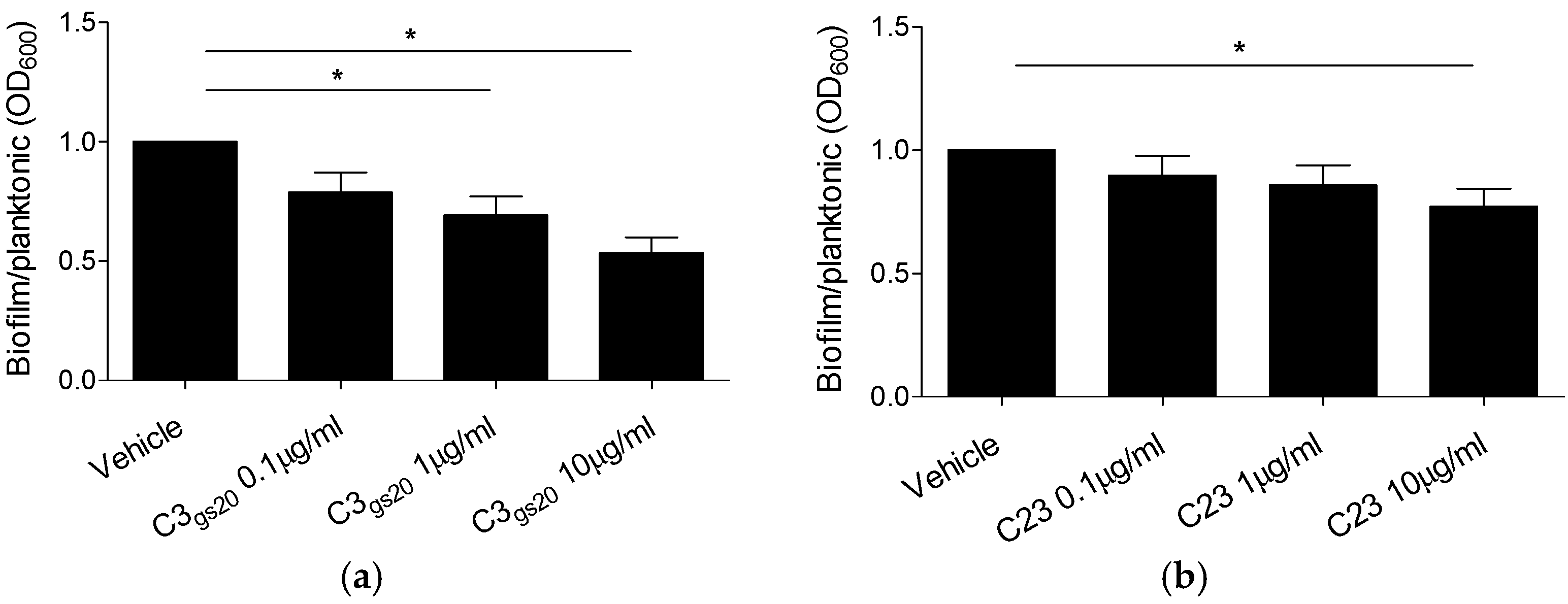
2.3. Efficacy of Synthesized HS Competitors in Mouse Models of Chronic P. aeruginosa Airways Infection
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Bacterial Strains
4.3. Mouse Strain
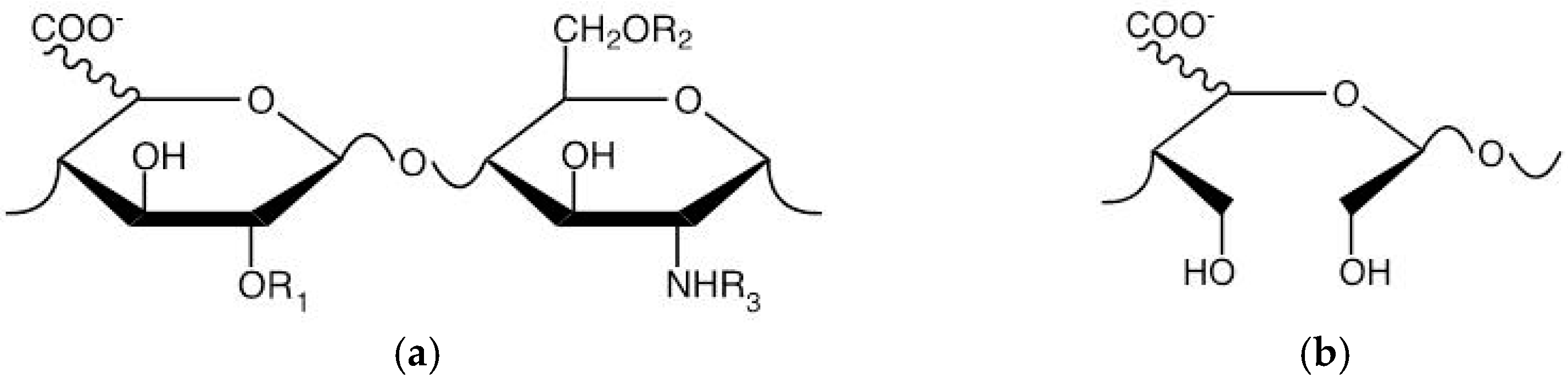
4.4. HS Competitors Synthesis and Characterization
4.5. Mouse Models of Acute and Chronic P. aeruginosa Infection
4.6. HS/heparin Analysis in Murine Lungs
4.7. Evaluation of Cytokines/Chemokines
4.8. Evaluation of P. aeruginosa Biofilm Formation
4.9. Statistics
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hassett, D.J.; Borchers, M.T.; Panos, R.J. Chronic obstructive pulmonary disease (COPD): Evaluation from clinical, immunological and bacterial pathogenesis perspectives. J. Microbiol. 2014, 52, 211–226. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.S.; Prince, A. Cystic fibrosis: A mucosal immunodeficiency syndrome. Nat. Med. 2012, 18, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Kondoh, Y. The clinical impact of major comorbidities on idiopathic pulmonary fibrosis. Respir. Investig. 2017, 55, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Reeves, E.P.; Bergin, D.A.; Murray, M.A.; McElvaney, N.G. The involvement of glycosaminoglycans in airway disease associated with cystic fibrosis. Sci. World J. 2011, 11, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.; Handel, T.M.; Johnson, Z.; Lau, E.K.; LiWang, P.; Clark-Lewis, I.; Borlat, F.; Wells, T.N.; Kosco-Vilbois, M.H. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. USA 2003, 100, 1885–1890. [Google Scholar] [CrossRef] [PubMed]
- Sadir, R.; Forest, E.; Lortat-Jacob, H. The heparan sulfate binding sequence of interferon-gamma increased the on rate of the interferon-gamma-interferon-gamma receptor complex formation. J. Biol. Chem. 1998, 273, 10919–10925. [Google Scholar] [CrossRef] [PubMed]
- Solic, N.; Wilson, J.; Wilson, S.J.; Shute, J.K. Endothelial activation and increased heparan sulfate expression in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2005, 172, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Papakonstantinou, E.; Klagas, I.; Roth, M.; Tamm, M.; Stolz, D. Acute Exacerbations of COPD Are Associated With Increased Expression of Heparan Sulfate and Chondroitin Sulfate in BAL. Chest 2016, 149, 685–695. [Google Scholar] [CrossRef] [PubMed]
