Genome-Wide Association Analysis of Mucilage and Hull Content in Flax (Linum usitatissimum L.) Seeds

 ,
, 
Abstract
1. Introduction
2. Results
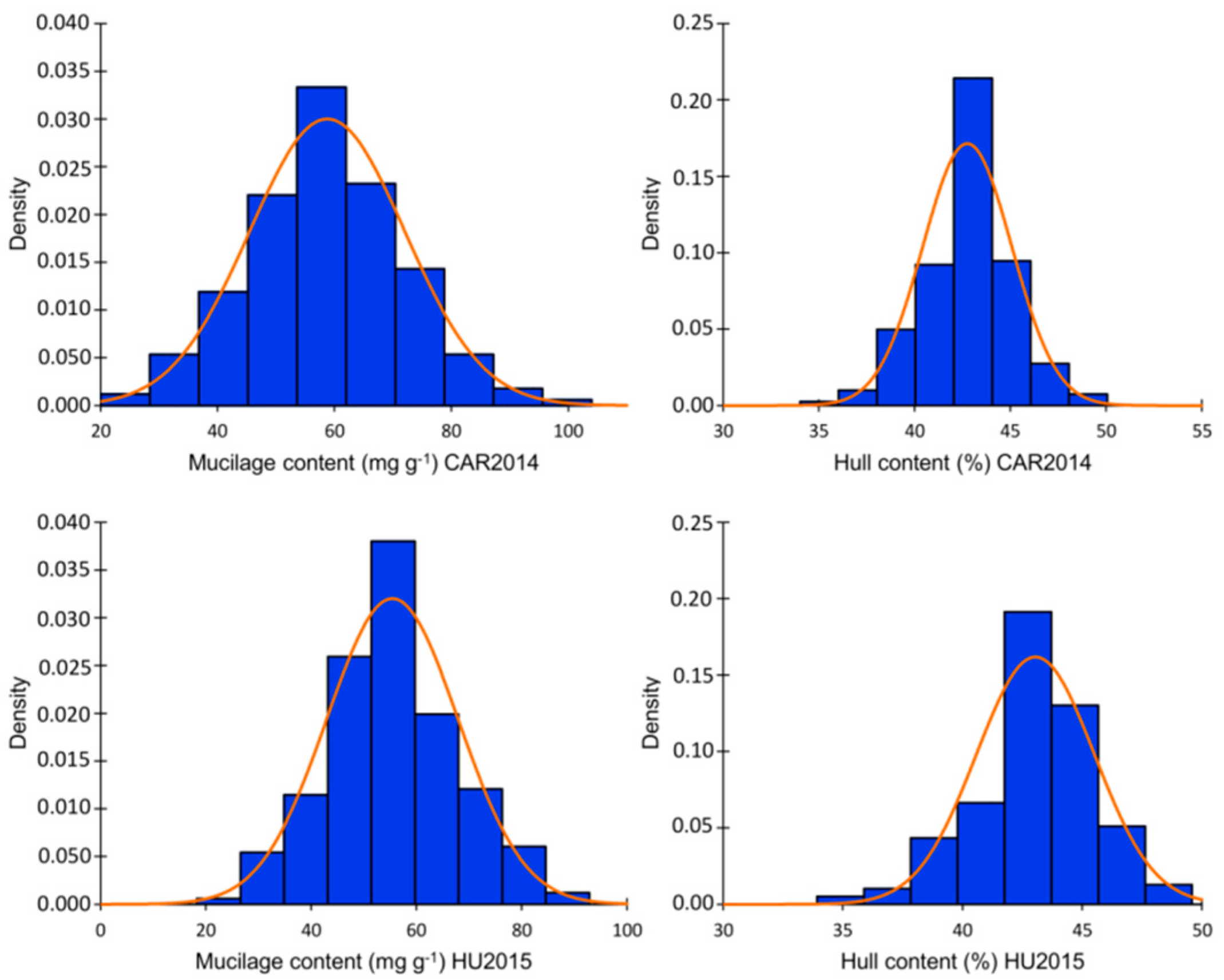
2.1. Phenotypic Evaluation
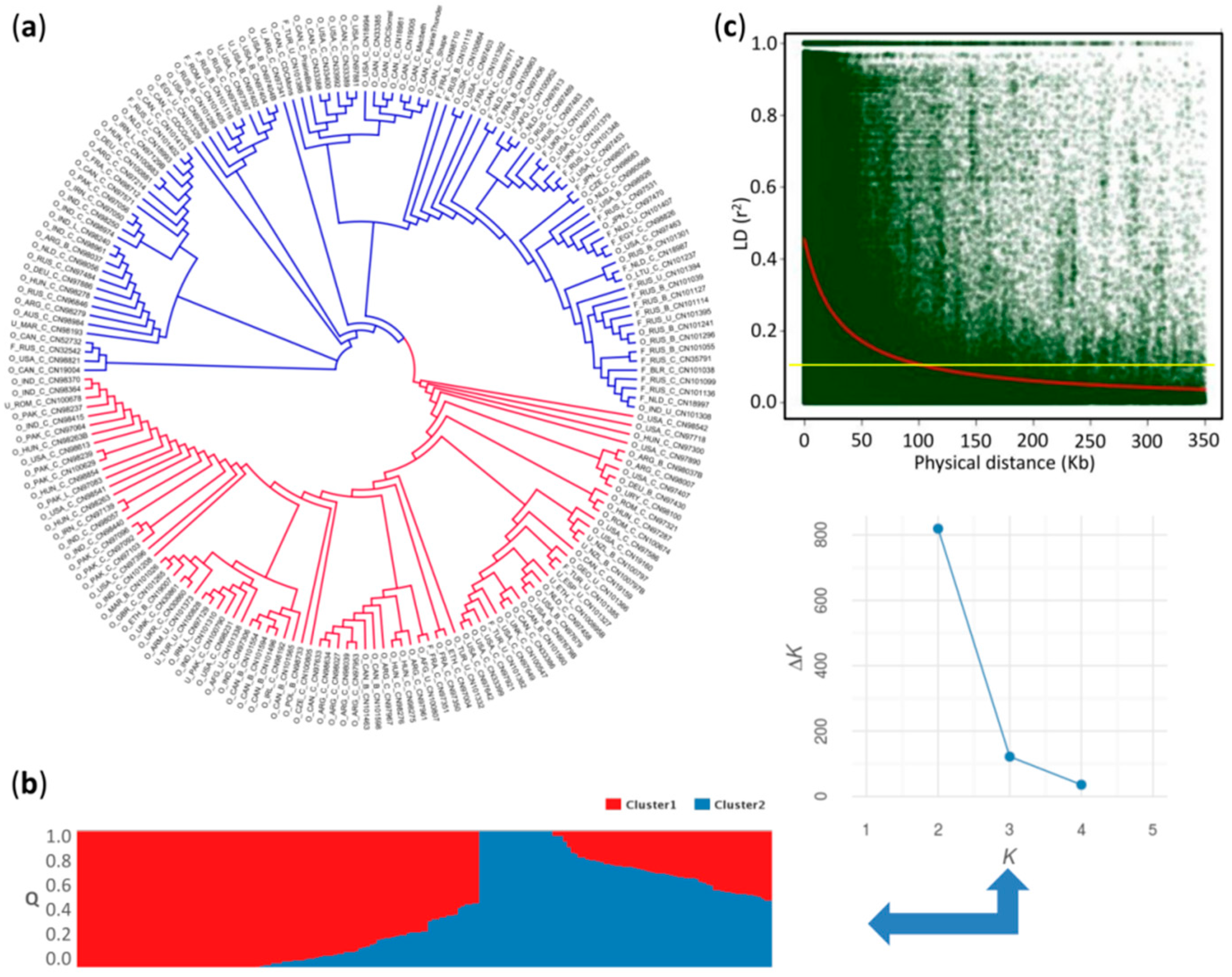
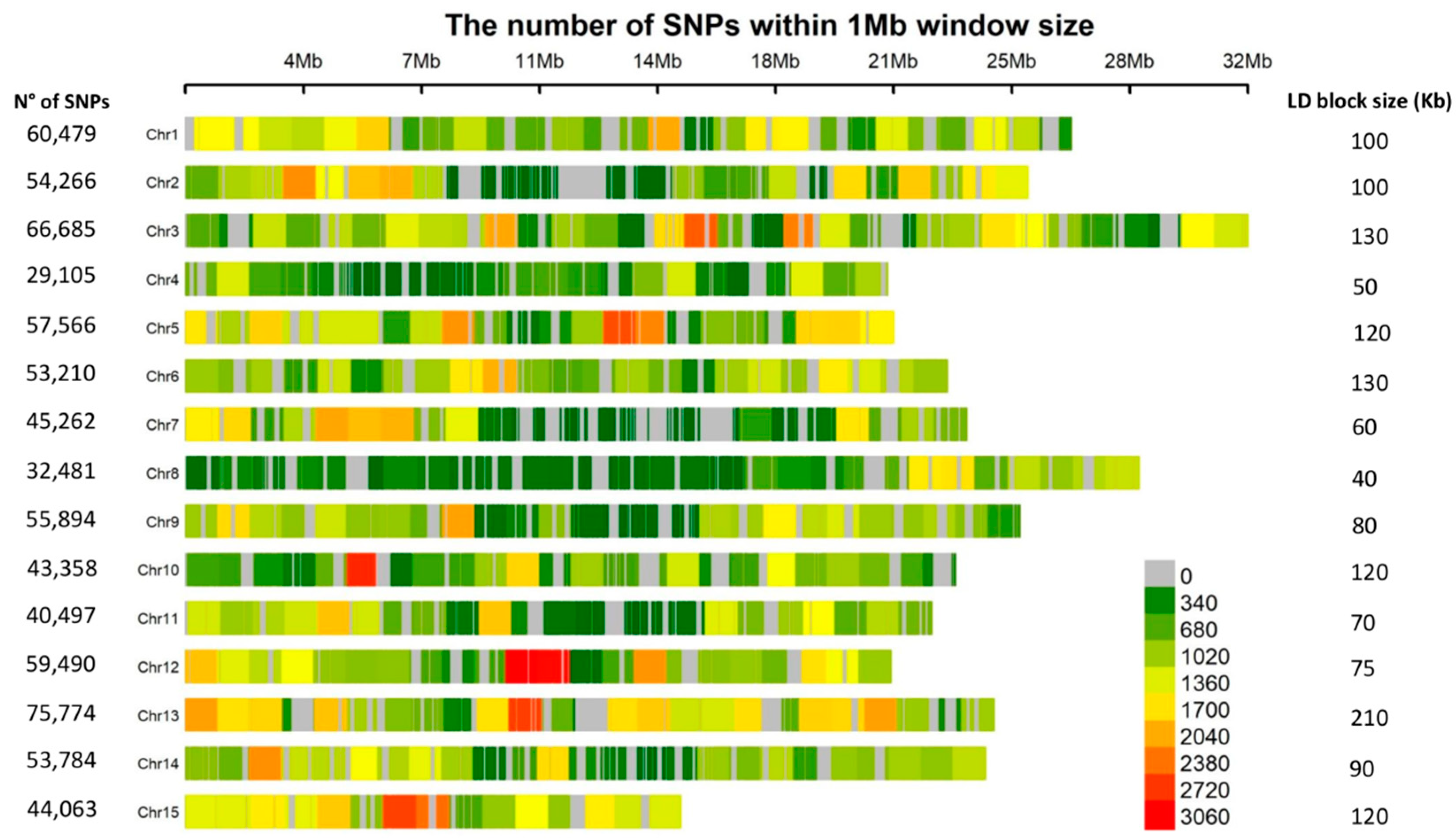
2.2. Population Structure and Linkage Disequilibrium
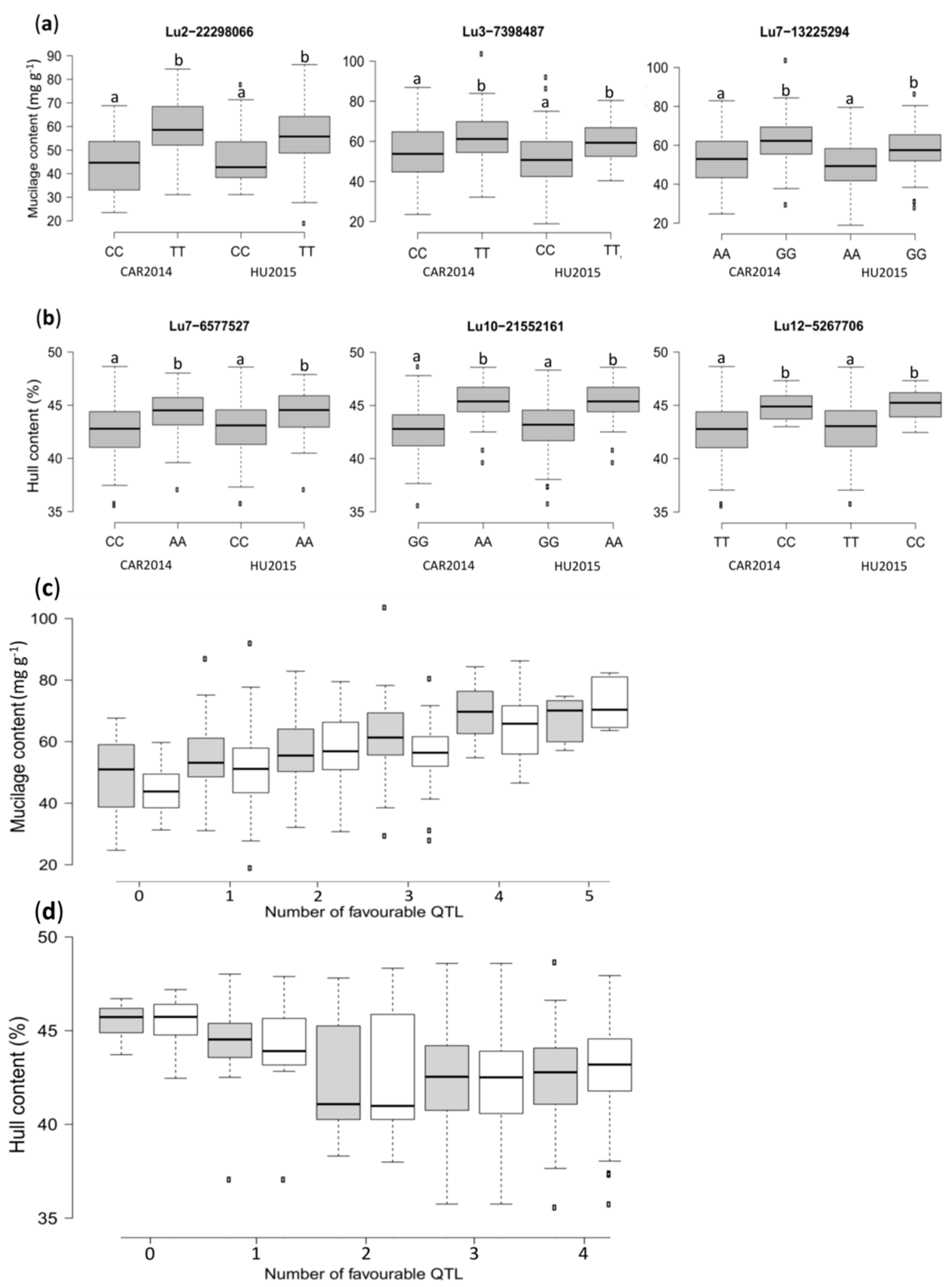
2.3. Genome-Wide Association Analysis
2.4. Identification of Candidate Genes
3. Discussion
3.1. Phenotypic Variation of Mucilage and Hull Contents
3.2. Population Structure and Linkage Disequilibrium
3.3. Genome-Wide Association Analysis
4. Materials and Methods
4.1. Plant Material, Field Trials, Phenotyping, and Statistical Analyses
4.2. Whole Genome Resequencing and SNP Calling
4.3. Population Structure, LD, Genome-Wide Association Study, and Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| MC | mucilage content |
| HC | gull content |
| GWAS | genome-wide association study |
| LD | linkage disequilibrium |
| kb | kilobase |
| SNP | single nucleotide polymorphism |
| SSR | simple sequence repeat |
| CAR2014 | Vilcún 2014 |
| HU2015 | Huichahue 2015 |
| REML | restricted maximum likelihood |
| AIC | Akaike information criterion |
| BIC | Bayesian information criterion |
| BLUE | best linear unbiased estimation |
| GLM | general linear model |
| MLM | mixed linear model |
| PCA | principal component analysis |
| Q–Q | quantile–quantile |
References
- Rabetafika, H.N.; van Remoortel, V.; Danthine, S.; Paquot, M.; Beckler, C. Flaxseed proteins: Food uses and health benefits. Int. J. Food Sci. Technol. 2011, 46, 221–228. [Google Scholar] [CrossRef]
- Kristensen, M.; Jensen, M.G.; Aarestrup, J.; Petersen, K.; Søndergaard, L.; Mikkelsen, M.S.; Astrup, A. Flaxseed dietary fibers lower cholesterol and increase fecal fat excretion, but magnitude of effect depend on food type. Nutr. Metab. 2012, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hunt, K.; Jones, J.K.N. The structure of linseed mucilage: Part II. Can. J. Chem. 1962, 40, 1266–1279. [Google Scholar] [CrossRef]
- Kaewmanee, T.; Bagnasco, L.; Benjakul, S.; Lanteri, S.; Morelli, C.F.; Speranza, G.; Cosulich, M.E. Characterisation of mucilages extracted from seven Italian cultivars of flax. Food Chem. 2014, 148, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Haughn, G.; Chaudhury, A. Genetic analysis of seed coat development in Arabidopsis. Trends Plant Sci. 2005, 10, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Bhatty, R.S.; Cherdkiatgumchai, P. Compositional analysis of laboratory-prepared and commercial samples of linseed meal and of hull isolated from flax. J. Am. Oil Chem. Soc. 1990, 67, 79–84. [Google Scholar] [CrossRef]
