Thioredoxin Confers Intrinsic Resistance to Cytostatic Drugs in Human Glioma Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
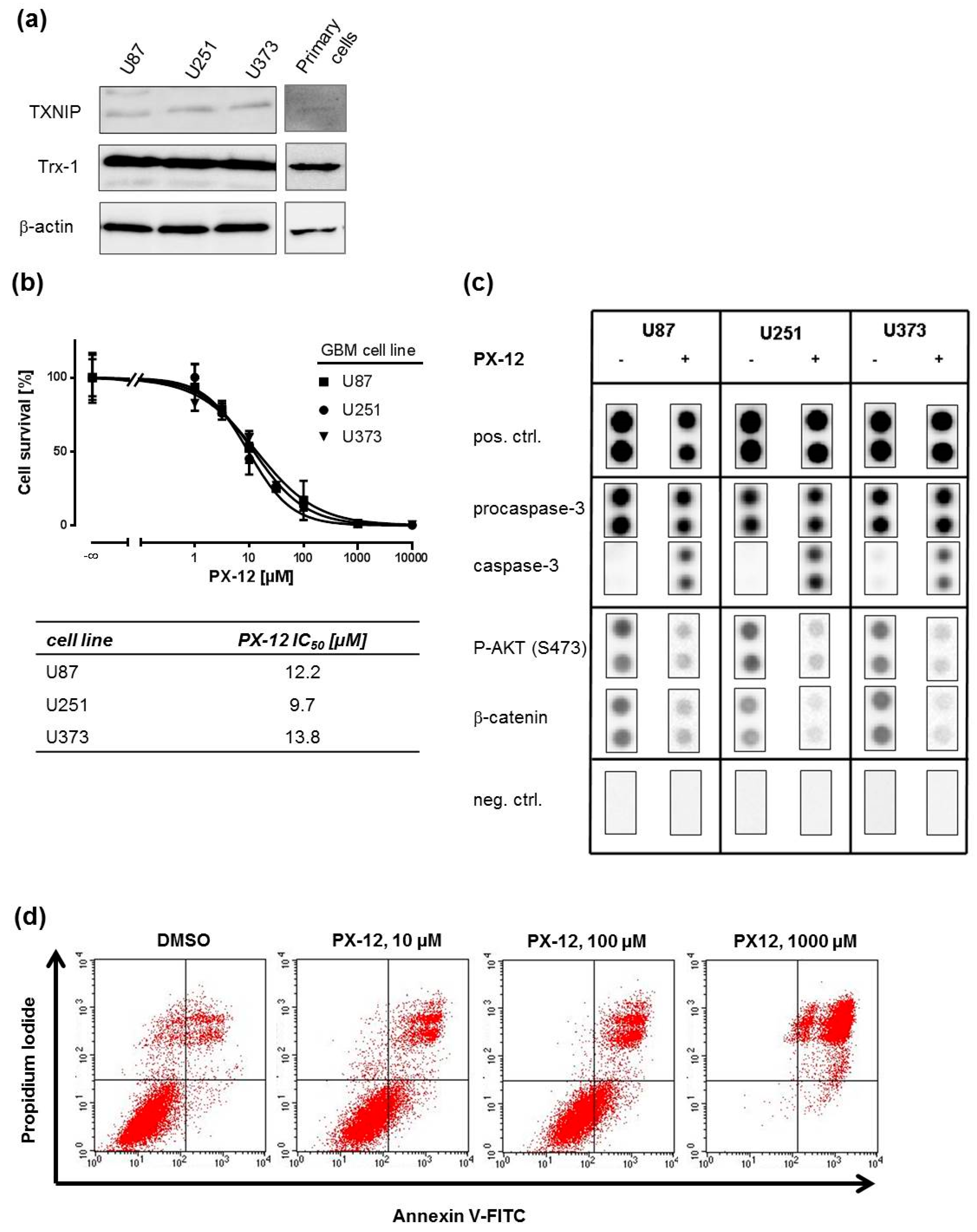
2.1. Trx-Inhibition Triggers Apoptosis in Human GBM Cell Lines
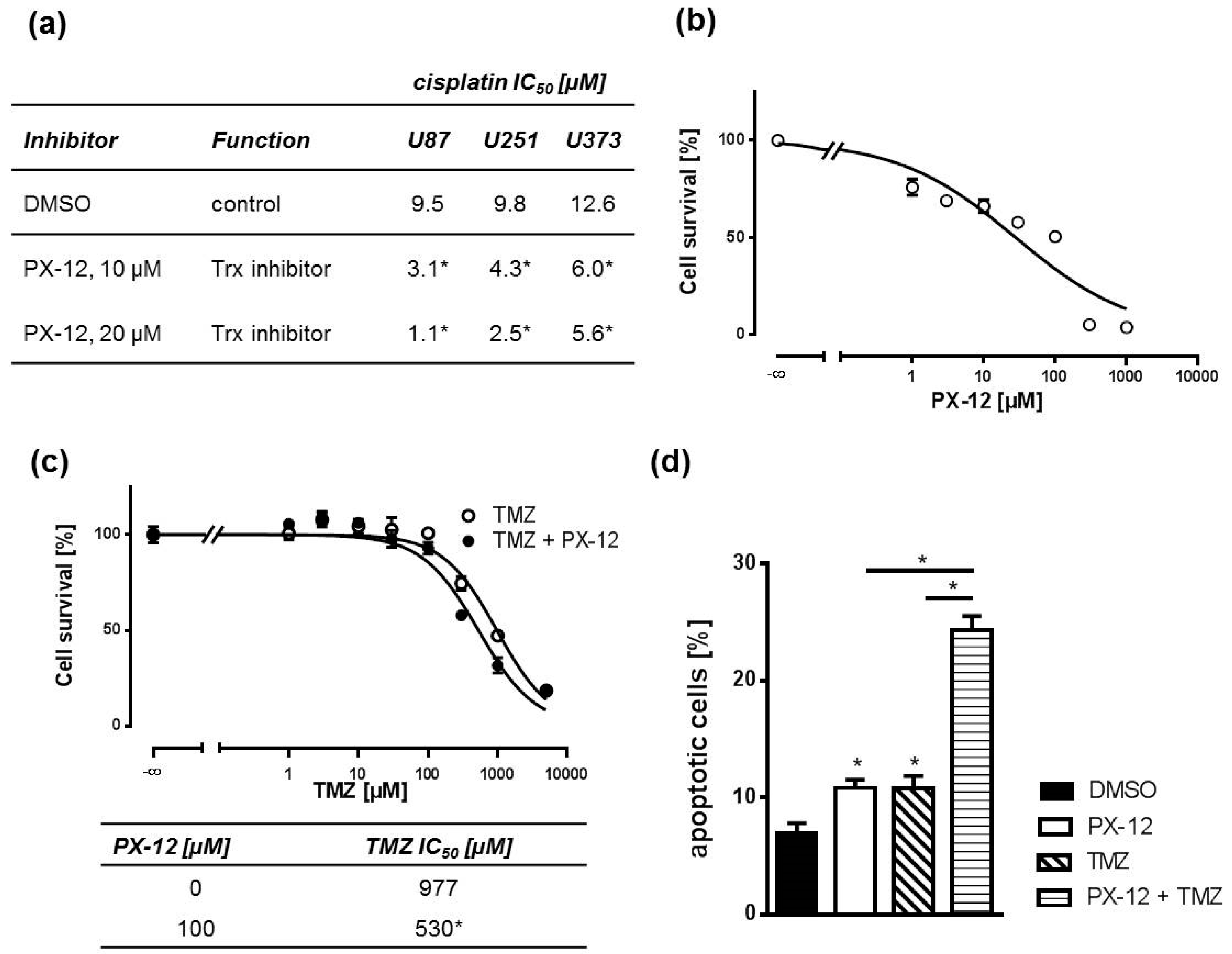
2.2. Pharmacological Trx Inhibition Sensitizes Human GBM Cells to Cytostatic Drugs
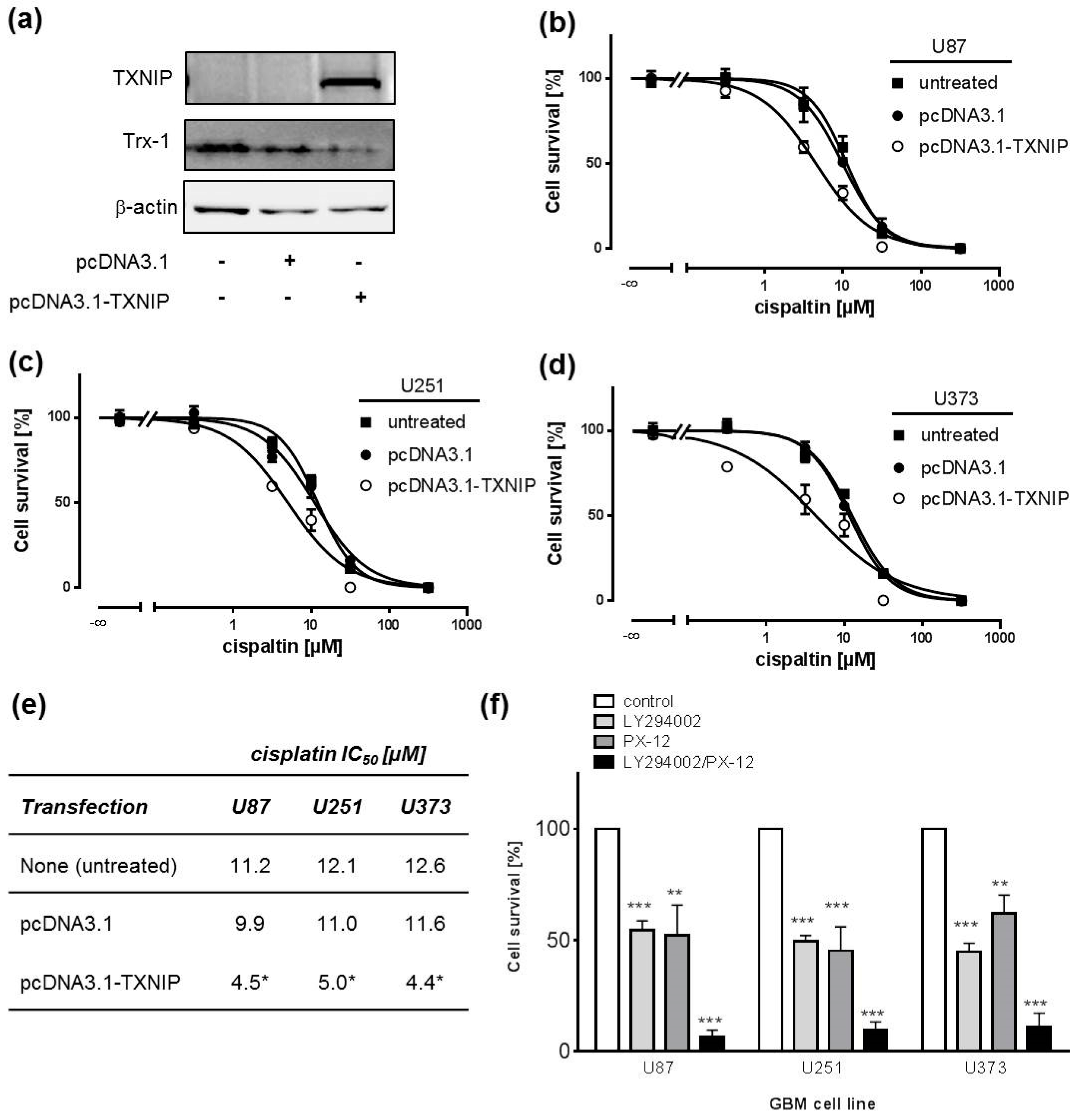
2.3. TXNIP Overexpression Chemosensitizes Human GBM Cell Lines
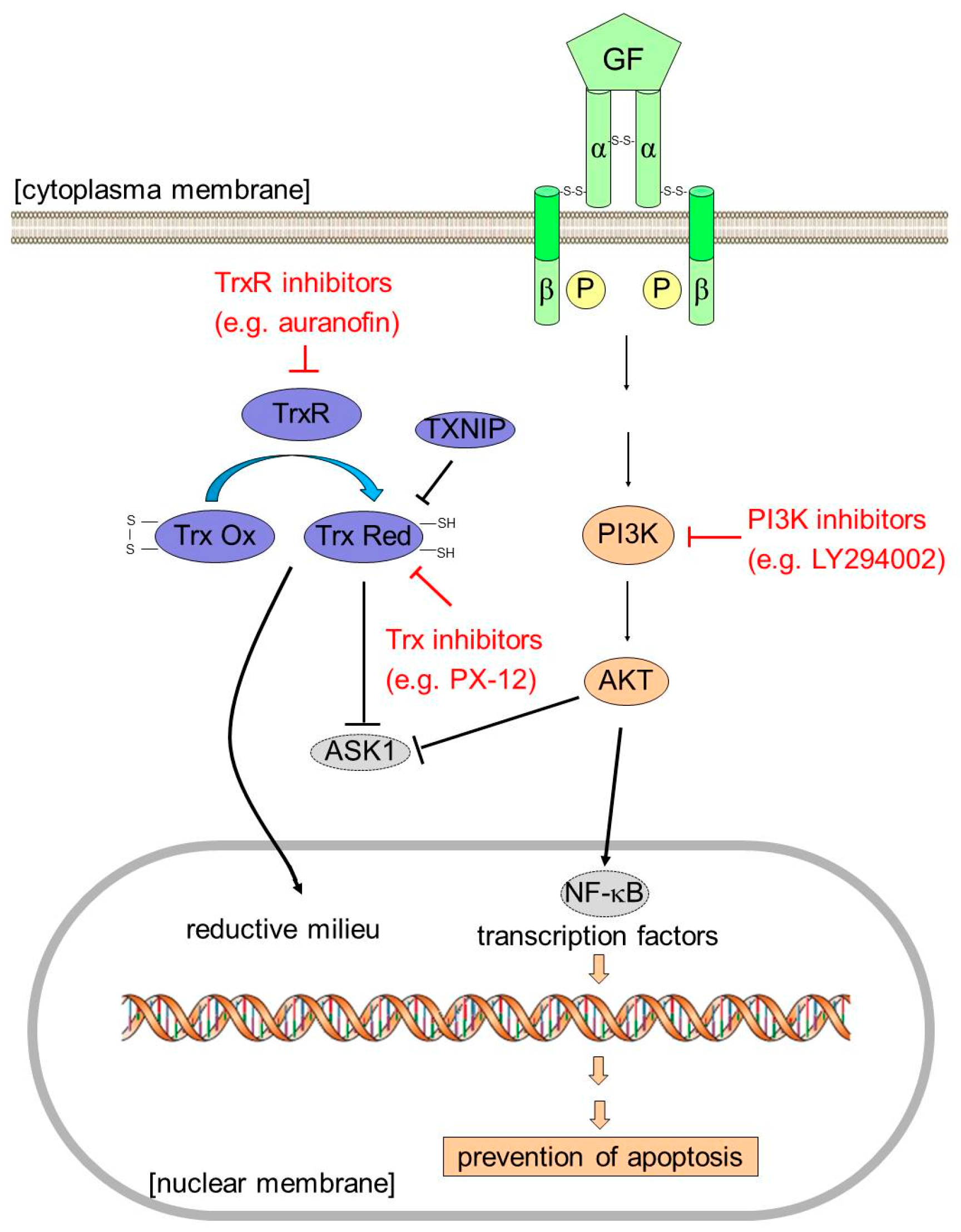
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Transfection, and Preparation of Cell Lysates
4.2. Proteome Profiling
4.3. Western Blot Analysis
4.4. MTT Assay
4.5. Annexin V Apoptosis Assay
4.6. Data Analysis and Statistical Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Disclaimer
Abbreviations
| AKT | protein kinase B (PKB) |
| ATP | adenosine triphosphate |
| ASK1 | apoptosis signal-regulating kinase 1 |
| GBM | glioblastoma multiforme |
| GF | growth factor |
| HIF-1 α | hypoxia-inducible factor-1 α |
| JNK | c-Jun N-terminal kinase |
| MAP kinase | mitogen-activated protein kinase |
| PI3K | phosphatidylinositide 3-kinase |
| PTEN | phosphatase and tensin homolog |
| ROS | reactive oxygen species |
| TMZ | temozolomide |
| Trx | thioredoxin |
| TrxR | thioredoxin reductase |
| TXNIP | Trx-interacting protein |
| VDUP1 | vitamin-D3-upregulated protein 1 |
| VEGF | vascular endothelial growth factor |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-Year analysis of the EORTC-NCIC trial. Lancet. Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Gittleman, H.; Xu, J.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2009–2013. Neuro Oncol. 2016, 18, v1–v75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrasco-Garcia, E.; Saceda, M.; Martinez-Lacaci, I. Role of receptor tyrosine kinases and their ligands in glioblastoma. Cells 2014, 3, 199–235. [Google Scholar] [CrossRef] [PubMed]
