Her2-Targeted Therapy Induces Autophagy in Esophageal Adenocarcinoma Cells

 , and
, and
Abstract
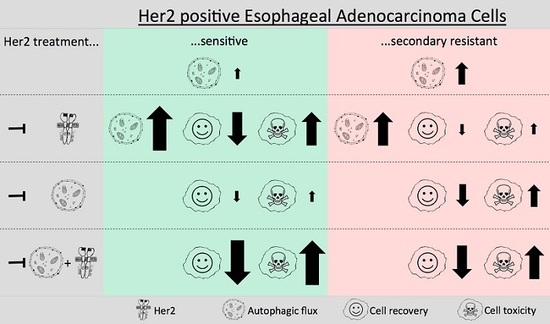
:
1. Introduction
2. Results
2.1. Validation of Lapatinib Sensitivity and Her2 Status of OE19 Cells
2.2. Induction of Autophagic Flux in OE19 Cells upon Lapatinib Treatment
2.3. Her2-Inhibition-Resistant OE19 Daughter Cells Show Increased Autophagic Activity Compared to Their Normal Counterparts
2.4. Blocking Autophagy in Combination with Her2 Inhibition Significantly Reduces OE19 Cell Viability and Colony Formation
2.5. Assessment of Her2 Status and Autophagy Marker Levels in a Treatment-Naïve EAC Patient Cohort
3. Discussion
4. Materials and Methods
4.1. Drugs and Inhibitors
4.2. Cell Culture and Generation of Resistant OE19 Cells
4.3. Western Blotting
4.4. Autophagic Flux Analysis by Flow Cytometry
4.5. alamarBlue® Assay
4.6. Colony Forming Assay and Cell Counting
4.7. Caspase-Glo® 3/7 Assay
4.8. Statistical Analysis
4.9. Patients and Tissue Samples
4.10. Immunohistochemistry for FFPE Tumor Tissue
4.11. Evaluation of Her2 Status and Classification of Her2-Positive Tumors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lagergren, J.; Smyth, E.; Cunningham, D.; Lagergren, P. Oesophageal cancer. Lancet 2017, 390, 2383–2396. [Google Scholar] [CrossRef] [Green Version]
- Coleman, H.G.; Xie, S.-H.; Lagergren, J. The Epidemiology of Esophageal Adenocarcinoma. Gastroenterology 2018, 154, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Dulak, A.M.; Stojanov, P.; Peng, S.; Lawrence, M.S.; Fox, C.; Stewart, C.; Bandla, S.; Imamura, Y.; Schumacher, S.E.; Shefler, E.; et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat. Genet. 2013, 45, 478–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bang, Y.-J.; Van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. ToGA Trial Investigators Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet Lond. Engl. 2010, 376, 687–697. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Gerson, J.N.; Skariah, S.; Denlinger, C.S.; Astsaturov, I. Perspectives of HER2-targeting in gastric and esophageal cancer. Expert Opin. Investig. Drugs 2017, 26, 531–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Pedro, J.M.B.-S.; Kroemer, G. Defective Autophagy Initiates Malignant Transformation. Mol. Cell 2016, 62, 473–474. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cufí, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Corominas-Faja, B.; Urruticoechea, A.; Martin-Castillo, B.; Menendez, J.A. Autophagy-related gene 12 (ATG12) is a novel determinant of primary resistance to HER2-targeted therapies: Utility of transcriptome analysis of the autophagy interactome to guide breast cancer treatment. Oncotarget 2012, 3, 1600–1614. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, C.E.; Reidel, S.I.; Bal de Kier Joffé, E.D.; Jasnis, M.A.; Fiszman, G.L. Autophagy Protects from Trastuzumab-Induced Cytotoxicity in HER2 Overexpressing Breast Tumor Spheroids. PLoS ONE 2015, 10, e0137920. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Martin, A.; Oliveras-Ferraros, C.; Menendez, J.A. Autophagy facilitates the development of breast cancer resistance to the anti-HER2 monoclonal antibody trastuzumab. PLoS ONE 2009, 4, e6251. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wu, L.; Qiao, H.; Han, T.; Chen, S.; Liu, X.; Jiang, R.; Wei, Y.; Feng, D.; Zhang, Y.; et al. Autophagy stimulates apoptosis in HER2-overexpressing breast cancers treated by lapatinib. J. Cell. Biochem. 2013, 114, 2643–2653. [Google Scholar] [CrossRef] [PubMed]