- Gajardo, H.A.; Quian, R.; Soto-Cerda, B. Agronomic and quality assessment of linseed advanced breeding lines varying in seed mucilage content and their use for food and feed. Crop Sci. 2017, 57, 2979–2990. [Google Scholar] [CrossRef]
- Cherian, G.; Quezada, N. Egg quality, fatty acid composition and immunoglobulin Y content in eggs from laying hens fed full fat camelina or flax seed. J. Anim. Sci. Biotechnol. 2016, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Oomah, B.D.; Mazza, G. Processing of flaxseed meal: Effect of solvent extraction on physicochemical characteristics. LWT-Food Sci. Technol. 1993, 26, 312–317. [Google Scholar] [CrossRef]
- Sosulski, F.W.; Bakal, A. Isolated proteins from rapeseed, flax and sunflower meals. Can. Inst. Food Sci. Technol. J. 1969, 2, 28–32. [Google Scholar] [CrossRef]
- Kessler, R.W.; Kohler, R. New strategies for exploiting flax and hemp. Chemtech 1996, 26, 34–42. [Google Scholar]
- Sosulski, F.; Zadernowski, R. Fractionation of rapeseed meal into hour and hull components. J. Am. Oil Chem. Soc. 1981, 58, 96–98. [Google Scholar] [CrossRef]
- Daun, J.K.; DeClercq, D.R. Quality of yellow and dark seeds in Brassica campentris canola varieties Candle and Tobin. J. Am. Oil Chem. Soc. 1988, 65, 122–126. [Google Scholar] [CrossRef]
- Francoz, E.; Ranocha, P.; Burlat, V.; Dunand, C. Arabidopsis seed mucilage secretory cells: Regulation and dynamics. Trends Plant Sci. 2015, 8, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Venglat, P.; Xiang, D.; Qiu, S.; Stone, S.L.; Tibiche, C.; Cram, D.; Alting-Mees, M.; Nowak, J.; Cloutier, S.; Deyholos, M.; et al. Gene expression analysis of flax seed development. BMC Plant Biol. 2011, 11, 74. [Google Scholar] [CrossRef] [PubMed]
- Oomah, B.D.; Kenaschuk, E.O.; Cui, W.; Mazza, G. Variation in the composition of water-soluble polysaccharides in flaxseed. J. Agric. Food Chem. 1995, 43, 1484–1488. [Google Scholar] [CrossRef]
- Oomah, B.D.; Mazza, G. Effect of dehulling on chemical composition and physical properties of flaxseed. LWT-Food Sci. Technol. 1997, 30, 135–140. [Google Scholar] [CrossRef]
- Diederichsen, A.; Raney, J.P.; Duguid, S.D. Variation of mucilage in flax seed and its relationship with other seed characters. Crop Sci. 2006, 46, 365–371. [Google Scholar] [CrossRef]
