Artificial Heme Enzymes for the Construction of Gold-Based Biomaterials

 ,
,  , ,
, , 
Abstract
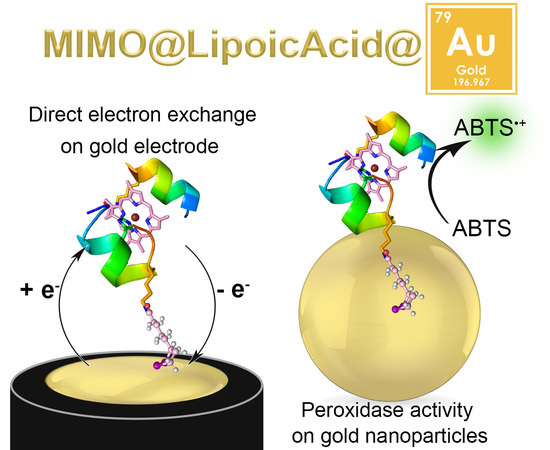
:
1. Introduction
2. Results and Discussion
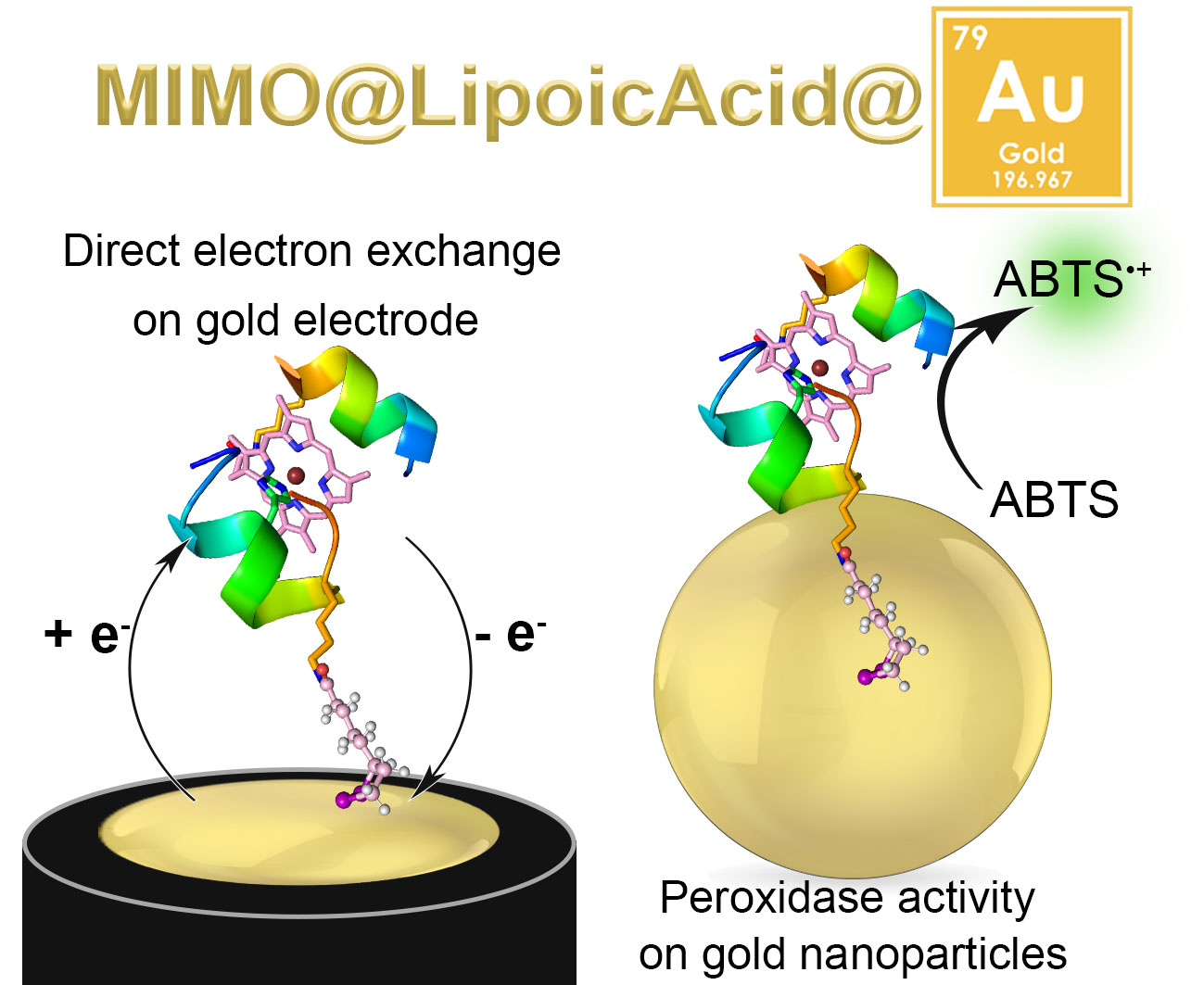
2.1. Synthesis of MIMO@LA
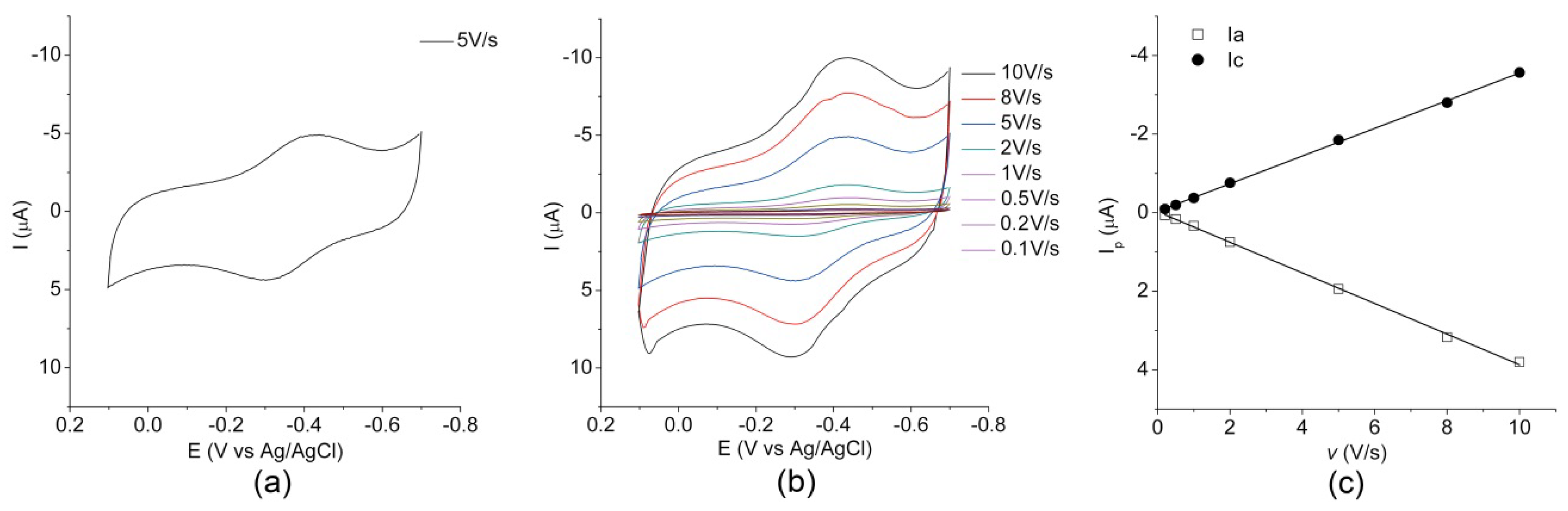
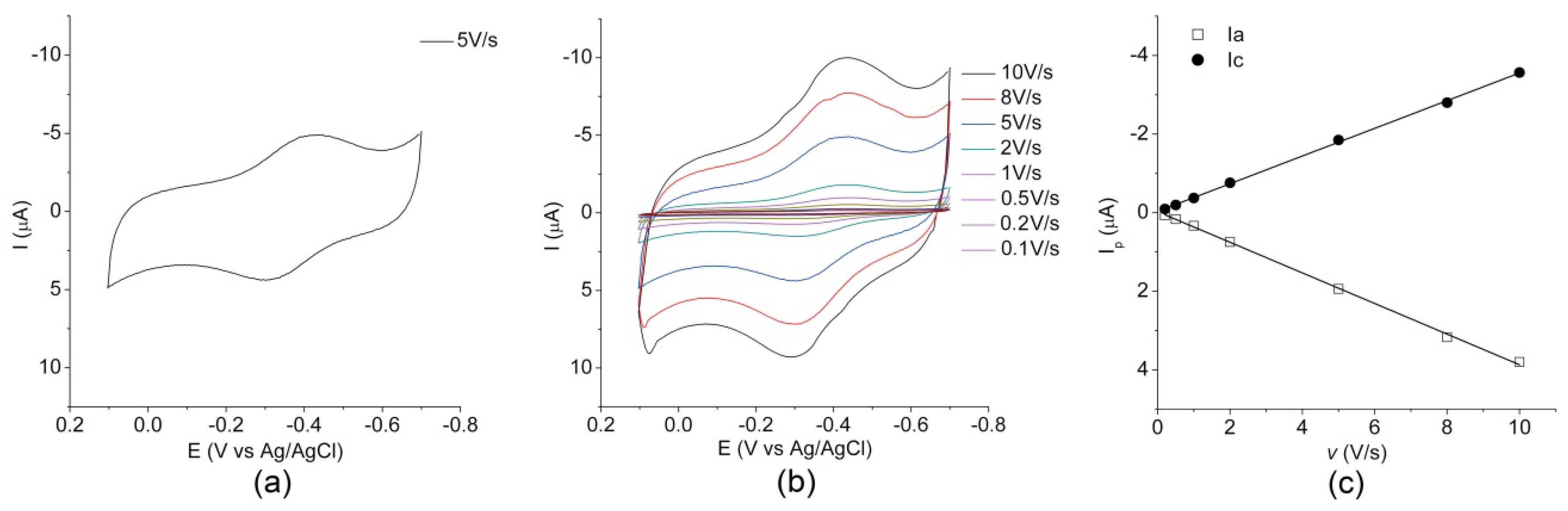
2.2. Electrochemical Characterization
2.3. MIMO@LA Catalytic Activity on AuNPs
2.3.1. Synthesis and Characterization of MIMO@LA@AuNPs
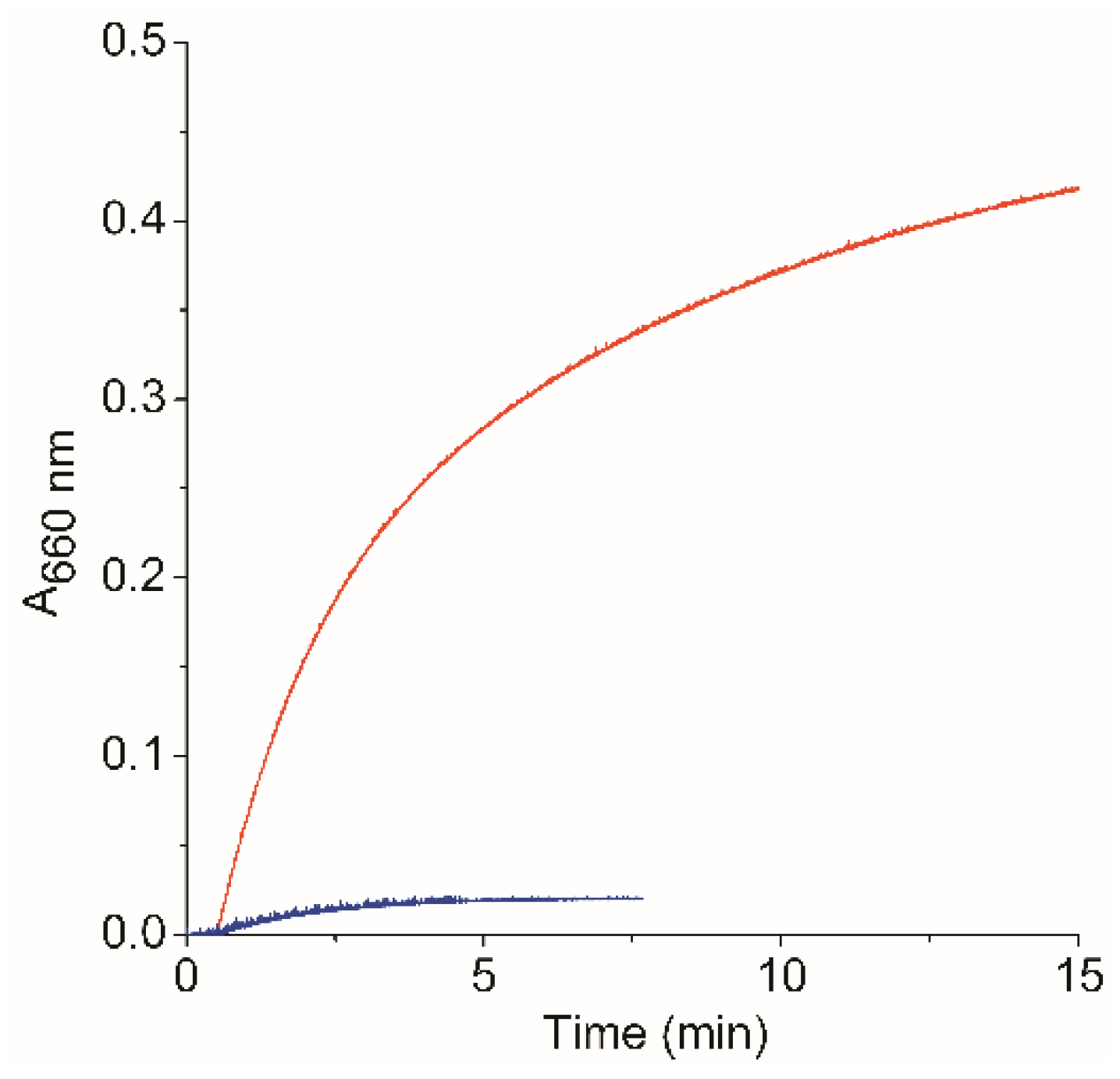
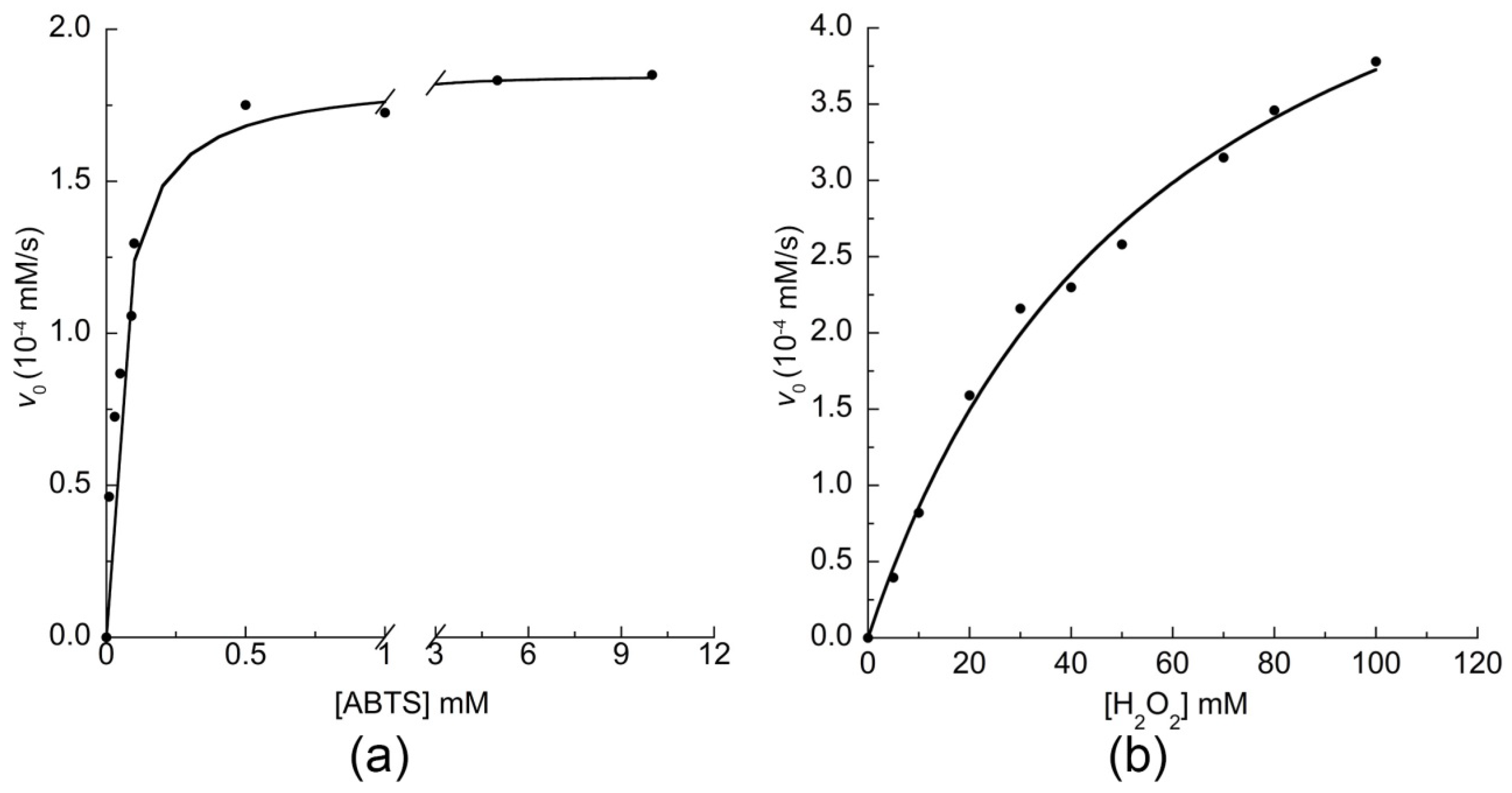
2.3.2. Catalytic Activity of MIMO@LA@AuNPs
3. Materials and Methods
3.1. Synthesis of MIMO@LA
3.2. Synthesis and Characterization of MIMO@LA@AuNPs
3.3. MIMO Radius of Gyration Calculation
3.4. AuNPs Physico-Chemical Characterization
3.5. Voltammetric Analysis
3.6. Catalytic Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nastri, F.; Chino, M.; Maglio, O.; Bhagi-Damodaran, A.; Lu, Y.; Lombardi, A. Design and engineering of artificial oxygen-activating metalloenzymes. Chem. Soc. Rev. 2016, 45, 5020–5054. [Google Scholar] [CrossRef] [PubMed]
- Grayson, K.J.; Anderson, J.L.R. The ascent of man (made oxidoreductases). Curr. Opin. Struct. Biol. 2018, 51, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Hyster, T.K.; Ward, T.R. Genetic optimization of metalloenzymes: Enhancing enzymes for non-natural reactions. Angew. Chem. Int. Ed. 2016, 55, 7344–7357. [Google Scholar] [CrossRef] [PubMed]
- Schwizer, F.; Okamoto, Y.; Heinisch, T.; Gu, Y.; Pellizzoni, M.M.; Lebrun, V.; Reuter, R.; Köhler, V.; Lewis, J.C.; Ward, T.R. Artificial metalloenzymes: Reaction scope and optimization strategies. Chem. Rev. 2017, 118, 142–231. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.C. Artificial metalloenzymes and metallopeptide catalysts for organic synthesis. ACS Catal. 2013, 3, 2954–2975. [Google Scholar] [CrossRef]
- Mocny, C.S.; Pecoraro, V.L. De novo protein design as a methodology for synthetic bioinorganic chemistry. Acc. Chem. Res. 2015, 48, 2388–2396. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Cangelosi, V.M.; Zastrow, M.L.; Tegoni, M.; Plegaria, J.S.; Tebo, A.G.; Mocny, C.S.; Ruckthong, L.; Qayyum, H.; Pecoraro, V.L. Protein design: Toward functional metalloenzymes. Chem. Rev. 2014, 114, 3495–3578. [Google Scholar] [CrossRef] [PubMed]
- Bhagi-Damodaran, A.; Hosseinzade, P.; Mirts, E.; Reed, J.; Petrik, I.D.; Lu, Y. Design of heteronuclear metalloenzymes. Methods Enzymol. 2016, 580, 501–537. [Google Scholar] [CrossRef] [PubMed]