- Westergren-Thorsson, G.; Hedstrom, U.; Nybom, A.; Tykesson, E.; Ahrman, E.; Hornfelt, M.; Maccarana, M.; van Kuppevelt, T.H.; Dellgren, G.; Wildt, M.; et al. Increased deposition of glycosaminoglycans and altered structure of heparan sulfate in idiopathic pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2017, 83, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Veraldi, N.; Hughes, A.J.; Rudd, T.R.; Thomas, H.B.; Edwards, S.W.; Hadfield, L.; Skidmore, M.A.; Siligardi, G.; Cosentino, C.; Shute, J.K.; et al. Heparin derivatives for the targeting of multiple activities in the inflammatory response. Carbohydr. Polym. 2015, 117, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Cigana, C.; Lore, N.I.; Riva, C.; De Fino, I.; Spagnuolo, L.; Sipione, B.; Rossi, G.; Nonis, A.; Cabrini, G.; Bragonzi, A. Tracking the immunopathological response to Pseudomonas aeruginosa during respiratory infections. Sci. Rep. 2016, 6, 21465. [Google Scholar] [CrossRef] [PubMed]
- Linhardt, R.J.; Turnbull, J.E.; Wang, H.M.; Loganathan, D.; Gallagher, J.T. Examination of the substrate specificity of heparin and heparan sulfate lyases. Biochemistry 1990, 29, 2611–2617. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, V.; Capila, I.; Raman, R.; Srinivasan, A.; Bosques, C.J.; Pojasek, K.; Wrick, M.A.; Sasisekharan, R. The catalytic machinery of chondroitinase ABC I utilizes a calcium coordination strategy to optimally process dermatan sulfate. Biochemistry 2006, 45, 11130–11139. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Yoshida, K.; Sugiura, M.; Sugahara, K.; Khoo, K.H.; Morris, H.R.; Dell, A. Structural studies on the bacterial lyase-resistant tetrasaccharides derived from the antithrombin III-binding site of porcine intestinal heparin. J. Biol. Chem. 1993, 268, 4780–4787. [Google Scholar] [PubMed]
- Casu, B.; Guerrini, M.; Guglieri, S.; Naggi, A.; Perez, M.; Torri, G.; Cassinelli, G.; Ribatti, D.; Carminati, P.; Giannini, G.; Penco, S.; et al. Undersulfated and glycol-split heparins endowed with antiangiogenic activity. J. Med. Chem. 2004, 47, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.L.; Stone, P.J.; Nugent, M.A. New insights into the inhibition of human neutrophil elastase by heparin. Biochemistry 2006, 45, 9104–9120. [Google Scholar] [CrossRef] [PubMed]
- Spillmann, D.; Witt, D.; Lindahl, U. Defining the interleukin-8-binding domain of heparan sulfate. J. Biol. Chem. 1998, 273, 15487–15493. [Google Scholar] [CrossRef] [PubMed]
- Casu, B.; Naggi, A.; Torri, G. Heparin-derived heparan sulfate mimics to modulate heparan sulfate-protein interaction in inflammation and cancer. Matrix Biol. 2010, 29, 442–452. [Google Scholar] [CrossRef] [PubMed]