- Spielmeyer, W.; Green, A.G.; Bittisnish, D.; Mendham, N.; Lagudah, E.S. Identification of quantitative trait loci contributing to Fusarium wilt resistance on an AFLP linkage map of flax (Linum usitatissimum). Theor. Appl. Genet. 1998, 97, 633–641. [Google Scholar] [CrossRef]
- Asgarinia, P.; Cloutier, S.; Duguid, S.; Rashid, K.; Mirlohi, A.F.; Banik, M.; Saeidi, G. Mapping quantitative trait loci for powdery mildew resistance in flax (Linum usitatissimum L.). Crop Sci. 2013, 53, 2462–2472. [Google Scholar] [CrossRef]
- Cloutier, S.; Ragupathy, R.; Niu, Z.; Duguid, S. SSR-based linkage map of flax (Linum usitatissimum L.) and mapping of QTL underlying fatty acid composition traits. Mol. Breed. 2011, 28, 437–451. [Google Scholar] [CrossRef]
- Kumar, S.; You, F.M.; Duguid, S.; Booker, H.; Rowland, G.; Cloutier, S. QTL for fatty acid composition and yield in linseed (Linum usitatissimum L.). Theor. Appl. Genet. 2015, 128, 965–984. [Google Scholar] [CrossRef] [PubMed]
- Sudarshan, G.P.; Kulkarni, M.; Akhov, L.; Ashe, P.; Shaterian, H.; Cloutier, S.; Rowland, G.; Wei, Y.; Selvaraj, G. QTL mapping and molecular characterization of the classical D locus controlling seed and flower color in Linum usitatissimum (flax). Sci. Rep. 2017, 7, 15751. [Google Scholar] [CrossRef] [PubMed]
- Soto-Cerda, B.J.; Duguid, S.; Booker, H.; Rowland, G.; Diederichsen, A.; Cloutier, S. Genomic regions underlying agronomic traits in linseed (Linum usitatissimum L.) as revealed by association mapping. J. Integr. Plant Biol. 2014, 56, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Soto-Cerda, B.J.; Duguid, S.; Booker, H.; Rowland, G.; Diederichsen, A.; Cloutier, S. Association mapping of seed quality traits using the Canadian flax (Linum usitatissimum L.) core collection. Theor. Appl. Genet. 2014, 127, 881–896. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Dai, Z.; Yang, Z.; Sun, J.; Zhao, D.; Yang, X.; Zhang, L.; Tang, Q.; Su, J. Genome-wide association study identifying candidate genes influencing important agronomic traits of flax (Linum usitatissimum L.) using SLAF-seq. Front. Plant Sci. 2018, 8, 2232. [Google Scholar] [CrossRef] [PubMed]
- You, F.M.; Xiao, J.; Li, P.; Yao, Z.; Jia, G.; He, L.; Kumar, S.; Soto-Cerda, B.; Duguid, S.D.; Booker, H.M.; et al. Genome-Wide Association Study and Selection Signatures Detect Genomic Regions Associated with Seed Yield and Oil Quality in Flax. Int. J. Mol. Sci. 2018, 19, 2303. [Google Scholar] [CrossRef] [PubMed]
