Attempt to Untangle the Prion-Like Misfolding Mechanism for Neurodegenerative Diseases


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
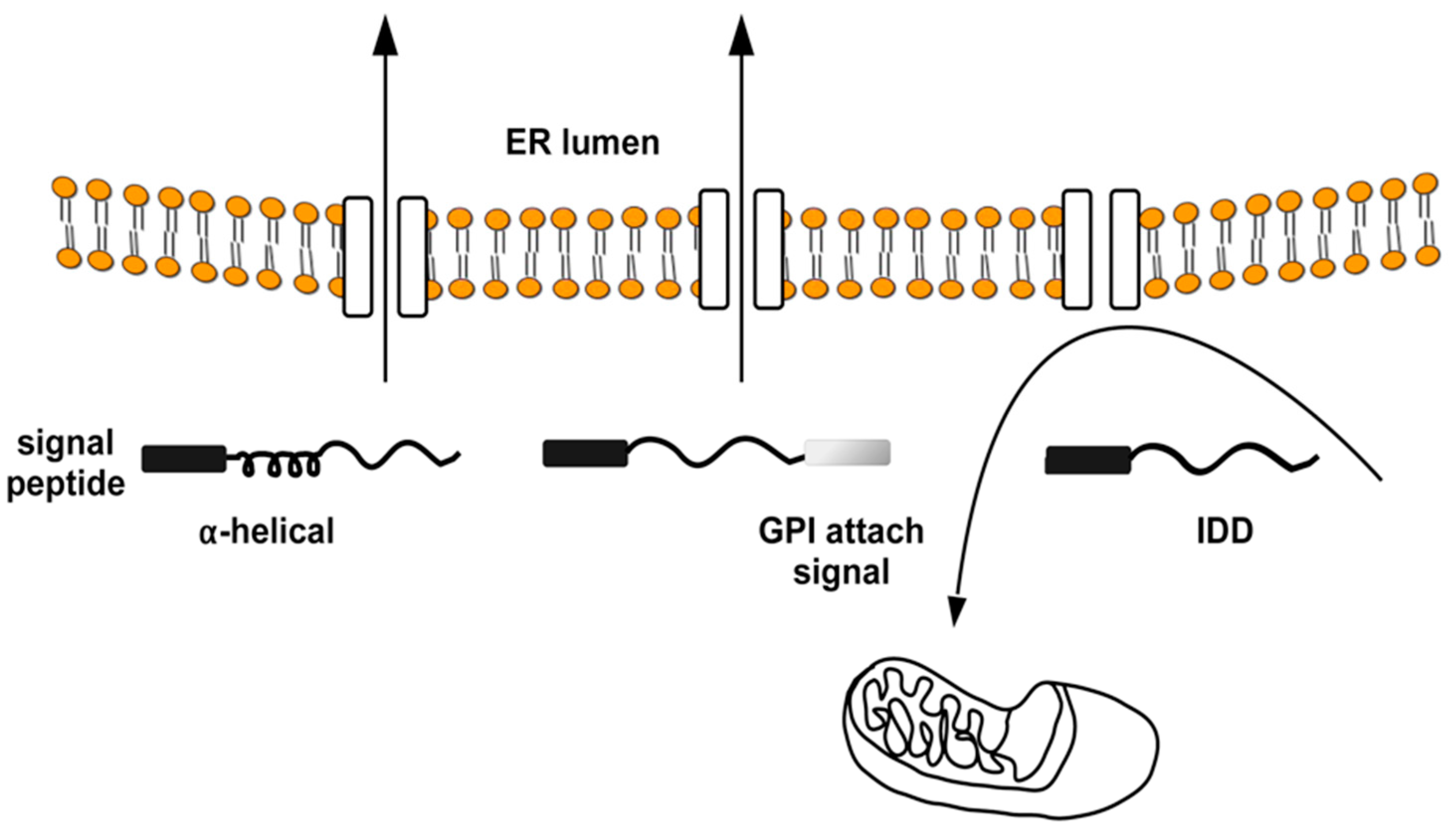
