Paired CRISPR/Cas9 Nickases Mediate Efficient Site-Specific Integration of F9 into rDNA Locus of Mouse ESCs

 , ,
, , 
Abstract
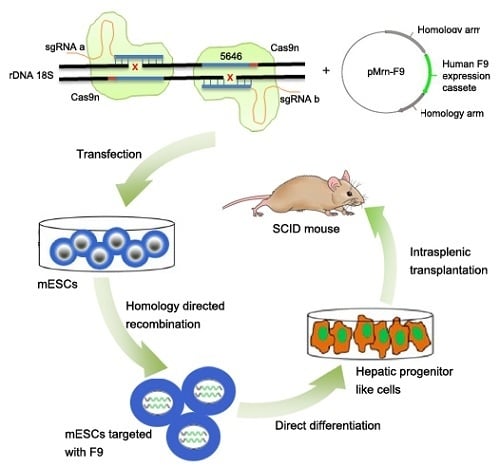
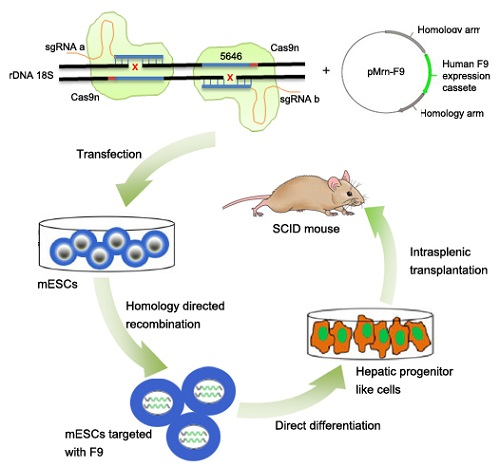
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
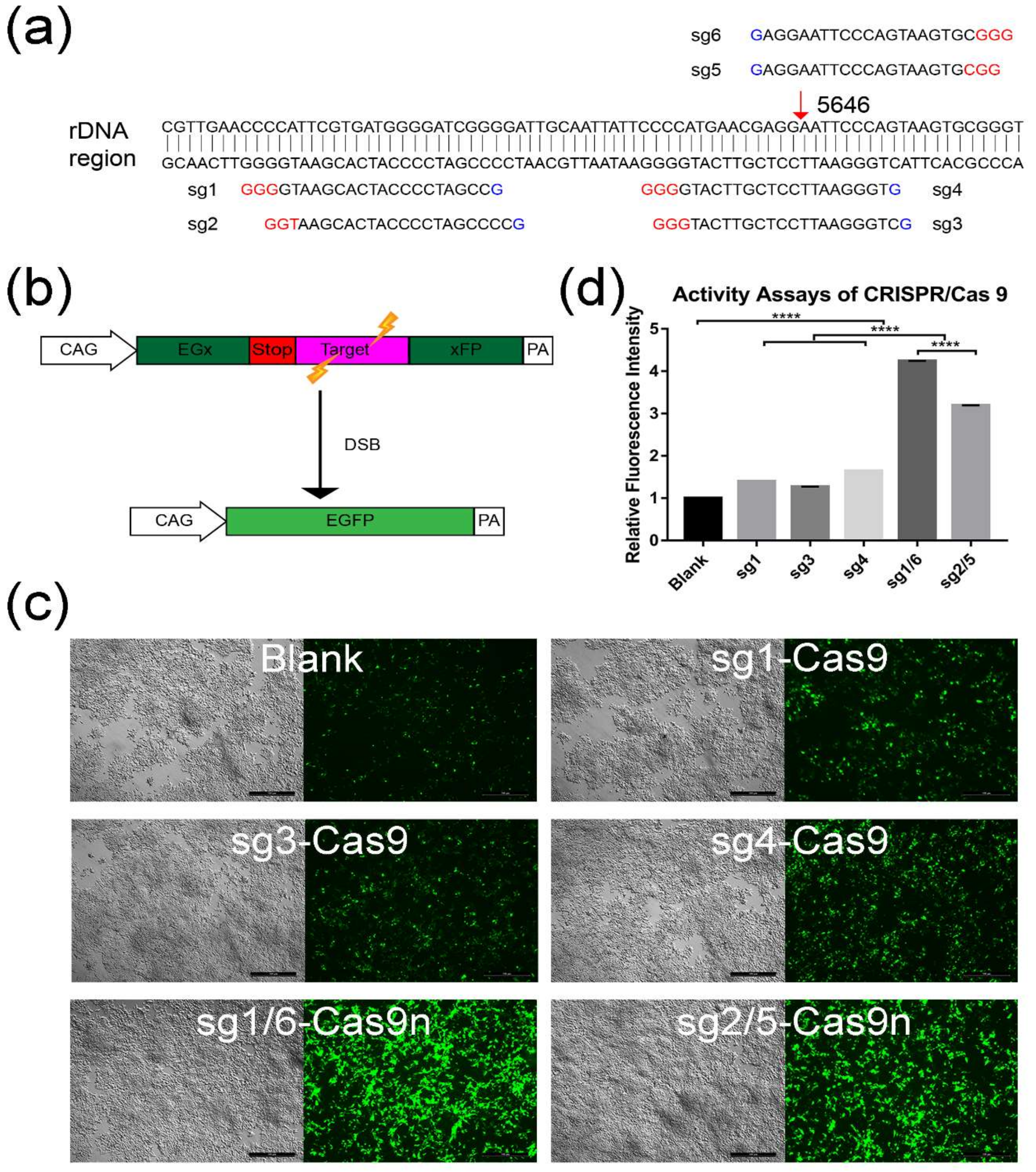
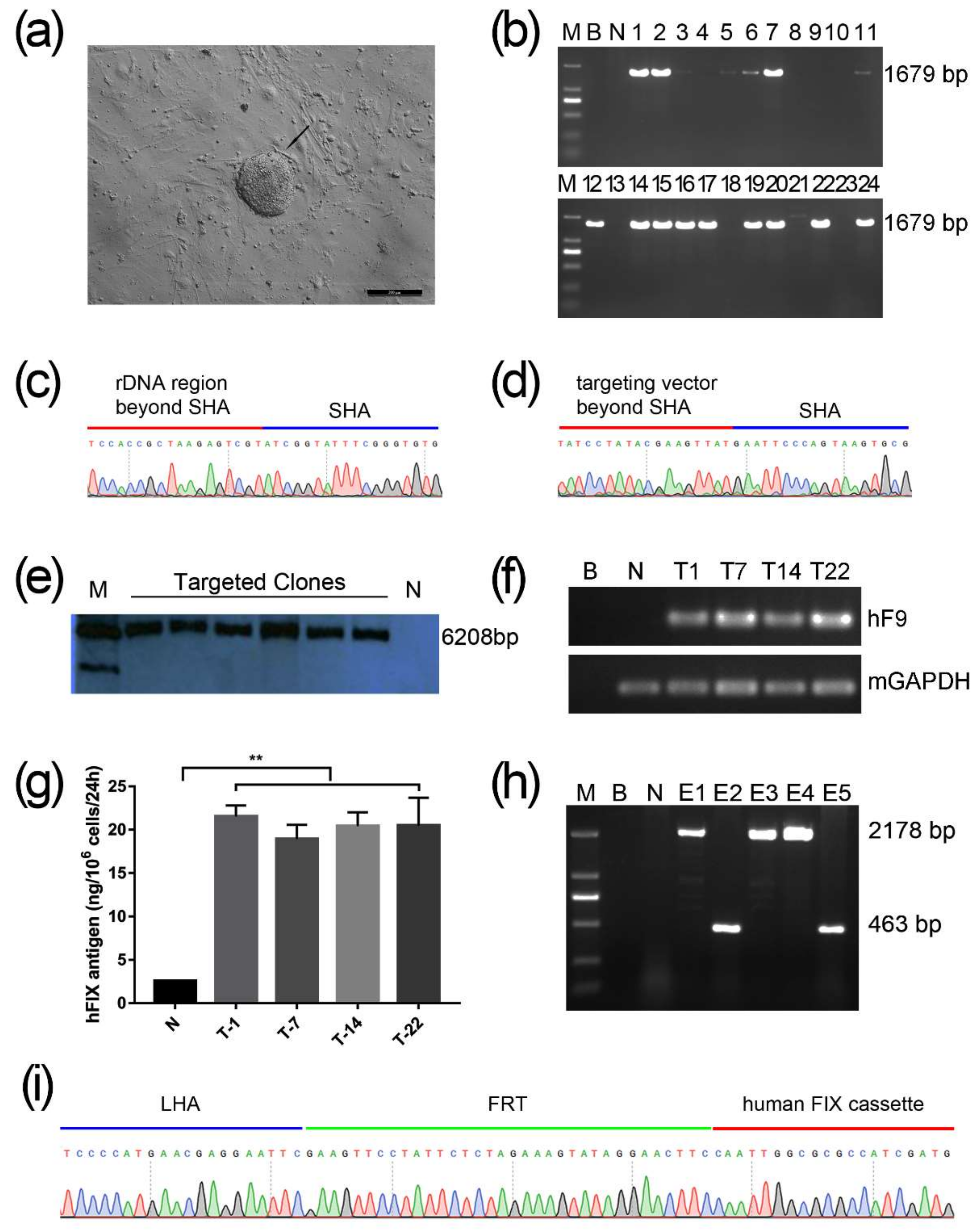
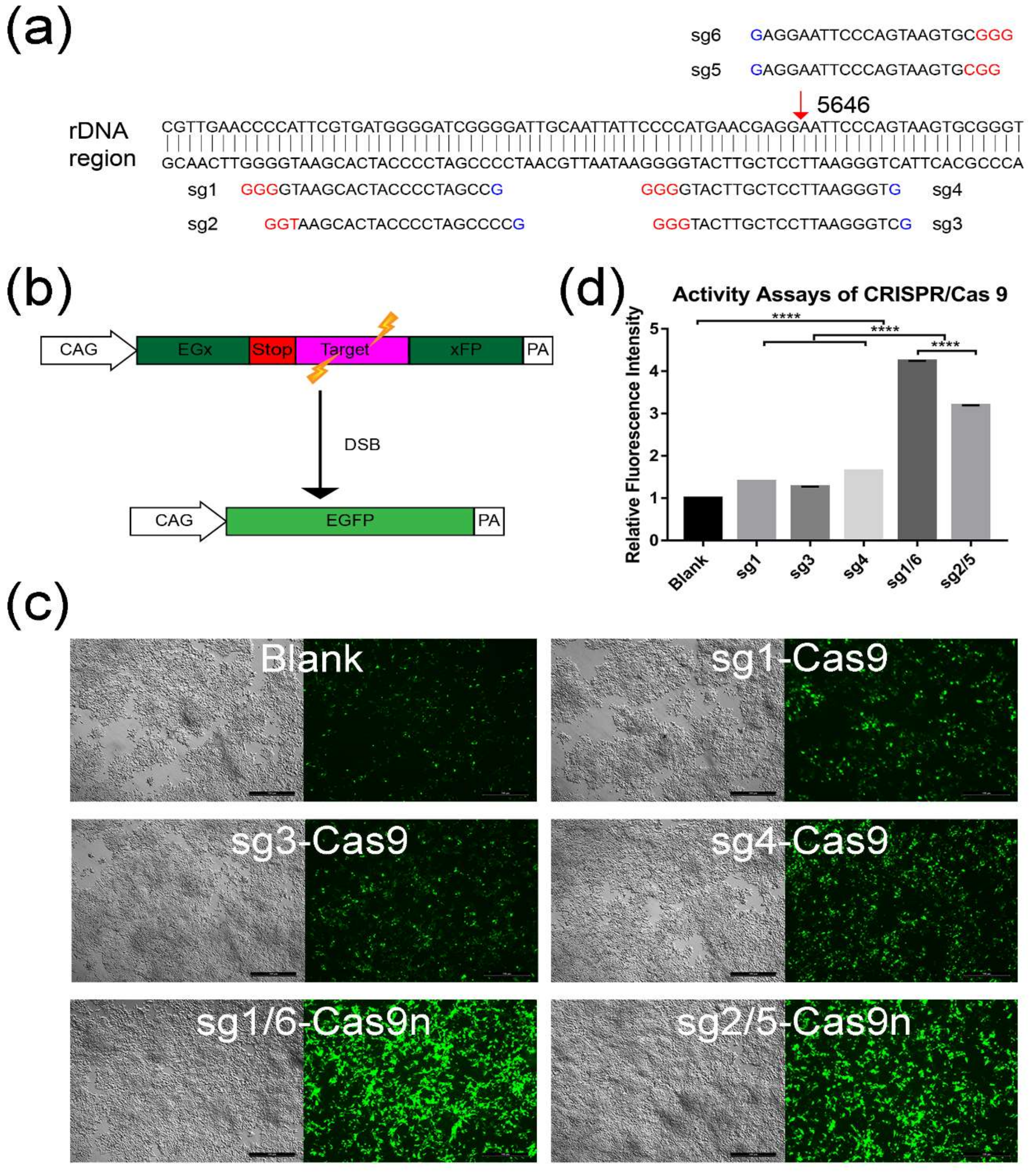
2.1. Screening for An Efficient rDNA-Targeted sgRNA-CRISPR/Cas9 System
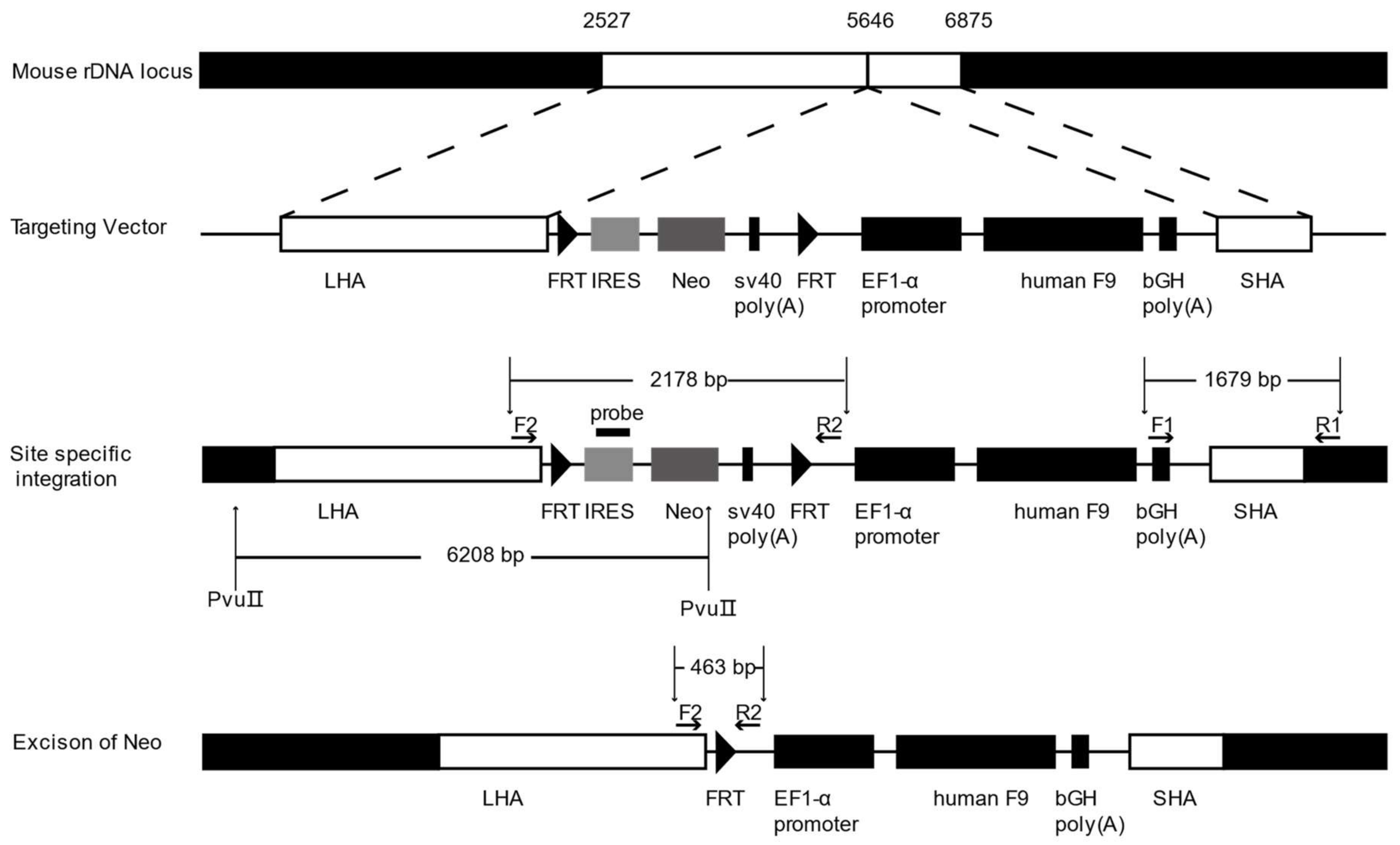
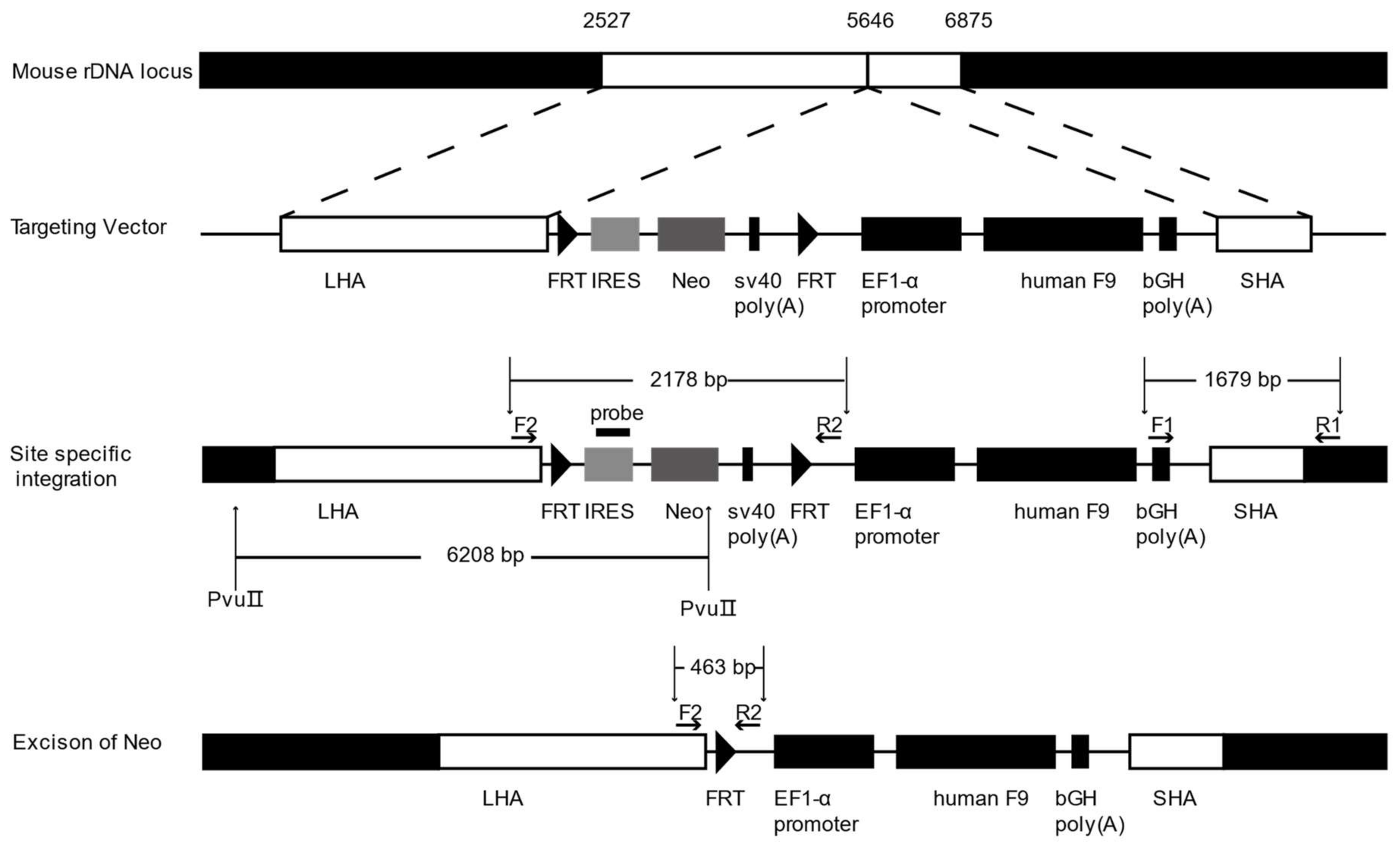
2.2. Gene Targeting of F9 into the rDNA Region of mESCs by Paired Cas9n and Non-Viral Targeting Vector pMrnF9
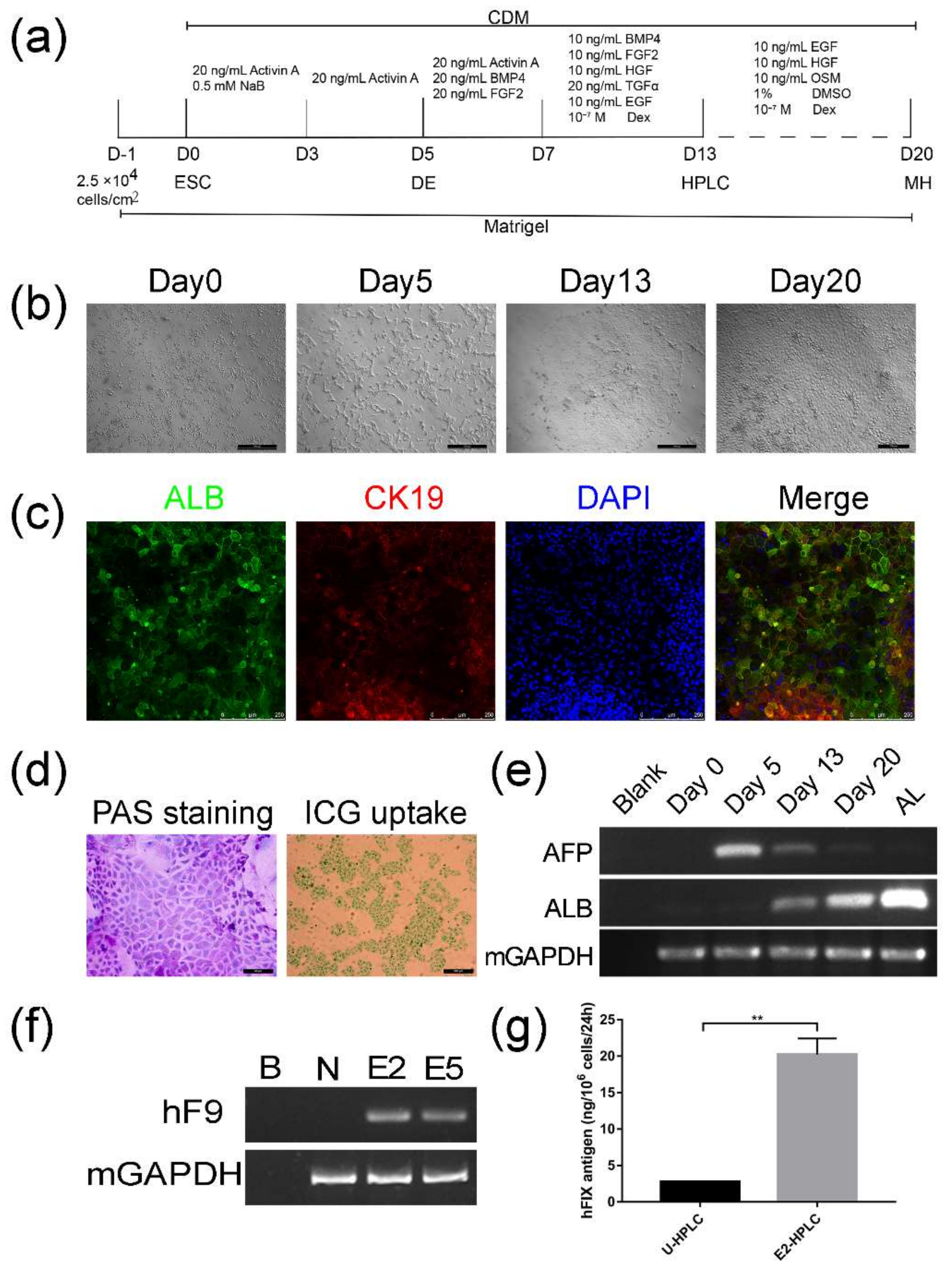
2.3. Differentiation of Targeted mESCs into HPLCs and Mature Hepatocytes
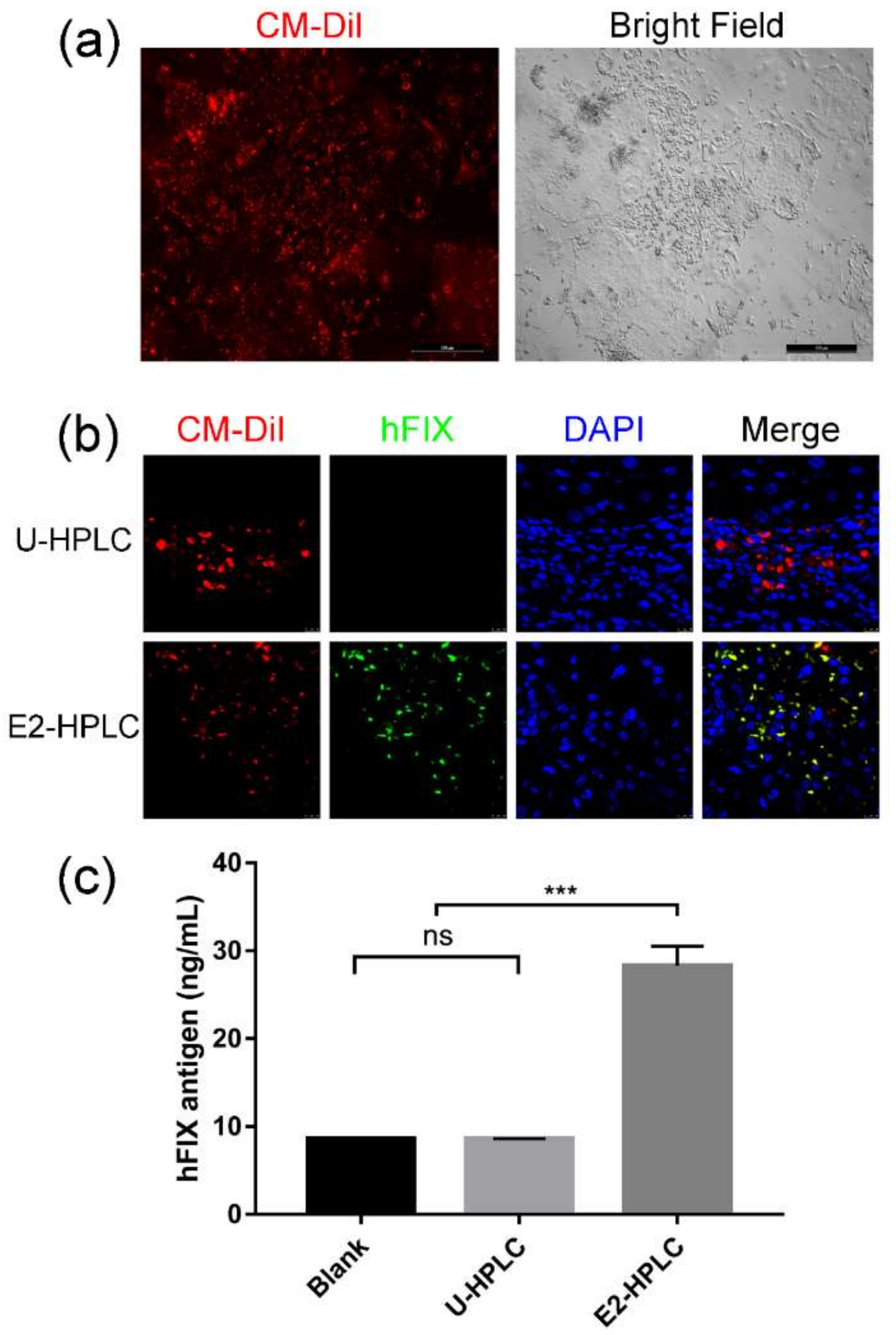
2.4. Transplantation of HPLCs in SCID Mice
3. Discussion
4. Materials and Methods
4.1. Design and Selection for sgRNAs-CRISPR/Cas9 Using Plasmid pCAG-EGx-Target-xFP
4.2. Construction of Non-Viral Targeting Vector pMrnF9
4.3. mESCs Culture and Gene Targeting
4.4. Southern Blotting
4.5. Excision of the Neo Cassette
4.6. ELISA for Human FIX Antigen
4.7. Differentiation of mESCs into HPLCs and Maturation of HPLCs
4.8. Periodic Acid Schiff Stain and Indocyanin Green Uptake Assay
4.9. Intrasplenic Transplantation of HPLCs into SCID Mice
4.10. Immunofluorescence Staining
4.11. Data Analysis and Statistics