- Jeschek, M.; Panke, S.; Ward, T.R. Artificial metalloenzymes on the verge of new-to-nature metabolism. Trends Biotechnol. 2018, 36, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Petrik, I.D.; Liu, J.; Lu, Y. Metalloenzyme design and engineering through strategic modifications of native protein scaffolds. Curr. Opin. Chem. Biol. 2014, 19, 67–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Hu, C.; Xia, L.; Wang, J. Artificial metalloenzyme design with unnatural amino acids and non-native cofactors. ACS Catal. 2018, 8, 1851–1863. [Google Scholar] [CrossRef]
- Jeschek, M.; Reuter, R.; Heinisch, T.; Trindler, C.; Klehr, J.; Panke, S.; Ward, T.R. Directed evolution of artificial metalloenzymes for in vivo metathesis. Nature 2016, 537, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Chino, M.; Leone, L.; Zambrano, G.; Pirro, F.; D’Alonzo, D.; Firpo, V.; Aref, D.; Lista, L.; Maglio, O.; Nastri, F.; et al. Oxidation catalysis by iron and manganese porphyrins within enzyme-like cages. Biopolymers 2018. [CrossRef] [PubMed]
- Yu, Y.; Cui, C.; Liu, X.; Petrik, I.D.; Wang, J.; Lu, Y. A designed metalloenzyme achieving the catalytic rate of a native enzyme. J. Am. Chem. Soc. 2015, 137, 11570–11573. [Google Scholar] [CrossRef] [PubMed]
- Oohora, K.; Meichin, H.; Kihira, Y.; Sugimoto, H.; Shiro, Y.; Hayashi, T. Manganese(V) porphycene complex responsible for inert c–h bond hydroxylation in a myoglobin matrix. J. Am. Chem. Soc. 2017, 139, 18460–18463. [Google Scholar] [CrossRef] [PubMed]
- Oohora, K.; Meichin, H.; Zhao, L.; Wolf, M.W.; Nakayama, A.; Hasegawa, J.; Lehnert, N.; Hayashi, T. Catalytic cyclopropanation by myoglobin reconstituted with iron porphycene: Acceleration of catalysis due to rapid formation of the carbene species. J. Am. Chem. Soc. 2017, 139, 17265–17268. [Google Scholar] [CrossRef] [PubMed]
- Dydio, P.; Key, H.M.; Nazarenko, A.; Rha, J.Y.; Seyedkazemi, V.; Clark, D.S.; Hartwig, J.F. An artificial metalloenzyme with the kinetics of native enzymes. Science 2016, 354, 102–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Key, H.M.; Dydio, P.; Clark, D.S.; Hartwig, J.F. Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature 2016, 534, 534–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, F.H. Directed evolution: Bringing new chemistry to life. Angew. Chem. Int. Ed. 2017, 56, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Hammer, S.C.; Kubik, G.; Watkins, E.; Huang, S.; Minges, H.; Arnold, F.H. Anti-Markovnikov alkene oxidation by metal-oxo–mediated enzyme catalysis. Science 2017, 358, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.B.J.; Lewis, R.D.; Chen, K.; Arnold, F.H. Directed evolution of cytochrome c for carbon-silicon bond formation: Bringing silicon to life. Science 2016, 354, 1048–1051. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.B.J.; Huang, X.; Gumulya, Y.; Chen, K.; Arnold, F.H. Genetically programmed chiral organoborane synthesis. Nature 2017, 552, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Huang, X.; Kan, S.B.J.; Zhang, R.K.; Arnold, F.H. Enzymatic construction of highly strained carbocycles. Science 2018, 360, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Caserta, G.; Chino, M.; Firpo, V.; Zambrano, G.; Leone, L.; D’Alonzo, D.; Nastri, F.; Maglio, O.; Pavone, V.; Lombardi, A. Enhancement of peroxidase activity in the artificial Mimochrome VI catalysts through rational design. ChemBioChem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Watkins, D.W.; Jenkins, J.M.X.; Grayson, K.J.; Wood, N.; Steventon, J.W.; Vay, K.K.L.; Goodwin, M.I.; Mullen, A.S.; Bailey, H.J.; Crump, M.P.; et al. Construction and in vivo assembly of a catalytically proficient and hyperthermostable de novo enzyme. Nat. Commun. 2017, 8, 358. [Google Scholar] [CrossRef] [PubMed]
- Bilal, M.; Iqbalb, H.M.N.; Guoa, S.; Hua, H.; Wanga, W.; Zhang, X. State-of-the-art protein engineering approaches using biological macromolecules: A review from immobilization to implementation view point. Int. J. Biol. Macromol. 2018, 108, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A.; van Pelt, S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.A.; Husain, Q. Potential applications of enzymes immobilized on/in nano materials: A review. Biotechnol. Adv. 2012, 30, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Algar, W.R.; Prasuhn, D.E.; Stewart, M.H.; Jennings, T.L.; Blanco-Canosa, J.B.; Dawson, P.E.; Medintz, I.L. The Controlled Display of Biomolecules on Nanoparticles: A Challenge Suited to Bioorthogonal Chemistry. Bioconjug. Chem. 2011, 22, 825–858. [Google Scholar] [CrossRef] [PubMed]
- Katz, E.; Willner, I. Integrated nanoparticle-biomolecule hybrid systems: Synthesis, properties, and applications. Angew. Chem. Int. Ed. Engl. 2004, 43, 6042–6108. [Google Scholar] [CrossRef] [PubMed]
- Zdarta, J.; Meyer, A.S.; Jesionowski, T.; Pinelo, M. A General overview of support materials for enzyme immobilization: Characteristics, properties, practical utility. Catalysts 2018, 8, 92. [Google Scholar] [CrossRef]
- Chen, M.; Zeng, G.; Xu, P.; Lai, C.; Tang, L. How do enzymes ‘meet’ nanoparticles and nanomaterials? Trends Biochem. Sci. 2017, 42, 914–930. [Google Scholar] [CrossRef] [PubMed]
- Cipolatti, E.P.; Silva, M.J.A.; Klein, M.; Feddern, V.; Feltes, M.C.; Oliveira, J.V.; Ninow, J.L.; de Oliveira, D. Current status and trends in enzymatic nanoimmobilization. J. Mol. Cat. B 2014, 99, 56–67. [Google Scholar] [CrossRef]
- Auriemma, F.; de Rosa, C.; Malafronte, A.; Di Girolamo, R.; Santillo, C.; Gerelli, Y.; Fragneto, G.; Barker, R.; Pavone, V.; Maglio, O.; et al. Nano-in-nano approach for enzyme immobilization based on block copolymers. ACS Appl. Mater. Interfaces 2017, 9, 29318–29327. [Google Scholar] [CrossRef] [PubMed]