- Ersoz, E.S.; Yu, J.; Buckler, E.S. Applications of linkage disequilibrium and association mapping in maize. In Molecular Genetic Approaches to Maize Improvement; Kriz, A., Larkins, B., Eds.; Springer: Berlin, Germany, 2009; pp. 173–195. [Google Scholar]
- Cloutier, S.; Niu, Z.; Datla, R.; Duguid, S. Development and analysis of EST-SSRs for flax (Linum usitatissimum L.). Theor. Appl. Genet. 2009, 119, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, S.; Miranda, E.; Ward, K.; Radovanovic, N.; Reimer, E.; Walichnowski, A.; Datla, R.; Rowland, G.; Duguid, S.; Ragupathy, R. Simple sequence repeat marker development from bacterial artificial chromosome end sequences and expressed sequence tags of flax (Linum usitatissimum L.). Theor. Appl. Genet. 2012, 125, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, S.; Ragupathy, R.; Miranda, E.; Radovanovic, N.; Reimer, E.; Walichnowski, A.; Ward, K.; Rowland, G.; Duguid, S.; Banik, M. Integrated consensus genetic and physical maps of flax (Linum usitatissimum L.). Theor. Appl. Genet. 2012, 125, 1783–1795. [Google Scholar] [CrossRef] [PubMed]
- Ragupathy, R.; Rathinavelu, R.; Cloutier, S. Physical mapping and BAC-end sequence analysis provide initial insights into the flax (Linum usitatissimum L.) genome. BMC Genom. 2011, 12, 217. [Google Scholar] [CrossRef] [PubMed]
- Altunkaya, A. Dermal Lubricant and Moisturizer. WO 2006/075236 A1, 20 July 2006. [Google Scholar]
- Anttila, M.; Kankaanpää-Anttila, B.; Sepponen, M.; Timonen, H.; Autio, K. Improving of Texture of Dairy Products. WO 2008/000913 A1, 3 January 2008. [Google Scholar]
- Kracht, W.; Dänicke, S.; Kluge, H.; Keller, K.; Matzke, W.; Henning, U.; Schumann, W. Effect of dehulling of rapeseed on feed value and nutrient digestibility of rape products in pigs. Arch. Anim. Nutr. 2004, 58, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Oomah, B.D.; Mazza, G. Fractionation of flaxseed with a batch dehuller. Ind. Crop Prod. 1998, 9, 19–27. [Google Scholar] [CrossRef]
- Yan, X.Y.; Li, J.N.; Fu, F.Y.; Jin, M.Y.; Chen, L.; Liu, L.Z. Co-location of seed oil content, seed hull content and seed coat color QTL in three different environments in Brassica napus L. Euphytica 2009, 170, 355–364. [Google Scholar] [CrossRef]
- Saedi, G.; Rowland, G.G. Seed color and linolenic acid effects on agronomic traits in flax. Can. J. Plant Sci. 1999, 79, 521–526. [Google Scholar] [CrossRef]