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| CRISPR/Cas9 | Clustered regularly-interspaced short palindromic repeats/CRISPR associated protein 9 |
| CDM | Chemically defined medium |
| DSB | Double strand break |
| DE | Definitive endoderm |
| EMCV-IRES | Encephalomyocarditis virus internal ribosomal entry site |
| FIX | Coagulation factor IX |
| FRT | Flippase recognition target |
| HDR | Homology directed recombination |
| HPLCs | Hepatic progenitor like cells |
| ICG | Indocyanine green |
| LHA | Long homologous arm |
| mESCs | Mouse embryonic stem cells |
| MH | Maturated hepatocyte |
| ORF | Open reading frame |
| PAS staining | Periodic acid-Schiff’s staining |
| rDNA | Ribosomal DNA |
| SCID | Severe combined immune deficiency |
| SHA | Short homologous arm |
| SSA | Single strand annealing |
References
- Mannucci, P.; Tuddenham, E. Medical progress. The hemophilias—From royal genes to gene therapy. N. Engl. J. Med. 2001, 344, 1773–1779. [Google Scholar] [CrossRef] [PubMed]
- Castaldo, G.; Nardiello, P.; Bellitti, F.; Santamaria, R.; Rocino, A.; Coppola, A.; di Minno, G.; Salvatore, F. Haemophilia B: From molecular diagnosis to gene therapy. Clin. Chem. Lab. Med. 2003, 41, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Brewer, A.K.; Mauser-Bunschoten, E.P.; Key, N.S.; Kitchen, S.; Llinas, A.; Ludlam, C.A.; Mahlangu, J.N.; Mulder, K.; Poon, M.C.; et al. Guidelines for the management of hemophilia. Haemophilia 2013, 19. [Google Scholar] [CrossRef] [PubMed]