- Daniel, M.C.; Astruc, D. Gold nanoparticles: Assembly, supramolecular chemistry, quantum-size-related properties, and applications toward biology, catalysis, and nanotechnology. Chem. Rev. 2004, 104, 293–346. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.; Zhu, G.; Wang, P. Catalytic behaviors of enzymes attached to nanoparticles: The effect of particle mobility. Biotechnol. Bioeng. 2003, 84, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Noll, T.; Noll, G. Strategies for “wiring” redox-active proteins to electrodes and applications in biosensors, biofuel cells, and nanotechnology. Chem. Soc. Rev. 2011, 40, 3564–3576. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.; Clark, D.S. Cytochrome P450 (CYP) enzymes and the development of CYP biosensors. Biosens. Bioelectron. 2013, 39, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, S.J.; Fantuzzi, G.; Gilardi, G. Breakthrough in P450 bioelectrochemistry and future perspectives. Biochim. Biophys. Acta 2011, 1814, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Netto, C.G.C.M.; Toma, H.E.; Andrade, L.H. Superparamagnetic nanoparticles as versatile carriers and supporting materials for enzymes. J. Mol. Cat. B 2013, 85–86, 71–92. [Google Scholar] [CrossRef]
- Lu, D.; Pang, G. A novel tetrahydrocannabinol electrochemical nano immunosensor based on horseradish peroxidase and double-layer gold nanoparticles. Molecules 2016, 21, 1377. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Tang, J.; Su, B.; Li, Q.; Chen, G. Electrochemical detection of hepatitis C virus with signal amplification using BamHI endonuclease and horseradish peroxidase-encapsulated nanogold hollow spheres. Chem. Commun. 2011, 47, 9477–9479. [Google Scholar] [CrossRef] [PubMed]
- Nastri, F.; Lombardi, A.; Morelli, G.; Maglio, O.; D’Auria, G.; Pedone, C.; Pavone, V. Hemoprotein Models Based on a Covalent Helix–Heme–Helix Sandwich: 1. Design, Synthesis, and Characterization. Chem. Eur. J. 1997, 3, 340–349. [Google Scholar] [CrossRef]
- D’Auria, G.; Maglio, O.; Nastri, F.; Lombardi, A.; Mazzeo, M.; Morelli, G.; Paolillo, L.; Pedone, C.; Pavone, V. Hemoprotein Models Based on a Covalent Helix–Heme–Helix Sandwich: 2. Structural Characterization of CoIIIMimochrome I δ and δ Isomers. Chem. Eur. J. 1997, 3, 350–362. [Google Scholar] [CrossRef]
- Nastri, F.; Lombardi, A.; Morelli, G.; Pedone, C.; Pavone, V.; Chottard, G.; Battioni, P.; Mansuy, D. Hemoprotein models based on a covalent helix-heme-helix sandwich. 3. Coordination properties, reactivity and catalytic application of Fe(III)- and Fe(II)-mimochrome I. J. Biol. Inorg. Chem. 1998, 3, 671–681. [Google Scholar] [CrossRef]