- Lore, N.I.; Cigana, C.; De Fino, I.; Riva, C.; Juhas, M.; Schwager, S.; Eberl, L.; Bragonzi, A. Cystic fibrosis-niche adaptation of Pseudomonas aeruginosa reduces virulence in multiple infection hosts. PLoS ONE 2012, 7, e35648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lore, N.I.; Cigana, C.; Riva, C.; De Fino, I.; Nonis, A.; Spagnuolo, L.; Sipione, B.; Cariani, L.; Girelli, D.; Rossi, G.; et al. IL-17A impairs host tolerance during airway chronic infection by Pseudomonas aeruginosa. Sci. Rep. 2016, 6, 25937. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.C.; Shum, D.K.; Ip, M.S. Sputum sol neutrophil elastase activity in bronchiectasis: Differential modulation by syndecan-1. Am. J. Respir. Crit. Care Med. 2003, 168, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Spagnuolo, L.; De Simone, M.; Lore, N.I.; De Fino, I.; Basso, V.; Mondino, A.; Cigana, C.; Bragonzi, A. The host genetic background defines diverse immune-reactivity and susceptibility to chronic Pseudomonas aeruginosa respiratory infection. Sci. Rep. 2016, 6, 36924. [Google Scholar] [CrossRef] [PubMed]
- De Simone, M.; Spagnuolo, L.; Lore, N.I.; Rossi, G.; Cigana, C.; De Fino, I.; Iraqi, F.A.; Bragonzi, A. Host genetic background influences the response to the opportunistic Pseudomonas aeruginosa infection altering cell-mediated immunity and bacterial replication. PLoS ONE 2014, 9, e106873. [Google Scholar] [CrossRef] [PubMed]
- Lore, N.I.; Iraqi, F.A.; Bragonzi, A. Host genetic diversity influences the severity of Pseudomonas aeruginosa pneumonia in the Collaborative Cross mice. BMC Genet. 2015, 16, 106. [Google Scholar] [CrossRef] [PubMed]
- Papakonstantinou, E.; Karakiulakis, G. The ‘sweet’ and ‘bitter’ involvement of glycosaminoglycans in lung diseases: Pharmacotherapeutic relevance. Br. J. Pharmacol. 2009, 157, 1111–1127. [Google Scholar] [CrossRef] [PubMed]
- Polverino, E.; Rosales-Mayor, E.; Dale, G.E.; Dembowsky, K.; Torres, A. The Role of Neutrophil Elastase Inhibitors in Lung Diseases. Chest 2017, 152, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Walsh, R.L.; Dillon, T.J.; Scicchitano, R.; McLennan, G. Heparin and heparan sulphate are inhibitors of human leucocyte elastase. Clin. Sci. 1991, 81, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Doring, G.; Bragonzi, A.; Paroni, M.; Akturk, F.F.; Cigana, C.; Schmidt, A.; Gilpin, D.; Heyder, S.; Born, T.; Smaczny, C.; et al. BIIL 284 reduces neutrophil numbers but increases P. aeruginosa bacteremia and inflammation in mouse lungs. J. Cyst. Fibros. 2014, 13, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Yonker, L.M.; Cigana, C.; Hurley, B.P.; Bragonzi, A. Host-pathogen interplay in the respiratory environment of cystic fibrosis. J. Cyst. Fibros. 