- George, L.A.; Sullivan, S.K.; Giermasz, A.; Rasko, J.E.J.; Samelson-Jones, B.J.; Ducore, J.; Cuker, A.; Sullivan, L.M.; Majumdar, S.; Teitel, J.; et al. Hemophilia B gene therapy with a high-specific-activity factor IX variant. N. Engl. J. Med. 2017, 377, 2215–2227. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; High, K. Immune responses to AAV vectors: Overcoming barriers to successful gene. Blood 2013, 122, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Moss, T.; Stefanovsky, V.Y. Promotion and regulation of ribosomal transcription in eukaryotes by RNA polymerase. Prog. Nucleic Acid Res. Mol. Biol. 1995, 50, 25–66. [Google Scholar] [CrossRef] [PubMed]
- Lisowski, L.; Lau, A.; Wang, Z.; Zhang, Y.; Zhang, F.; Grompe, M.; Kay, M.A. Ribosomal DNA integrating rAAV-rDNA vectors allow for stable transgene expression. Mol. Ther. 2012, 20, 1912–1923. [Google Scholar] [CrossRef] [PubMed]
- Schenkwein, D.; Turkki, V.; Ahlroth, M.K.; Timonen, O.; Airenne, K.J.; Ylä-Herttuala, S. rDNA-directed integration by an HIV-1 integrase—I-PpoI fusion protein. Nucleic Acids Res. 2013, 41. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, Y.; Li, Z.; Yang, J.; Xue, J.; Hu, Y.; Feng, M.; Niu, W.; Yang, Q.; Lei, M.; et al. Targeting of the human coagulation factor IX gene at rDNA locus of human embryonic stem cells. PLoS ONE 