- Lombardi, A.; Nastri, F.; Sanseverino, M.; Maglio, O.; Pedone, C.; Pavone, V. Miniaturized hemoproteins: Design, synthesis and characterization of mimochrome II. Inorg. Chim. Acta 1998, 301, 275–276. [Google Scholar] [CrossRef]
- Lombardi, A.; Nastri, F.; Pavone, V. Peptide-based heme—protein models. Chem. Rev. 2001, 101, 3165–3189. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, A.; Nastri, F.; Marasco, D.; Maglio, O.; De Sanctis, G.; Sinibaldi, F.; Santucci, R.; Coletta, M.; Pavone, V. Design of a new mimochrome with unique topology. Chem. Eur. J. 2003, 9, 5643–5654. [Google Scholar] [CrossRef] [PubMed]
- Di Costanzo, L.; Geremia, S.; Randaccio, L.; Nastri, F.; Maglio, O.; Lombardi, A.; Pavone, V. Miniaturized heme proteins: Crystal structure of Co(III)-mimochrome IV. J. Biol. Inorg. Chem. 2004, 9, 1017–1027. [Google Scholar] [CrossRef] [PubMed]
- Nastri, F.; Lista, L.; Ringhieri, P.; Vitale, R.; Faiella, M.; Andreozzi, C.; Travascio, P.; Maglio, O.; Lombardi, A.; Pavone, V. A Heme-peptide metalloenzyme mimetic with natural peroxidase-like activity. Chem. Eur. J. 2011, 17, 4444–4453. [Google Scholar] [CrossRef] [PubMed]
- Vicari, C.; Saraiva, I.H.; Maglio, O.; Nastri, F.; Pavone, V.; Louro, R.O.; Lombardi, A. Artificial heme-proteins: Determination of axial ligand orientations through paramagnetic NMR shifts. Chem. Commun. 2014, 50, 3852–3855. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, A.; Monari, S.; Sola, M.; Borsari, M.; Battistuzzi, G.; Ringhieri, P.; Nastri, F.; Pavone, V.; Lombardi, A. Redox and electrocatalytic properties of mimochrome VI, a synthetic heme peptide adsorbed on gold. Langmuir 2010, 26, 17831–17835. [Google Scholar] [CrossRef] [PubMed]
- Vitale, R.; Lista, L.; Lau-Truong, S.; Tucker, R.T.; Brett, M.J.; Limoges, B.; Pavone, V.; Lombardi, A.; Balland, V. Spectroelectrochemistry of FeIII- and CoIII-mimochrome VI artificial enzymes immobilized on mesoporous ITO electrodes. Chem. Commun. 2014, 50, 1894–1896. [Google Scholar] [CrossRef] [PubMed]
- Vitale, R.; Lista, L.; Cerrone, C.; Caserta, G.; Chino, M.; Maglio, O.; Nastri, F.; Pavone, V.; Lombardi, A. An artificial heme-enzyme with enhanced catalytic activity: Evolution, functional screening and structural characterization. Org. Biomol. Chem. 2015, 13, 4859–4868. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Cargill, A.A.; Medintz, I.L.; Claussen, J.C. Increasing the activity of immobilized enzymes with nanoparticle conjugation. Curr. Opin. Biotech. 2015, 34, 242–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walper, S.A.; Turner, K.B.; Medintz, I.L. Enzymatic bioconjugation of nanoparticles: Developing specificity and control. Curr. Opin. Biotech. 2015, 34, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Sardar, M. Enzyme Immobilization: An Overview on Nanoparticles as Immobilization Matrix. Anal. Biochem. 2015, 4. [Google Scholar] [CrossRef]