- Ishii, N.; Maier, D.; Merlo, A.; Tada, M.; Sawamura, Y.; Diserens, A.C.; van Meir, E.G. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999, 9, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Iwadate, Y.; Fujimoto, S.; Tagawa, M.; Namba, H.; Sueyoshi, K.; Hirose, M.; Sakiyama, S. Association of p53 gene mutation with decreased chemosensitivity in human malignant gliomas. Int. J. Cancer 1996, 69, 236–240. [Google Scholar] [CrossRef]
- Holmgren, A. Thioredoxin structure and mechanism: Conformational changes on oxidation of the active-site sulfhydryls to a disulfide. Structure 1995, 3, 239–243. [Google Scholar] [CrossRef]
- Nishinaka, Y.; Nishiyama, A.; Masutani, H.; Oka, S.; Ahsan, K.M.; Nakayama, Y.; Ishii, Y.; Nakamura, H.; Maeda, M.; Yodoi, J. Loss of thioredoxin-binding protein-2/vitamin D3 up-regulated protein 1 in human T-cell leukemia virus type I-dependent T-cell transformation: Implications for adult T-cell leukemia leukemogenesis. Cancer Res. 2004, 64, 1287–1292. [Google Scholar] [CrossRef] [PubMed]
- Powis, G.; Montfort, W.R. Properties and biological activities of thioredoxins. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 261–295. [Google Scholar] [CrossRef] [PubMed]
- Sasada, T.; Nakamura, H.; Ueda, S.; Sato, N.; Kitaoka, Y.; Gon, Y.; Takabayashi, A.; Spyrou, G.; Holmgren, A.; Yodoi, J. Possible involvement of thioredoxin reductase as well as thioredoxin in cellular sensitivity to cis-diamminedichloroplatinum (II). Free Radic. Biol. Med. 1999, 27, 504–514. [Google Scholar] [CrossRef]
- Yokomizo, A.; Ono, M.; Nanri, H.; Makino, Y.; Ohga, T.; Wada, M.; Okamoto, T.; Yodoi, J.; Kuwano, M.; Kohno, K. Cellular levels of thioredoxin associated with drug sensitivity to cisplatin, mitomycin C, doxorubicin, and etoposide. Cancer Res. 1995, 55, 4293–4296. [Google Scholar] [PubMed]