2012, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Liu, X.; Long, P.; Xiao, D.; Cun, J.; Li, Z.; Xue, J.; Wu, Y.; Luo, S.; Wu, L.; et al. Nonviral gene targeting at rDNA locus of human mesenchymal stem cells. BioMed Res. Int. 2013, 2013, 135189. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, F.; Wu, Y.; Wang, X.; Feng, M.; Li, Z.; Zhou, M.; Wang, Y.; Wu, L.; Liu, X.; et al. Enhanced tumor growth inhibition by mesenchymal stem cells derived from iPSCs with targeted integration of interleukin24 into rDNA loci. Oncotarget 2017, 8, 40791–40803. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.; Wu, Y.; Li, Z.; Hu, Z.; Wang, X.; Hu, X.; Wang, X.; Liu, X.; Zhou, M.; Liu, B.; et al. Targeting of the human F8 at the multicopy rDNA locus in Hemophilia a patient-derived iPSCs using TALENickases. Biochem. Biophys. Res. Commun. 2016, 472, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Gao, T.; Wang, X.; Hu, Y.; Hu, X.; Hu, Z.; Pang, J.; Li, Z.; Xue, J.; Feng, M.; et al. TALE nickase mediates high efficient targeted transgene integration at the human multi-copy ribosomal DNA locus. Biochem. Biophys. Res. Commun. 2014, 446, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Reh, W.A.; Vasquez, K.M. Gene targeting by homologous recombination. eLS 2014. [Google Scholar] [CrossRef]