- Zhao, K.; Tung, C.W.; Eizenga, G.C.; Wright, M.H.; Ali, M.L.; Price, A.H.; Norton, G.J.; Islam, M.R.; Reynolds, A.; Mezey, J.; et al. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat. Commun. 2011, 2, 467. [Google Scholar] [CrossRef] [PubMed]
- Smỳkal, P.; Bačová-Kerteszová, N.; Kalendar, R.; Corander, J.; Schulman, A.H.; Pavelek, M. Genetic diversity of cultivated flax (Linum usitatissimum L.) germplasm assessed by retrotransposon-based markers. Theor. Appl. Genet. 2011, 122, 1385–1397. [Google Scholar] [CrossRef] [PubMed]
- Soto-Cerda, B.J.; Diederichsen, A.; Ragupathy, R.; Cloutier, S. Genetic characterization of a core collection of flax (Linum usitatissimum L.) suitable for association mapping studies and evidence of divergent selection between fiber and linseed types. BMC Plant Biol. 2013, 13, 78. [Google Scholar] [CrossRef] [PubMed]
- Chandrawati, N.S.; Kumar, R.; Kumar, S.; Singh, P.K.; Yadav, V.K.; Ranade, S.A.; Yadav, H.K. Genetic diversity, population structure and association analysis in linseed (Linum usitatissimum L.). Physiol. Mol. Biol. Plants 2017, 23, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Abdurakhmonov, I.; Abdukarimov, A. Application of association mapping to understanding the genetic diversity of plant germplasm resources. Int. J. Plant Genom. 2008, 2008, 574297. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ranc, N.; Munos, S.; Rolland, S.; Bouchet, J.P.; Desplant, N.; Le Paslier, M.C.; Liang, Y.; Brunel, D.; Causse, M. Phenotypic diversity and association mapping for fruit quality traits in cultivated tomato and related species. Theor. Appl. Genet. 2013, 126, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Ching, A.; Bhattramakki, D.; Dolan, M.; Tingey, S.; Morgante, M.; Rafalski, A. Linkage disequilibrium and sequence diversity in a 500-kbp region around the adh1 locus in elite maize germplasm. Theor. Appl. Genet. 2004, 109, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Hatzig, S.V.; Frisch, M.; Breuer, F.; Nesi, N.; Ducournau, S.; Wagner, M.H.; Leckband, G.; Abbadi, A.; Snowdon, R.J. Genome-wide association mapping unravels the genetic control of seed germination and vigor in Brassica napus. Front. Plant Sci. 2015, 6, 221. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Rosenberg, N.A.; Donnelly, P. Association mapping in structured populations. Am. J. Hum. Genet. 2000, 67, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Pressoir, G.; Briggs, W.; Vroh, B.; Yamasaki, M.; Doebley, J.