2015, 14, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Lore, N.I.; Bragonzi, A.; Cigana, C. The IL-17A/IL-17RA axis in pulmonary defence and immunopathology. Cytokine Growth Factor Rev. 2016, 30, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Guragain, M.; King, M.M.; Williamson, K.S.; Perez-Osorio, A.C.; Akiyama, T.; Khanam, S.; Patrauchan, M.A.; Franklin, M.J. The Pseudomonas aeruginosa PAO1 Two-Component Regulator CarSR Regulates Calcium Homeostasis and Calcium-Induced Virulence Factor Production through Its Regulatory Targets CarO and CarP. J. Bacteriol. 2016, 198, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Plotkowski, M.C.; Costa, A.O.; Morandi, V.; Barbosa, H.S.; Nader, H.B.; de Bentzmann, S.; Puchelle, E. Role of heparan sulphate proteoglycans as potential receptors for non-piliated Pseudomonas aeruginosa adherence to non-polarised airway epithelial cells. J. Med. Microbiol. 2001, 50, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Bucior, I.; Pielage, J.F.; Engel, J.N. Pseudomonas aeruginosa pili and flagella mediate distinct binding and signaling events at the apical and basolateral surface of airway epithelium. PLoS Pathog. 2012, 8, e1002616. [Google Scholar] [CrossRef] [PubMed]
- Cigana, C.; Curcuru, L.; Leone, M.R.; Ierano, T.; Lore, N.I.; Bianconi, I.; Silipo, A.; Cozzolino, F.; Lanzetta, R.; Molinaro, A.; et al. Pseudomonas aeruginosa exploits lipid A and muropeptides modification as a strategy to lower innate immunity during cystic fibrosis lung infection. PLoS ONE 2009, 4, e8439. [Google Scholar] [CrossRef] [PubMed]
- Cigana, C.; Lore, N.I.; Bernardini, M.L.; Bragonzi, A. Dampening Host Sensing and Avoiding Recognition in Pseudomonas aeruginosa Pneumonia. J. Biomed. Biotechnol. 2011, 2011, 852513. [Google Scholar] [CrossRef] [PubMed]
- Bragonzi, A.; Paroni, M.; Nonis, A.; Cramer, N.; Montanari, S.; Rejman, J.; Di Serio, C.; Doring, G.; Tummler, B. Pseudomonas aeruginosa microevolution during cystic fibrosis lung infection establishes clones with adapted virulence. Am. J. Respir. Crit. Care Med. 2009, 180, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Naggi, A.; Casu, B.; Perez, M.; Torri, G.; Cassinelli, G.; Penco, S.; Pisano, C.; Giannini, G.; Ishai-Michaeli, R.; Vlodavsky, I. Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded N-acetylation, and glycol splitting. J. Biol. Chem. 2005, 280, 12103–12113. [Google Scholar] [CrossRef] [PubMed]
- Bertini, S.; Bisio, A.; Torri, G.; Bensi, D.; Terbojevich, M. Molecular weight determination of heparin and dermatan sulfate by size exclusion chromatography with a triple detector array. Biomacromolecules 2005, 6, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Kukavica-Ibrulj, I.; Facchini, M.; Cigana, C.; Levesque, R.C.; Bragonzi, A. Assessing Pseudomonas aeruginosa virulence and the host response using murine models of acute and chronic lung infection. Methods Mol. Biol. 2014, 1149, 757–771. [Google Scholar] [PubMed]