- Nemudryi, A.A.; Valetdinova, K.R.; Medvedev, S.P.; Zakian, S.M. TALEN and CRISPR/CAS genome editing systems: Tools of discovery. Acta Naturae 2014, 6, 19–40. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Qiu, Z.; Shao, Y.; Chen, Y.; Guan, Y.; Liu, M.; Li, Y.; Gao, N.; Wang, L.; Lu, X.; et al. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 681–683. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Aach, J.; Stranges, P.B.; Esvelt, K.M.; Moosburner, M.; Kosuri, S.; Yang, L.; Church, G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013, 31, 833–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Mashiko, D.; Young, S.A.M.; Muto, M.; Kato, H.; Nozawa, K.; Ogawa, M.; Noda, T.; Kim, Y.J.; Satouh, Y.; Fujihara, Y.; et al. Feasibility for a large scale mouse mutagenesis by injecting CRISPR/Cas plasmid into zygotes. Dev. Growth Differ. 2014, 56, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liu, P.; Liu, C.; Xiang, D.; Deng, L.; Li, W.; Wangensteen, K.; Song, J.; Ma, Y.; Hui, L.; et al. Hepatoblast-Like progenitor cells derived from embryonic stem cells can repopulate livers of mice. Gastroenterology 2010, 139, 2158–2169. [Google Scholar] [CrossRef] [PubMed]
- Xiao-Jie, L.; Hui-Ying, X.; Zun-Ping, K.; Jin-Lian, C.; Li-Juan, J. CRISPR-Cas9: A new and promising player in gene therapy. J. Med. Genet. 2015, 52, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wen, Y.; Guo, X. CRISPR/Cas9 for genome editing: Progress, implications and challenges. Hum. Mol. Genet. 