- Ulman, A. Formation and structure of self-assembled monolayers. Chem. Rev. 1996, 96, 1533–1554. [Google Scholar] [CrossRef] [PubMed]
- Wink, T.; van Zuilen, S.J.; Bult, A.; van Bennekom, W.P. Self-assembled monolayers for biosensors. Analyst 1997, 122, 43R–50R. [Google Scholar] [CrossRef]
- Mahl, D.; Greulich, C.; Meyer-Zaika, W.; Koller, M.; Epple, M. Gold nanoparticles: Dispersibility in biological media and cell-biological effect. J. Mater. Chem. 2010, 20, 6176–6181. [Google Scholar] [CrossRef]
- Pérez-Rentero, S.; Grijalvo, S.; Peñuelas, G.; Fàbrega, C.; Eritja, R. Thioctic acid derivatives as building blocks to incorporate DNA oligonucleotides onto gold nanoparticles. Molecules 2014, 19, 10495–10523. [Google Scholar] [CrossRef] [PubMed]
- Koufaki, M.; Detsi, A.; Kiziridi, C. Multifunctional lipoic acid conjugates. Curr. Med. Chem. 2009, 16, 4728–4742. [Google Scholar] [CrossRef] [PubMed]
- Maglio, O.; Costanzo, S.; Cercola, R.; Zambrano, G.; Mauro, M.; Battaglia, R.; Ferrini, G.; Nastri, F.; Pavone, V.; Lombardi, A. QCM immunosensor for stem cell selection and extraction. Sensors 2017, 17, 2747. [Google Scholar] [CrossRef] [PubMed]
- Ahirwal, G.K.; Mitra, C.K. Direct electrochemistry of horseradish peroxidase-gold nanoparticles conjugate. Sensors 2009, 9, 881–894. [Google Scholar] [CrossRef] [PubMed]
- Onoda, A.; Ueya, Y.; Sakamoto, T.; Uematsua, T.; Hayashi, T. Supramolecular hemoprotein—gold nanoparticle conjugates. Chem. Commun. 2010, 46, 9107–9109. [Google Scholar] [CrossRef] [PubMed]
- Dougan, J.A.; Karlsson, C.; Smith, W.E.; Graham, D. Enhanced oligonucleotide—nanoparticle conjugate stability using thioctic acid modified oligonucleotides. Nucleic Acids Res. 2007, 35, 3668–3675. [Google Scholar] [CrossRef] [PubMed]
- Sharp, M.; Petersson, M.; Edstrom, K.J. Preliminary determinations of electron transfer kinetics involving ferrocene covalently attached to a platinum surface. J. Electroanal. Chem. 1979, 95, 123–130. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods, Fundamentals and Applications, 2nd ed.; John Wiley & Sons Inc.: New York, NY, USA, 2001; ISBN 0471043729. [Google Scholar]
- Zhao, P.; Li, N.; Astruc, D. State of the art in gold nanoparticle synthesis. Coord. Chem. Rev. 2013, 257, 638–665. [Google Scholar] [CrossRef]
- Turkevich, J.; Stevenson, P.C.; Hillier, J. A study of the nucleation and growth processes in the synthesis of colloidal gold. Discuss. Faraday Soc. 1951, 11, 55–75. [Google Scholar] [CrossRef]
- Templeton, A.C.; Pietron, J.J.; Murray, R.W.; Mulvaney, P. Solvent refractive index and core charge influences on the surface plasmon absorbance of alkanethiolate monolayer-protected gold clusters. J. Phys. Chem. B 2000, 104, 564–570. [Google Scholar] [CrossRef]