- Yang, Y.; MacLeod, V.; Dai, Y.; Khotskaya-Sample, Y.; Shriver, Z.; Venkataraman, G.; Sasisekharan, R.; Naggi, A.; Torri, G.; Casu, B.; et al. The syndecan-1 heparan sulfate proteoglycan is a viable target for myeloma therapy. Blood 2007, 110, 2041–2048. [Google Scholar] [CrossRef] [PubMed]
- Poli, M.; Asperti, M.; Naggi, A.; Campostrini, N.; Girelli, D.; Corbella, M.; Benzi, M.; Besson-Fournier, C.; Coppin, H.; Maccarinelli, F.; et al. Glycol-split nonanticoagulant heparins are inhibitors of hepcidin expression in vitro and in vivo. Blood 2014, 123, 1564–1573. [Google Scholar] [CrossRef] [PubMed]
- Cassinelli, G.; Lanzi, C.; Tortoreto, M.; Cominetti, D.; Petrangolini, G.; Favini, E.; Zaffaroni, N.; Pisano, C.; Penco, S.; Vlodavsky, I.; et al. Antitumor efficacy of the heparanase inhibitor SST0001 alone and in combination with antiangiogenic agents in the treatment of human pediatric sarcoma models. Biochem. Pharmacol. 2013, 85, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Bragonzi, A.; Worlitzsch, D.; Pier, G.B.; Timpert, P.; Ulrich, M.; Hentzer, M.; Andersen, J.B.; Givskov, M.; Conese, M.; Doring, G. Nonmucoid Pseudomonas aeruginosa expresses alginate in the lungs of patients with cystic fibrosis and in a mouse model. J. Infect. Dis. 2005, 192, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Baldan, R.; Cigana, C.; Testa, F.; Bianconi, I.; De Simone, M.; Pellin, D.; Di Serio, C.; Bragonzi, A.; Cirillo, D.M. Adaptation of Pseudomonas aeruginosa in Cystic Fibrosis airways influences virulence of Staphylococcus aureus in vitro and murine models of co-infection. PLoS ONE 2014, 9, e89614. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HS Competitors | MW (kDa) | N-acetyl (%) | Glycol-Split (%) |
|---|---|---|---|
| C23 | 17.2 | 100 | 0 |
| C3gs20 | 16.5 | 14.6 | 20 |
| LMW C23 | 8 | 91 | 0 |
| LMW C3gs20 | 8 | 14.6 | 18 |
| MMW C3gs45 | 12.6 | 14.6 | 45 |
| MMW C3gs90 | 9.6 | 14.6 | 90 |
| Cytokine/Chemokine | Level (pg/500 µg Lung) | p Value | |||
|---|---|---|---|---|---|
| Vehicle | C3gs20 | C23 | C3gs20 vs. Vehicle | C23 vs. Vehicle | |
| IL-4 | 3.31 ± 0.27 | 3.54 ± 0.19 | 3.37 ± 0.63 | ns | ns |
| IL-6 | 232.9 ± 8.53 | 176.9 ± 15.95 | 159.7 ± 7.14 | ** | *** |
| IL-12p40 | 10.97 ± 0.79 | 9.21 ± 0.76 | 7.86 ± 0.68 | ns | * |
| IL-12p70 | 83.98 ± 3.46 | 93.98 ± 9.67 | 87.04 ± 6.45 | ns | ns |
| IL-13 | 124.68 ± 8.49 | 140.26 ± 6.32 | 114.37 ± 5.33 | ns | ns |
| IL-17A | 6.77 ± 0.78 | 6.85 ± 0.21 | 6.66 ± 0.99 | ns | ns |
| Eotaxin | 353.62 ± 22.52 | 450.79 ± 33.85 | 341.3 ± 71.56 | ns | ns |
| G-CSF | 542 ± 10.56 | 465.8 ± 37.58 | 426.9 ± 36.53 | ns | * |