- Yamada, M.; Tomida, A.; Yoshikawa, H.; Taketani, Y.; Tsuruo, T. Overexpression of thioredoxin does not confer resistance to cisplatin in transfected human ovarian and colon cancer cell lines. Cancer Chemother. Pharmacol. 1997, 40, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Gao, J.; Wang, X.; Wen, W.; Yang, H.; Tian, Y.; Liu, N.; Wang, Z.; Liu, H.; Zhang, Y.; et al. A novel indication of thioredoxin-interacting protein as a tumor suppressor gene in malignant glioma. Oncol. Lett. 2017, 14, 2053–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gollapalli, K.; Ghantasala, S.; Atak, A.; Rapole, S.; Moiyadi, A.; Epari, S.; Srivastava, S. Tissue Proteome Analysis of Different Grades of Human Gliomas Provides Major Cues for Glioma Pathogenesis. OMICS 2017, 21, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M.; McGrath, K.L.; Di Trapani, G.; Charoentong, P.; Shah, F.; King, M.M.; Clarke, F.M.; Tonissen, K.F. The thioredoxin system in breast cancer cell invasion and migration. Redox Biol. 2016, 8, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the Thioredoxin System for Cancer Therapy. Trends Pharmacol. Sci. 2017, 38, 794–808. [Google Scholar] [CrossRef] [PubMed]
- Stafford, W.C.; Peng, X.; Olofsson, M.H.; Zhang, X.; Luci, D.K.; Lu, L.; Cheng, Q.; Tresaugues, L.; Dexheimer, T.S.; Coussens, N.P.; et al. Irreversible inhibition of cytosolic thioredoxin reductase 1 as a mechanistic basis for anticancer therapy. Sci. Transl. Med. 2018, 10, 428. [Google Scholar] [CrossRef] [PubMed]
- Fiskus, W.; Saba, N.; Shen, M.; Ghias, M.; Liu, J.Y.; Das Gupta, S.; Chauhan, L.; Rao, R.; Gunewardena, S.; Schorno, K.; et al. Auranofin Induces Lethal Oxidative and Endoplasmic Reticulum Stress and Exerts Potent Preclinical Activity against Chronic Lymphocytic Leukemia. Cancer Res. 2014, 74, 2520–2532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang-Bo, H.; Jeong, J.W.; Han, M.H.; Park, C.; Hong, S.H.; Kim, G.Y.; Moon, S.K.; Cheong, J.; Kim, W.J.; Yoo, Y.H.; et al. Auranofin, an inhibitor of thioredoxin reductase, induces apoptosis in hepatocellular carcinoma Hep3B cells by generation of reactive oxygen species. Gen. Physiol. Biophys. 