- De Carlo, S.; Harris, J.R. Negative staining and cryo-negative staining of macromolecules and viruses for TEM. Micron 2011, 42, 117–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michen, B.; Geers, C.; Vanhecke, D.; Endes, C.; Rothen-Rutishauser, B.; Balog, S.; Petri-Fink, A. Avoiding drying-artifacts in transmission electron microscopy: Characterizing the size and colloidal state of nanoparticles. Sci. Rep. 2015, 5, 9793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattoussi, H.; Mauro, J.M.; Goldman, E.R.; Anderson, G.P.; Sundar, V.C.; Mikulec, F.V.; Bawendi, M.G. Self-assembly of CdSe-ZnS quantum dot bioconjugates using an engineered recombinant protein. J. Am. Chem. Soc. 2000, 122, 12142–12150. [Google Scholar] [CrossRef]
- Cebula, J.; Ottewill, R.H.; Ralston, J.; Pusey, P.N. Investigations of microemulsions by light scattering and neutron scattering. J. Chem. Soc. Faraday Trans. 1 1981, 77, 2585–2612. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of Gyration as an Indicator of Protein Structure Compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Dewald, I.; Isakin, O.; Schubert, J.; Kraus, T.; Chanana, M. Protein identity and environmental parameters determine the final physicochemical properties of protein-coated metal nanoparticles. J. Phys. Chem. C 2015, 119, 25482–25492. [Google Scholar] [CrossRef]
- Rodrigues, R.C.; Ortiz, C.; Berenguer-Murcia, A.; Torres, R.; Fernández-Lafuente, R. Modifying enzyme activity and selectivity by immobilization. Chem. Soc. Rev. 2013, 42, 6290–6307. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.C.S.D.; Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Rodrigues, R.C.; Fernandez-Lafuente, R. Importance of the support properties for immobilization or purification of enzymes. ChemCatChem. 2015, 7, 2413–2432. [Google Scholar] [CrossRef]
- Männel, M.J.; Kreuzer, L.P.; Goldhahn, C.; Schubert, J.; Hart, M.J.; Chanana, M. Catalytically active protein coatings: Toward enzymatic cascade reactions at the intercolloidal level. ACS Catal. 2017, 7, 1664–1672. [Google Scholar] [CrossRef]
- Ni, Y.; Li, Y.; Huang, Z.; He, K.; Zhuang, J.; Yang, W. Improved activity of immobilized horseradish peroxidase on gold nanoparticles in the presence of bovine serum albumin. J. Nanopart. Res. 2013, 15, 2038. [Google Scholar] [CrossRef]
- Tadepalli, S.; Wang, Z.; Slocik, J.; Naik, R.R.; Singamanen, S. Effect of size and curvature on the enzyme activity of bionanoconjugates. Nanoscale 2017, 9, 15666–15672. [Google Scholar] [CrossRef] [PubMed]
- Lata, J.P.; Gao, L.; Mukai, C.; Cohen, R.; Nelson, J.L.; Anguish, L.; Coonrod, S.; Travis, A.J. Effects of nanoparticle size on multilayer formation and kinetics of tethered enzymes. Bioconjug. Chem. 2015, 26, 1931–1938. [Google Scholar] [CrossRef] [PubMed]