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Western, T.L.; Young, D.S.; Dean, G.H.; Tan, W.L.; Samuels, A.L.; Haughn, G.W. MUCILAGE-MODIFIED4 encondes a putative pectin biosynthetic enzyme developmentally regulated by APETALA2, TRANSPARENT TESTA GLABRA1, and GLABRA2 in the Arabidopsis seed coat. Plant Physiol. 2004, 134, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Zhou, G.; Abdeen, A.A.; Schafhauser, J.; Richardson, B.; Atmodjo, M.A.; Jung, J.; Wicker, L.; Mohnen, D.; Western, T.; Hahn, M.G. GALACTURONOSYLTRANSFERASE-LIKE5 is involved in the production of Arabidopsis seed coat mucilage. Plant Physiol. 2013, 163, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Rautengarten, C.; Usadel, B.; Neumetzler, L.; Hartmann, J.; Büssis, D.; Altmann, T. A subtilisin-like serine protease essential for mucilage release from Arabidopsis seed coats. Plant J. 2008, 54, 466–480. [Google Scholar] [CrossRef] [PubMed]
- Saez-Aguayo, S.; Ralet, M.C.; Berger, A.; Botran, L.; Ropartz, D.; Marion-Poll, A.; North, H.M. PECTIN METHYLESTERASE INHIBITOR6 promotes Arabidopsis mucilage release by limiting methylesterification of homogalacturonan in seed coat epidermal cells. Plant Cell 2013, 25, 308–323. [Google Scholar] [CrossRef] [PubMed]
- Louvet, R.; Cavel, E.; Gutierrez, L.; Guénin, S.; Roger, D.; Gillet, F.; Guerineau, F.; Pelloux, J. Comprehensive expression profiling of the pectin methylesterase gene family during silique development in Arabidopsis thaliana. Planta 2006, 224, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Katavic, V.; Yu, Y.; Kunst, L.; Haughn, G. Arabidopsis glabra2 mutant seeds deficient in mucilage biosynthesis produce more oil. Plant J. 2012, 69, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, J.N.; Chen, L.; Tang, Z.L.; Zhang, X.K. Genetic correlation analysis for main characters in yellow-seeded rapeseed lines (Brassica napus L.). Chin. J. Oil Crop Sci. 2003, 25, 8–11. [Google Scholar]
- Khan, N.A.; Booker, H.; Yu, P. Molecular structures and metabolic characteristics of protein in brown and yellow flaxseed with altered nutrient traits. J. Agri. Food Chem. 2014, 62, 6556–6564. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Hasan, M.; Lu, K.; Liu, L.; Zhang, K.; Fu, F.; Wang, M.; Liu, S.; Bu, H.; Wang, R.; et al. Identification of QTL for seed coat colour and oil content in Brassica napus by association mapping using SSR markers. Can. J. Plant Sci. 2015, 95, 387–395. [Google Scholar] [CrossRef]