2017, 36, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.W.; Kwon, Y.J.; Ye, D.J.; Baek, H.S.; Lee, J.E.; Chun, Y.J. Auranofin Suppresses Plasminogen Activator Inhibitor-2 Expression through Annexin A5 Induction in Human Prostate Cancer Cells. Biomol. Ther. 2017, 25, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Hu, J.; Wu, S.; Wang, L.; Cao, X.; Zhang, X.; Dai, B.; Cao, M.; Shao, R.; Zhang, R.; et al. Auranofin-mediated inhibition of PI3K/AKT/mTOR axis and anticancer activity in non-small cell lung cancer cells. Oncotarget 2016, 7, 3548–3558. [Google Scholar] [CrossRef] [PubMed]
- Hou, G.X.; Liu, P.P.; Zhang, S.; Yang, M.; Liao, J.; Yang, J.; Hu, Y.; Jiang, W.Q.; Wen, S.; Huang, P. Elimination of stem-like cancer cell side-population by auranofin through modulation of ROS and glycolysis. Cell. Death Dis. 2018, 9, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habermann, K.J.; Grunewald, L.; van Wijk, S.; Fulda, S. Targeting redox homeostasis in rhabdomyosarcoma cells: GSH-depleting agents enhance auranofin-induced cell death. Cell. Death Dis. 2017, 8, e3067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raninga, P.V.; Di Trapani, G.; Vuckovic, S.; Tonissen, K.F. Cross-talk between two antioxidants, thioredoxin reductase and heme oxygenase-1, and therapeutic implications for multiple myeloma. Redox Biol. 2016, 8, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Marzano, C.; Gandin, V.; Folda, A.; Scutari, G.; Bindoli, A.; Rigobello, M.P. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radic Biol. Med. 2007, 42, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Manchandia, T.; Ban, K.; Gao, S.; Miller, C.; Chandra, J. Adaphostin cytoxicity in glioblastoma cells is ROS-dependent and is accompanied by upregulation of heme oxygenase-1. Cancer Chemother. Pharmacol. 2007, 59, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Joseph, C.; Ghosh, S.; Agarwal, A.; Mishra, M.K.; Sen, E. Kaempferol induces apoptosis in glioblastoma cells through oxidative stress. Mol. Cancer Ther. 2007, 6, 2544–2553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.; Klinger, V.; Keksel, C.; Bonigut, V.; Kiefer, D.; Caspers, J.; Walther, J.; Wos-Maganga, M.; Weickhardt, S.; Rohn, G.; et al. Inhibition of the PI3K but not the MEK/ERK pathway sensitizes human glioma cells to alkylating drugs. Cancer Cell Int. 2018, 18, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welsh, S.J.; Bellamy, W.T.; Briehl, M.M.; Powis, G. The redox protein thioredoxin-1 (Trx-1) increases hypoxia-inducible factor 1alpha protein expression: Trx-1 overexpression results in increased vascular endothelial growth factor production and enhanced tumor angiogenesis. Cancer Res. 2002, 62, 5089–5095. [Google Scholar] [PubMed]
- Hedley, D.; Pintilie, M.; Woo, J.; Nicklee, T.; Morrison, A.; Birle, D.; Fyles, A.; Milosevic, M.; Hill, R. Up-regulation of the redox mediators thioredoxin and apurinic/apyrimidinic excision (APE)/Ref-1 in hypoxic microregions of invasive cervical carcinomas, mapped using multispectral, wide-field fluorescence image analysis. Am. J. Pathol. 2004, 164, 557–565. [Google Scholar] [CrossRef]
- Bhatia, M.; Lovitt, C.J.; Raninga, P.V.; Avery, V.M.; Di Trapani, G.; Tonissen, K.F. Expression of the thioredoxin system in an in vivo-like cancer cell environment upon auranofin treatment. Eur. J. Cell Biol. 2016, 95, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Powis, G.; Kirkpatrick, D.L.; Angulo, M.; Baker, A. Thioredoxin redox control of cell growth and death and the effects of inhibitors. Chem. Biol. Interact. 