- Lordick, F.; Mariette, C.; Haustermans, K.; Obermannová, R.; Arnold, D. ESMO Guidelines Committee Oesophageal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, v50–v57. [Google Scholar] [CrossRef] [PubMed]
- Wetterskog, D.; Shiu, K.-K.; Chong, I.; Meijer, T.; Mackay, A.; Lambros, M.; Cunningham, D.; Reis-Filho, J.S.; Lord, C.J.; Ashworth, A. Identification of novel determinants of resistance to lapatinib in ERBB2-amplified cancers. Oncogene 2014, 33, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Fichter, C.D.; Timme, S.; Braun, J.A.; Gudernatsch, V.; Schöpflin, A.; Bogatyreva, L.; Geddert, H.; Faller, G.; Klimstra, D.; Tang, L.; et al. EGFR, HER2 and HER3 dimerization patterns guide targeted inhibition in two histotypes of esophageal cancer. Int. J. Cancer 2014, 135, 1517–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlberg, P.S.; Jacobson, B.A.; Dahal, G.; Fink, J.M.; Kratzke, R.A.; Maddaus, M.A.; Ferrin, L.J. ERBB2 amplifications in esophageal adenocarcinoma. Ann. Thorac. Surg. 2004, 78, 1790–1800. [Google Scholar] [CrossRef] [PubMed]
- Langer, R.; Rauser, S.; Feith, M.; Nährig, J.M.; Feuchtinger, A.; Friess, H.; Höfler, H.; Walch, A. Assessment of ErbB2 (Her2) in oesophageal adenocarcinomas: Summary of a revised immunohistochemical evaluation system, bright field double in situ hybridisation and fluorescence in situ hybridisation. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc 2011, 24, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Gump, J.M.; Thorburn, A. Sorting cells for basal and induced autophagic flux by quantitative ratiometric flow cytometry. Autophagy 2014, 10, 1327–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bago, R.; Malik, N.; Munson, M.J.; Prescott, A.R.; Davies, P.; Sommer, E.; Shpiro, N.; Ward, R.; Cross, D.; Ganley, I.G.; et al. Characterization of VPS34-IN1, a selective inhibitor of Vps34, reveals that the phosphatidylinositol 3-phosphate-binding SGK3 protein kinase is a downstream target of class III phosphoinositide 3-kinase. Biochem. J. 2014, 463, 413–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chude, C.I.; Amaravadi, R.K. Targeting Autophagy in Cancer: Update on Clinical Trials and Novel Inhibitors. Int. J. Mol. Sci. 2017, 18, 1279. [Google Scholar] [CrossRef] [PubMed]
- Adams, O.; Dislich, B.; Berezowska, S.; Schläfli, A.M.; Seiler, C.A.; Kröll, D.; Tschan, M.P.; Langer, R. Prognostic relevance of autophagy markers LC3B and p62 in esophageal adenocarcinomas. Oncotarget 2016, 7, 39241–39255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhu, X.; Qiao, H.; Ye, M.; Lai, X.; Yu, S.; Ding, L.; Wen, A.; Zhang, J. Protective autophagy promotes the resistance of HER2-positive breast cancer cells to lapatinib. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2016, 37, 2321–2331. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, T.R.; O’Sullivan, G.C.; McKenna, S.L. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 2011, 7, 509–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, M.; Lee, K.-H.; Lee, H.S.; Jeong, C.W.; Kwak, C.; Kim, H.H.; Ku, J.H. Concurrent Autophagy Inhibition Overcomes the Resistance of Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Human Bladder Cancer Cells. Int. J. Mol. Sci. 2017, 18, 321. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.B.; Kerkhoff, A.; Lenz, G.; Kessler, T. Targeted therapies of HER2-positive gastric adenocarcinoma. Transl. Cancer Res. 2017, 6, S1177–S1180. [Google Scholar] [CrossRef]