2016, 46, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Liu, X.; Wu, Y.; Niu, W.; Long, P.; Liu, J.; Lei, M.; Hu, Y.; Wu, L.; Li, Z.; et al. Ectopic expression of factor VIII in MSCs and hepatocytes derived from rDNA targeted hESCs. Clin. Chim. Acta 2018, 0–1. [Google Scholar] [CrossRef] [PubMed]
- Stults, D.M.; Killen, M.W.; Pierce, H.H.; Pierce, A.J. Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Res. 2008, 18, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, P.; Djalali, M.; Hansmann, I.; Kattner, E.; Meisel-Stosiek, M.; Probeck, H.D.; Schmidt, A.; Wolf, M. The genetic significance of accessory bisatellited marker chromosomes. Hum. Genet. 1983, 65, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.P.; Vanneaux, V.; Branger, J.; Touboul, T.; Sentilhes, L.; Mainot, S.; Lainas, P.; Leclerc, P.; Uzan, G.; Mahieu-Caputo, D.; et al. The role of HGF on invasive properties and repopulation potential of human fetal hepatic progenitor cells. Exp. Cell Res. 2009, 315, 3396–3405. [Google Scholar] [CrossRef] [PubMed]
- Mahieu-Caputo, D.; Allain, J.-E.; Branger, J.; Coulomb, A.; Delgado, J.-P.; Andreoletti, M.; Mainot, S.; Frydman, R.; Leboulch, P.; Di Santo, J.P.; et al. Repopulation of athymic mouse liver by cryopreserved early human fetal hepatoblasts. Hum. Gene Ther. 2004, 15, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.A.; Wauthier, E.; Lozoya, O.; McClelland, R.; Bowsher, J.E.; Barbier, C.; Prestwich, G.; Hsu, E.; Gerber, D.A.; Reid, L.M. Successful