1998, 111, 23–34. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, H.; Cao, M.; Wang, L.; Wu, S.; Fang, B. Auranofin Enhances Ibrutinib’s Anticancer Activity in EGFR-Mutant Lung Adenocarcinoma. Mol. Cancer Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Liao, Y.; Liu, N.; Hua, X.; Cai, J.; Yang, C.; Long, H.; Zhao, C.; Chen, X.; Lan, X.; et al. Two clinical drugs deubiquitinase inhibitor auranofin and aldehyde dehydrogenase inhibitor disulfiram trigger synergistic anti-tumor effects in vitro and in vivo. Oncotarget 2016, 7, 2796–2808. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Lee, S.J.; Kim, J.T.; Kim, S.J.; Min, J.K.; Bae, K.H.; Jung, H.; Kim, B.Y.; Lim, J.S.; Yang, Y.; et al. Kallikrein-related peptidase 6 induces chemotherapeutic resistance by attenuating auranofin-induced cell death through activation of autophagy in gastric cancer. Oncotarget 2016, 7, 85332–85348. [Google Scholar] [CrossRef] [PubMed]
- Oommen, D.; Yiannakis, D.; Jha, A.N. BRCA1 deficiency increases the sensitivity of ovarian cancer cells to auranofin. Mutat. Res. 2016, 784, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Kwon, Y.J.; Baek, H.S.; Ye, D.J.; Cho, E.; Choi, H.K.; Oh, K.S.; Chun, Y.J. Synergistic induction of apoptosis by combination treatment with mesupron and auranofin in human breast cancer cells. Arch. Pharm. Res. 2017, 40, 746–759. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.L.; Jang, H.; Kim, E.H.; Shin, D. Targeting of the Glutathione, Thioredoxin, and Nrf2 Antioxidant Systems in Head and Neck Cancer. Antioxid. Redox Signal. 