- Curea, F.G.; Hebbar, M.; Ilie, S.M.; Bacinschi, X.E.; Trifanescu, O.G.; Botnariuc, I.; Anghel, R.M. Current Targeted Therapies in HER2-Positive Gastric Adenocarcinoma. Cancer Biother. Radiopharm. 2017, 32, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, M.; Tschan, M.P.; Britschgi, C.; Britschgi, A.; Hügli, B.; Grob, T.J.; Leupin, N.; Mueller, B.U.; Simon, H.-U.; Ziemiecki, A.; et al. The death-associated protein kinase 2 is up-regulated during normal myeloid differentiation and enhances neutrophil maturation in myeloid leukemic cells. J. Leukoc. Biol. 2007, 81, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Tschan, M.P.; Fischer, K.M.; Fung, V.S.; Pirnia, F.; Borner, M.M.; Fey, M.F.; Tobler, A.; Torbett, B.E. Alternative splicing of the human cyclin D-binding Myb-like protein (hDMP1) yields a truncated protein isoform that alters macrophage differentiation patterns. J. Biol. Chem. 2003, 278, 42750–42760. [Google Scholar] [CrossRef] [PubMed]
- Schläfli, A.M.; Berezowska, S.; Adams, O.; Langer, R.; Tschan, M.P. Reliable LC3 and p62 autophagy marker detection in formalin fixed paraffin embedded human tissue by immunohistochemistry. Eur. J. Histochem. 2015, 59, 2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, M.; Stoss, O.; Shi, D.; Büttner, R.; Vijver, M.V.D.; Kim, W.; Ochiai, A.; Rüschoff, J.; Henkel, T. Assessment of a HER2 scoring system for gastric cancer: Results from a validation study. Histopathology 2008, 52, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, N.; Gotoda, T.; Hara, M.; Hale, M.D.; Tsuchiya, T.; Matsubayashi, J.; Kono, S.; Kusano, C.; Itoi, T.; Fujimoto, K.; et al. Five biopsy specimens from the proximal part of the tumor reliably determine HER2 protein expression status in gastric cancer. Gastric Cancer 2016, 19, 553–560. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LC3B | Total | |||
|---|---|---|---|---|
| Low | High | |||
| Her2 | negative | 50 | 44 | 94 |
| positive | 8 | 10 | 18 | |
| total | 58 | 54 | 112 | |
| p62 | Total | |||
|---|---|---|---|---|
| Low | High | |||
| Her2 | negative | 21 | 73 | 94 |
| positive | 3 | 15 | 18 | |
| total | 24 | 88 | 112 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janser, F.A.; Adams, O.; Bütler, V.; Schläfli, A.M.; Dislich, B.; Seiler, C.A.; Kröll, D.; Langer, R.; Tschan, M.P. Her2-Targeted Therapy Induces Autophagy in Esophageal Adenocarcinoma Cells. Int. J. Mol. Sci. 2018, 19, 3069. https://doi.org/10.3390/ijms19103069
Janser FA, Adams O, Bütler V, Schläfli AM, Dislich B, Seiler CA, Kröll D, Langer R, Tschan MP. Her2-Targeted Therapy Induces Autophagy in Esophageal Adenocarcinoma Cells. International Journal of Molecular Sciences. 2018; 19(10):3069. https://doi.org/10.3390/ijms19103069
Chicago/Turabian StyleJanser, Félice A., Olivia Adams, Vanessa Bütler, Anna M. Schläfli, Bastian Dislich, Christian A. Seiler, Dino Kröll, Rupert Langer, and Mario P. Tschan. 2018. "Her2-Targeted Therapy Induces Autophagy in Esophageal Adenocarcinoma Cells" International Journal of Molecular Sciences 19, no. 10: 3069. https://doi.org/10.3390/ijms19103069