- Badani, A.G.; Snowdon, R.J.; Wittkop, B.; Lipsa, F.D.; Baetzel, R.; Horn, R.; De Haro, A.; Font, R.; Lühs, W.; Friedt, W. Colocalization of a partially dominant gene for yellow seed colour with a major QTL influencing acid detergent fibre (ADF) content in different crosses of oilseed rape (Brassica napus). Genome 2006, 49, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Sun, L.; Li, S.; Wang, W.; Ding, Y.; Swarm, S.A.; Li, L.; Wang, X.; Tang, X.; Zhang, Z.; et al. Elevation of soybean seed oil content through selection for seed coat shininess. Nat. Plants 2018, 4, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Roszak, P.; Köhler, C. Polycomb group proteins are required to couple seed coat initiation to fertilization. Proc. Natl. Acad. Sci. USA 2011, 108, 20826–20831. [Google Scholar] [CrossRef] [PubMed]
- Mizzotti, C.; Ezquer, I.; Paolo, D.; Rueda-Romero, P.; Guerra, R.F.; Battaglia, R.; Rogachev, I.; Aharoni, A.; Kater, M.M.; Caporali, E.; et al. SEEDSTICK is a master regulator of development and metabolism in the Arabidopsis seed coat. PLoS Genet. 2014, 10, e1004856. [Google Scholar] [CrossRef] [PubMed]
- Marinova, K.; Pourcel, L.; Weder, B.; Schwarz, M.; Barron, D.; Routaboul, J.M.; Debeaujon, I.; Klein, M. The Arabidopsis MATE transporter TT12 acts as a vacuolar flavonoid/H+-antiporter active in proanthocyanidin-accumulating cells of the seed coat. Plant Cell 2007, 19, 2023–2038. [Google Scholar] [CrossRef] [PubMed]
- Kovinich, N.; Saleem, A.; Arnason, J.T.; Miki, B. Functional characterization of a UDP-glucose:flavonoid 3-O-glucosyltransferase from the seed coat of black soybean (Glycine max (L.) Merr.). Phytochemistry 2010, 71, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Dubos, C.; Lepiniec, L. Transcriptional control of flavonoid biosynthesis by MYB–bHLH–WDR complexes. Trends Plant Sci. 2015, 20, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Mole, S.; Waterman, P.G. Tannic acid and proteolytic enzymes: Enzyme inhibition or substrate deprivation? Phytochemistry 1986, 26, 99–102. [Google Scholar] [CrossRef]
- Diederichsen, A.; Kusters, P.M.; Kessler, D.; Bainas, Z.; Gugel, R.K. Assembling a core collection from the flax world collection maintained by Plant Gene Resources of Canada. Genet. Resour. Crop Evol. 2013, 60, 1479–1485. [Google Scholar] [CrossRef]
- VSN International. Genstat for Windows, 18th ed.; VSN International: Hemel Hempstead, UK, 2015; Available online: http://www.Genstat.co.uk (accessed on 10 May 2015).