2017, 27, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Rodman, S.N.; Spence, J.M.; Ronnfeldt, T.J.; Zhu, Y.; Solst, S.R.; O’Neill, R.A.; Allen, B.G.; Guan, X.; Spitz, D.R.; Fath, M.A. Enhancement of Radiation Response in Breast Cancer Stem Cells by Inhibition of Thioredoxin- and Glutathione-Dependent Metabolism. Radiat. Res. 2016, 186, 385–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Bouzakoura, S.; de Mey, S.; Jiang, H.; Law, K.; Dufait, I.; Corbet, C.; Verovski, V.; Gevaert, T.; Feron, O.; et al. Auranofin radiosensitizes tumor cells through targeting thioredoxin reductase and resulting overproduction of reactive oxygen species. Oncotarget 2017, 8, 35728–35742. [Google Scholar] [CrossRef] [PubMed]
- Raninga, P.V.; Di Trapani, G.; Vuckovic, S.; Bhatia, M.; Tonissen, K.F. Inhibition of thioredoxin 1 leads to apoptosis in drug-resistant multiple myeloma. Oncotarget 2015, 6, 15410–15424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raninga, P.V.; Di Trapani, G.; Vuckovic, S.; Tonissen, K.F. TrxR1 inhibition overcomes both hypoxia-induced and acquired bortezomib resistance in multiple myeloma through NF-small ka, Cyrillicbeta inhibition. Cell. Cycle 2016, 15, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, D.L.; Kuperus, M.; Dowdeswell, M.; Potier, N.; Donald, L.J.; Kunkel, M.; Berggren, M.; Angulo, M.; Powis, G. Mechanisms of inhibition of the thioredoxin growth factor system by antitumor 2-imidazolyl disulfides. Biochem. Pharmacol. 1998, 55, 987–994. [Google Scholar] [CrossRef]
- Meuillet, E.J.; Mahadevan, D.; Berggren, M.; Coon, A.; Powis, G. Thioredoxin-1 binds to the C2 domain of PTEN inhibiting PTEN’s lipid phosphatase activity and membrane binding: A mechanism for the functional loss of PTEN’s tumor suppressor activity. Arch. Biochem. Biophys. 2004, 429, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Toledano, M.B.; Leonard, W.J. Modulation of transcription factor NF-kappa B binding activity by oxidation-reduction in vitro. Proc. Natl. Acad. Sci. USA 1991, 88, 4328–4332. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Guttridge, D.C.; Mayo, M.W.; Baldwin, A.S. NF-kappaB induces expression of the Bcl-2 homologue A1/Bfl-1 to preferentially suppress chemotherapy-induced apoptosis. Mol. Cell. Biol. 1999, 19, 5923–5929. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, A.H.; Khursigara, G.; Sun, X.; Franke, T.F.; Chao, M.V. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol. Cell. Biol. 2001, 21, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Ichijo, H.; Nishida, E.; Irie, K.; ten Dijke, P.; Saitoh, M.; Moriguchi, T.; Takagi, M.; Matsumoto, K.; Miyazono, K.; Gotoh, Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997, 275, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.F.; Wang, J.; Shao, W.; Wu, C.P.; Chen, Z.P.; To, S.T.; Li, W.P. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eckstein, N.; Servan, K.; Girard, L.; Cai, D.; von Jonquieres, G.; Jaehde, U.; Kassack, M.U.; Gazdar, A.F.; Minna, J.D.; Royer, H.D. Epidermal growth factor receptor pathway analysis identifies amphiregulin as a key factor for cisplatin resistance of human breast cancer cells. J. Biol. Chem. 2008, 283, 739–750. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haas, B.; Schütte, L.; Wos-Maganga, M.; Weickhardt, S.; Timmer, M.; Eckstein, N. Thioredoxin Confers Intrinsic Resistance to Cytostatic Drugs in Human Glioma Cells. Int. J. Mol. Sci. 2018, 19, 2874. https://doi.org/10.3390/ijms19102874
Haas B, Schütte L, Wos-Maganga M, Weickhardt S, Timmer M, Eckstein N. Thioredoxin Confers Intrinsic Resistance to Cytostatic Drugs in Human Glioma Cells. International Journal of Molecular Sciences. 2018; 19(10):2874. https://doi.org/10.3390/ijms19102874
Chicago/Turabian StyleHaas, Bodo, Lena Schütte, Maria Wos-Maganga, Sandra Weickhardt, Marco Timmer, and Niels Eckstein. 2018. "Thioredoxin Confers Intrinsic Resistance to Cytostatic Drugs in Human Glioma Cells" International Journal of Molecular Sciences 19, no. 10: 2874. https://doi.org/10.3390/ijms19102874