| IFN-γ | 538.19 ± 15.12 | 525.13 ± 72.72 | 462.34 ± 54.04 | ns | ns |
| MCP-1 | 1327 ± 89.7 | 1239 ± 111.1 | 864.2 ± 118.6 | ns | * |
| MIP-1β | 1302.61 ± 124.2 | 1308.63 ± 142.3 | 1003.01 ± 191.9 | ns | ns |
| RANTES | 195.83 ± 19.19 | 247.57 ± 39.04 | 248.55 ± 46.88 | ns | ns |
| Cytokine/Chemokine | Level (pg/500 µg Lung) | p Value | |||
|---|---|---|---|---|---|
| Vehicle | C3gs20 | C23 | C3gs20 vs. Vehicle | C23 vs. Vehicle | |
| IL-1α | 26 ± 3.14 | 6.91 ± 0.47 | 7.75 ± 0.75 | ns | ns |
| IL-1β | 24.04 ± 4.92 | 10.42 ± 2.11 | 15.93 ± 2.63 | * | ns |
| IL-2 | 4.49 ± 1.10 | nd | nd | n/a | n/a |
| IL-5 | 2.96 ± 0.37 | 2.85 ± 1.03 | 3.52 ± 0.60 | ns | ns |
| IL-9 | 99.56 ± 12.57 | 132.10 ± 24.21 | 116.00 ± 19.28 | ns | ns |
| IL-10 | 3.89 ± 0.25 | 3.10 ± 0.84 | 3.19 ± 0.47 | ns | ns |
| IL-12p40 | 11.27 ± 1.07 | 10.15 ± 1.13 | 10.20 ± 0.49 | ns | ns |
| IL-12p70 | 8.93 ± 1.17 | 4.62 ± 1.19 | 5.74 ± 0.90 | * | ns |
| IL-13 | 98.20 ± 14.41 | 76.03 ± 14.45 | 98.11 ± 14.37 | ns | ns |
| IL-17A | 7.94 ± 1.27 | 3.29 ± 0.66 | 4.41 ± 0.90 | ** | * |
| Eotaxin | 264.20 ± 30.39 | 135.20 ± 49.53 | 237.40 ± 34.22 | ns | ns |
| G-CSF | 4.62 ± 0.95 | 2.57 ± 0.54 | 3.89 ± 0.21 | * | ns |
| GM-CSF | 29.95 ± 2.66 | nd | 28.82 ± 10.48 | n/a | ns |
| IFN-γ | 5.22 ± 0.71 | 3.90 ± 1.03 | 6.13 ± 0.79 | ns | ns |
| KC | 13.92 ± 2.64 | 7.91 ± 0.69 | 11.71 ± 1.47 | * | ns |
| MCP-1 | 84.93 ± 9.12 | 70.74 ± 12.04 | 67.41 ± 7.78 | ns | ns |
| MIP-1β | 16.94 ± 1.99 | 15.80 ± 2.38 | 16.04 ± 2.05 | ns | ns |
| RANTES | 6.95 ± 0.59 | 6.86 ± 1.04 | 8.47 ± 1.07 | ns | ns |
| TNF-α | 6.59 ± 0.33 | 4.44 ± 0.63 | 7.55 ± 0.81 | ns | ns |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorè, N.I.; Veraldi, N.; Riva, C.; Sipione, B.; Spagnuolo, L.; De Fino, I.; Melessike, M.; Calzi, E.; Bragonzi, A.; Naggi, A.; et al. Synthesized Heparan Sulfate Competitors Attenuate Pseudomonas aeruginosa Lung Infection. Int. J. Mol. Sci. 2018, 19, 207. https://doi.org/10.3390/ijms19010207
Lorè NI, Veraldi N, Riva C, Sipione B, Spagnuolo L, De Fino I, Melessike M, Calzi E, Bragonzi A, Naggi A, et al. Synthesized Heparan Sulfate Competitors Attenuate Pseudomonas aeruginosa Lung Infection. International Journal of Molecular Sciences. 2018; 19(1):207. https://doi.org/10.3390/ijms19010207
Chicago/Turabian StyleLorè, Nicola Ivan, Noemi Veraldi, Camilla Riva, Barbara Sipione, Lorenza Spagnuolo, Ida De Fino, Medede Melessike, Elisa Calzi, Alessandra Bragonzi, Annamaria Naggi, and et al. 2018. "Synthesized Heparan Sulfate Competitors Attenuate Pseudomonas aeruginosa Lung Infection" International Journal of Molecular Sciences 19, no. 1: 207. https://doi.org/10.3390/ijms19010207