- Korkmaz, S.; Goksuluk, D.; Zararsiz, G. MVN: An R Package for Assessing Multivariate Normality. R J. 2014, 6, 151–162. [Google Scholar]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Hobson, N.; Galindo, L.; Zhu, S.; Shi, D.; McDill, J.; Yang, L.; Hawkins, S.; Neutelings, G.; Datla, R.; et al. The genome of flax (Linum usitatissimum) assembled de novo from short shotgun sequence reads. Plant J. 2012, 72, 461–473. [Google Scholar] [CrossRef] [PubMed]
- You, F.M.; Xiao, J.; Li, P.; Yao, Z.; Jia, G.; He, L.; Zhu, T.; Luo, M.C.; Wang, X.; Deyholos, M.K.; et al. Chromosome-scale pseudomolecules refined by optical, physical and genetic maps in flax. Plant J. 2018, 95, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; You, F.M.; Cloutier, S. Genome wide SNP discovery in flax through next generation sequencing of reduced representation libraries. BMC Genom. 2012, 13, 684. [Google Scholar] [CrossRef] [PubMed]
- You, F.M.; Deal, K.R.; Wang, J.; Britton, M.T.; Fass, J.N.; Lin, D.; Dandekar, A.M.; Leslie, C.A.; Aradhya, M.; Luo, M.C.; et al. Genome-wide SNP discovery in walnut with an AGSNP pipeline updated for SNP discovery in allogamous organisms. BMC Genom. 2012, 13, 354. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Francis, M.R. POPHELPER: An R package and web app to analyse and visualise population structure. Mol. Ecol. Resour. 2017, 1, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Hill, W.G.; Weir, B.S. Variances and covariances of squared linkage disequilibria in finite populations. Theor. Popul. Biol. 1998, 33, 54–78. [Google Scholar] [CrossRef]
- Turner, S.D. QQMAN: An R package for visualizing GWAS results using Q-Q and manhattan plots. biorXiv 2014. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Marker | Chromosome | Allele | MAF 1 | −log10 (P) | R2 (%) | ||
|---|---|---|---|---|---|---|---|---|
| CAR2014 | HU2015 | CAR2014 | HU2015 | |||||
| MC | Lu2-22298066 | 2 | T/C | 0.07 | 8.69 | 3.41ns 2 | 17.32 | ns 2 |
| Lu3-25559600 | 3 | G/T | 0.06 | 7.45 | 4.13ns 2 | 13.42 | ns 2 | |
| Lu3-26033342 | 3 | C/G | 0.07 | 7.68 | 4.23ns 2 | 13.25 | ns 2 | |
| Lu3-7398487 | 3 | C/T | 0.41 | 4.96ns 2 | 7.02 | ns 2 | 11.82 | |
| Lu5-3808878 | 5 | G/A | 0.10 | 8.03 | 10.21 | 14.97 | 16.52 | |
| Lu7-13225294 | 7 | G/A | 0.34 | 8.10 | 6.91 | 16.46 | 12.05 | |
| Lu11-2498303 | 11 | C/G | 0.16 | 7.05 | 7.47 | 14.25 | 13.18 | |
| HC | Lu7-6577527 | 7 | A/C | 0.13 | 6.90 | 7.36 | 14.66 | 15.79 |
| Lu10-21552161 | 10 | G/A | 0.09 | 6.90 | 6.16ns 2 | 16.32 | ns 2 | |
| Lu12-5267706 | 12 | C/T | 0.06 | 5.91ns 2 | 6.92 | ns 2 | 13.83 | |
| Lu13-2803224 | 13 | T/C | 0.06 | 7.83 | 8.45 | 17.43 | 18.20 | |
| Trait | Marker | Gene ID | Scaffold | A. Thaliana Ortholog | Gene Bank | Identity (%) | E-Value | Distance from Peak SNP (kb) |
|---|---|---|---|---|---|---|---|---|
| MC | Lu3-25559600 | Lus10009311 | 318 | GATL5 | at1g02720 | 27 | 7 × 10−27 | 64.41 |
| Lus10009288 | 318 | MUM4 | at1g53500 | 26 | 4 × 10−23 | 67.02 | ||
| Lus10009287 | 318 | PME36 | at3g60730 | 61 | 2 × 10−110 | 70.33 | ||
| Lus10009313 | 318 | SBT1.7 | at5g67360 | 45 | 0.0 | 75.66 | ||
| Lu3-26033342 | Lus10007101 | 772 | TT8 | at4g09820 | 38 | 5 × 10−15 | 58.92 | |
| Lus10007083 | 772 | SBT1.7 | at5g67360 | 39 | 1 × 10−152 | 49.60 | ||
| Lu5-3808878 | Lus10008285 | 489 | NARS1 | at3g15510 | 52 | 9 × 10−45 | 100.78 | |
| HC | Lu7-6577527 | Lus10035456 | 151 | AGL62 | at5g60440 | 43 | 6 × 10−39 | 11.40 |
| Lu12-5267706 | Lus10018306 | 163 | GH17 | at2g39640 | 34 | 9 × 10−86 | 39.93 | |
| Lu13-2803224 | Lus10026902 | 651 | DBR1 | at4g31770 | 68 | 0.0 | 96.87 | |
| Lus10026926 | 651 | UGT79B1 | at5g54060 | 25 | 2 × 10−32 | 238.19 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soto-Cerda, B.J.; Cloutier, S.; Quian, R.; Gajardo, H.A.; Olivos, M.; You, F.M. Genome-Wide Association Analysis of Mucilage and Hull Content in Flax (Linum usitatissimum L.) Seeds. Int. J. Mol. Sci. 2018, 19, 2870. https://doi.org/10.3390/ijms19102870
Soto-Cerda BJ, Cloutier S, Quian R, Gajardo HA, Olivos M, You FM. Genome-Wide Association Analysis of Mucilage and Hull Content in Flax (Linum usitatissimum L.) Seeds. International Journal of Molecular Sciences. 2018; 19(10):2870. https://doi.org/10.3390/ijms19102870
Chicago/Turabian StyleSoto-Cerda, Braulio J., Sylvie Cloutier, Rocío Quian, Humberto A. Gajardo, Marcos Olivos, and Frank M. You. 2018. "Genome-Wide Association Analysis of Mucilage and Hull Content in Flax (Linum usitatissimum L.) Seeds" International Journal of Molecular Sciences 19, no. 10: 2870. https://doi.org/10.3390/ijms19102870
APA StyleSoto-Cerda, B. J., Cloutier, S., Quian, R., Gajardo, H. A., Olivos, M., & You, F. M. (2018). Genome-Wide Association Analysis of Mucilage and Hull Content in Flax (Linum usitatissimum L.) Seeds. International Journal of Molecular Sciences, 19(10), 2870. https://doi.org/10.3390/ijms19102870