transplantation of human hepatic stem cells with restricted localization to liver using hyaluronan grafts. Hepatology 2013, 57, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, A.; Tesfaye, A.; Chu, V.; Nimgaonkar, I.; Zhang, F.; Lee, S.B.; Thorgeirsson, S.S.; Feinstone, S.M.; Liang, T.J. Engrafted human stem cell-derived hepatocytes establish an infectious HCV murine model. J. Clin. Invest. 2014, 124, 4953–4964. [Google Scholar] [CrossRef] [PubMed]
- Simioni, P.; Tormene, D.; Tognin, G.; Gavasso, S.; Bulato, C.; Iacobelli, N.P.; Finn, J.D.; Spiezia, L.; Radu, C.; Arruda, V.R. X-Linked thrombophilia with a mutant factor IX (factor IX Padua). N. Engl. J. Med. 2009, 361, 1671–1675. [Google Scholar] [CrossRef] [PubMed]
- Finn, J.D.; Nichols, T.C.; Svoronos, N.; Merricks, E.P.; Dwight, A.; Zhou, S.; Simioni, P.; High, K.A.; Arruda, V.R.; Dc, W.; et al. The efficacy and the risk of immunogenicity of FIX Padua (R338L) in hemophilia B dogs treated by AAV muscle gene therapy. Blood 2013, 120, 4521–4523. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.Y.; Yang, S.J.; Tao, M.H.; Jeng, Y.M.; Yu, I.S.; Lin, S.W. Incorporation of the factor IX Padua mutation into FIX-Triple improves clotting activity in vitro and in vivo. Thromb. Haemost. 2013, 110, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Li, P.; Tan, L.; Qu, S.; Ying, Q.L.; Song, H. Differentiation of mouse embryonic stem cells into hepatocytes induced by a combination of cytokines and sodium butyrate. J. Cell. Biochem. 