- Jamison, J.A.; Bryant, E.L.; Kadali, S.B.; Wong, M.S.; Colvin, V.L.; Matthews, K.S.; Calabretta, M.K. Altering protein surface charge with chemical modification modulates protein–gold nanoparticle aggregation. J. Nanopart. Res. 2011, 13, 625–636. [Google Scholar] [CrossRef]
- Frens, G. Controlled nucleation for the regulation of the particle size in monodisperse gold suspensions. Nat. Phys. Sci. 1973, 241, 20–22. [Google Scholar] [CrossRef]
- Haiss, W.; Thanh, N.T.K.; Aveyard, J.; Fernig, D.G. Determination of size and concentration of gold nanoparticles from UV-vis spectra. Anal. Chem. 2007, 79, 4215–4221. [Google Scholar] [CrossRef] [PubMed]
- Carvalhal, R.F.; Freire, R.S.; Kubota, L.T. Polycrystalline gold electrodes: A comparative study of pretreatment procedures used for cleaning and thiol self-assembly monolayer formation. Electroanalysis 2005, 17, 1251–1259. [Google Scholar] [CrossRef]
- Ranganathan, S.; Kuo, T.-C.; McCreery, R.L. Facile preparation of active glassy carbon electrodes with activated carbon and organic solvents. Anal. Chem. 1999, 71, 3574–3580. [Google Scholar] [CrossRef]
- Childs, R.E.; Bardsley, W.G. The steady-state kinetics of peroxidase with 2,2′-azino-di-(3-ethyl-benzthiazoline-6-sulphonic acid) as chromogen. Biochem. J. 1975, 145, 93–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleland, W.W. The kinetics of enzyme-catalyzed reactions with two or more substrates or products. I. Nomenclature and rate equations. Biochim. Biophys. Acta 1963, 67, 104–137. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | kcat (s−1) | KM ABTS (10−5 M) | kcat/KM (mM−1·s−1) |
|---|---|---|---|
| MIMO@LA@AuNPs | 1.7± 0.1 | 14.7 ± 0.1 | 11.5 |
| MIMO | 468 ± 27 | 12.5 ± 2 | 3743 ± 814 |
| MC6§ | 371 ± 14 | 8.4 ± 0.2 | 4417 ± 197 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zambrano, G.; Ruggiero, E.; Malafronte, A.; Chino, M.; Maglio, O.; Pavone, V.; Nastri, F.; Lombardi, A. Artificial Heme Enzymes for the Construction of Gold-Based Biomaterials. Int. J. Mol. Sci. 2018, 19, 2896. https://doi.org/10.3390/ijms19102896
Zambrano G, Ruggiero E, Malafronte A, Chino M, Maglio O, Pavone V, Nastri F, Lombardi A. Artificial Heme Enzymes for the Construction of Gold-Based Biomaterials. International Journal of Molecular Sciences. 2018; 19(10):2896. https://doi.org/10.3390/ijms19102896
Chicago/Turabian StyleZambrano, Gerardo, Emmanuel Ruggiero, Anna Malafronte, Marco Chino, Ornella Maglio, Vincenzo Pavone, Flavia Nastri, and Angela Lombardi. 2018. "Artificial Heme Enzymes for the Construction of Gold-Based Biomaterials" International Journal of Molecular Sciences 19, no. 10: 2896. https://doi.org/10.3390/ijms19102896