2010, 109, 606–614. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zhao, J.; Duan, N.; Liu, W.; Zhang, Y.; Zhou, M.; Hu, Z.; Feng, M.; Liu, X.; Wu, L.; et al. Paired CRISPR/Cas9 Nickases Mediate Efficient Site-Specific Integration of F9 into rDNA Locus of Mouse ESCs. Int. J. Mol. Sci. 2018, 19, 3035. https://doi.org/10.3390/ijms19103035
Wang Y, Zhao J, Duan N, Liu W, Zhang Y, Zhou M, Hu Z, Feng M, Liu X, Wu L, et al. Paired CRISPR/Cas9 Nickases Mediate Efficient Site-Specific Integration of F9 into rDNA Locus of Mouse ESCs. International Journal of Molecular Sciences. 2018; 19(10):3035. https://doi.org/10.3390/ijms19103035
Chicago/Turabian StyleWang, Yanchi, Junya Zhao, Nannan Duan, Wei Liu, Yuxuan Zhang, Miaojin Zhou, Zhiqing Hu, Mai Feng, Xionghao Liu, Lingqian Wu, and et al. 2018. "Paired CRISPR/Cas9 Nickases Mediate Efficient Site-Specific Integration of F9 into rDNA Locus of Mouse ESCs" International Journal of Molecular Sciences 19, no. 10: 3035. https://doi.org/10.3390/ijms19103035
APA StyleWang, Y., Zhao, J., Duan, N., Liu, W., Zhang, Y., Zhou, M., Hu, Z., Feng, M., Liu, X., Wu, L., Li, Z., & Liang, D. (2018). Paired CRISPR/Cas9 Nickases Mediate Efficient Site-Specific Integration of F9 into rDNA Locus of Mouse ESCs. International Journal of Molecular Sciences, 19(10), 3035. https://doi.org/10.3390/ijms19103035