2. ER/Golgi Quality Control and the Role of GPI-Anchor in Protein Conformation
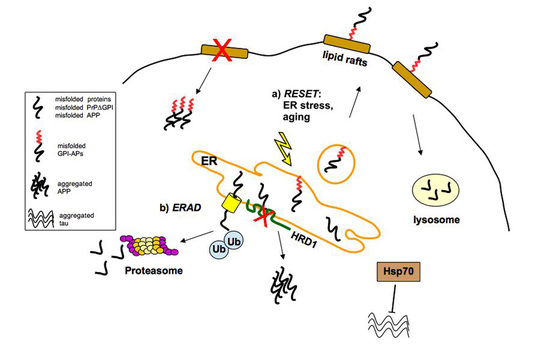
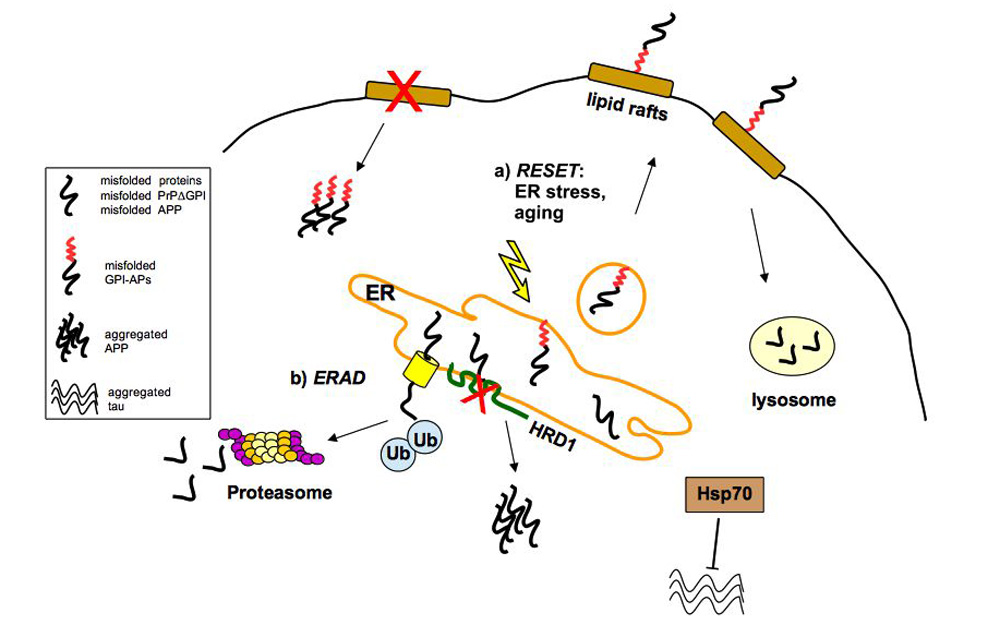
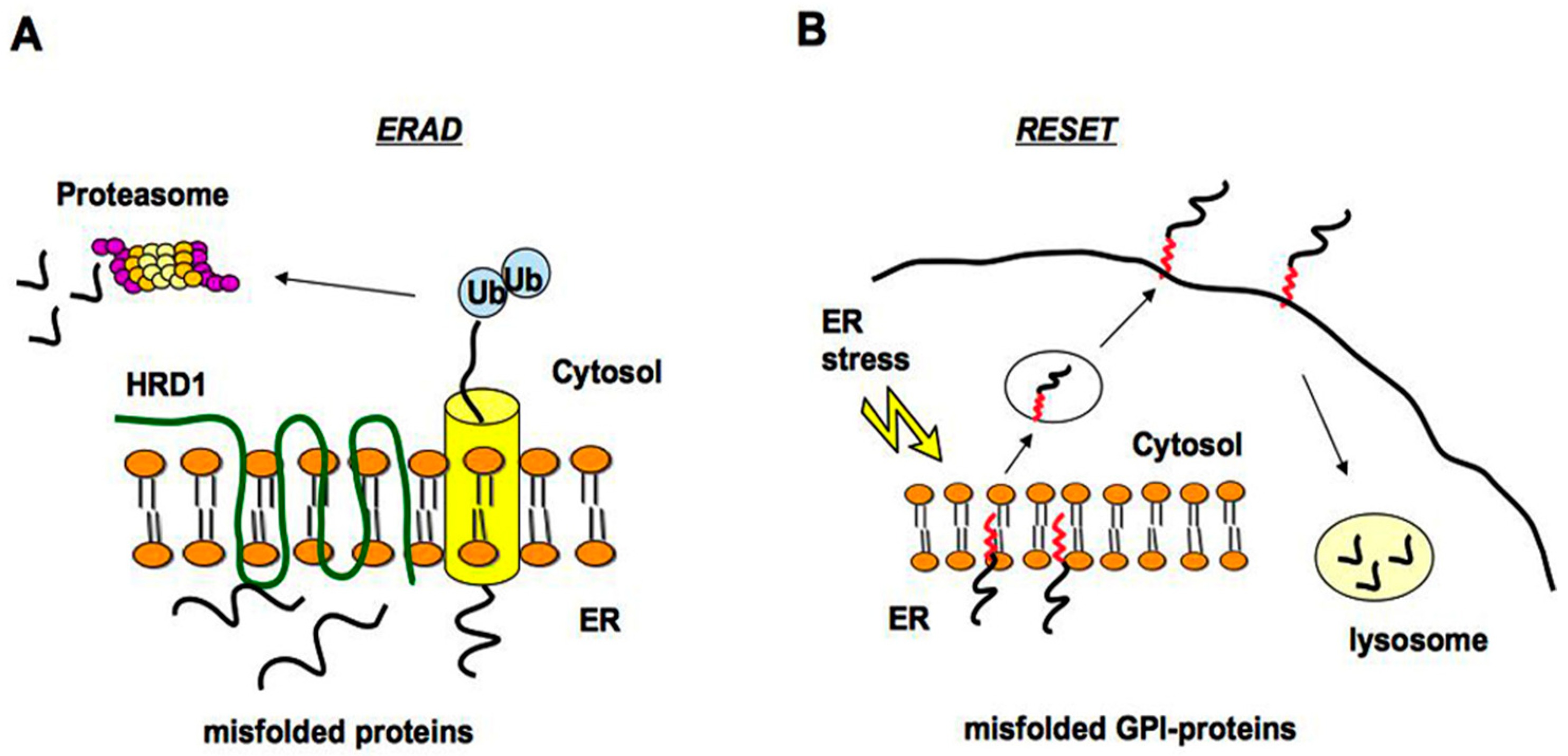
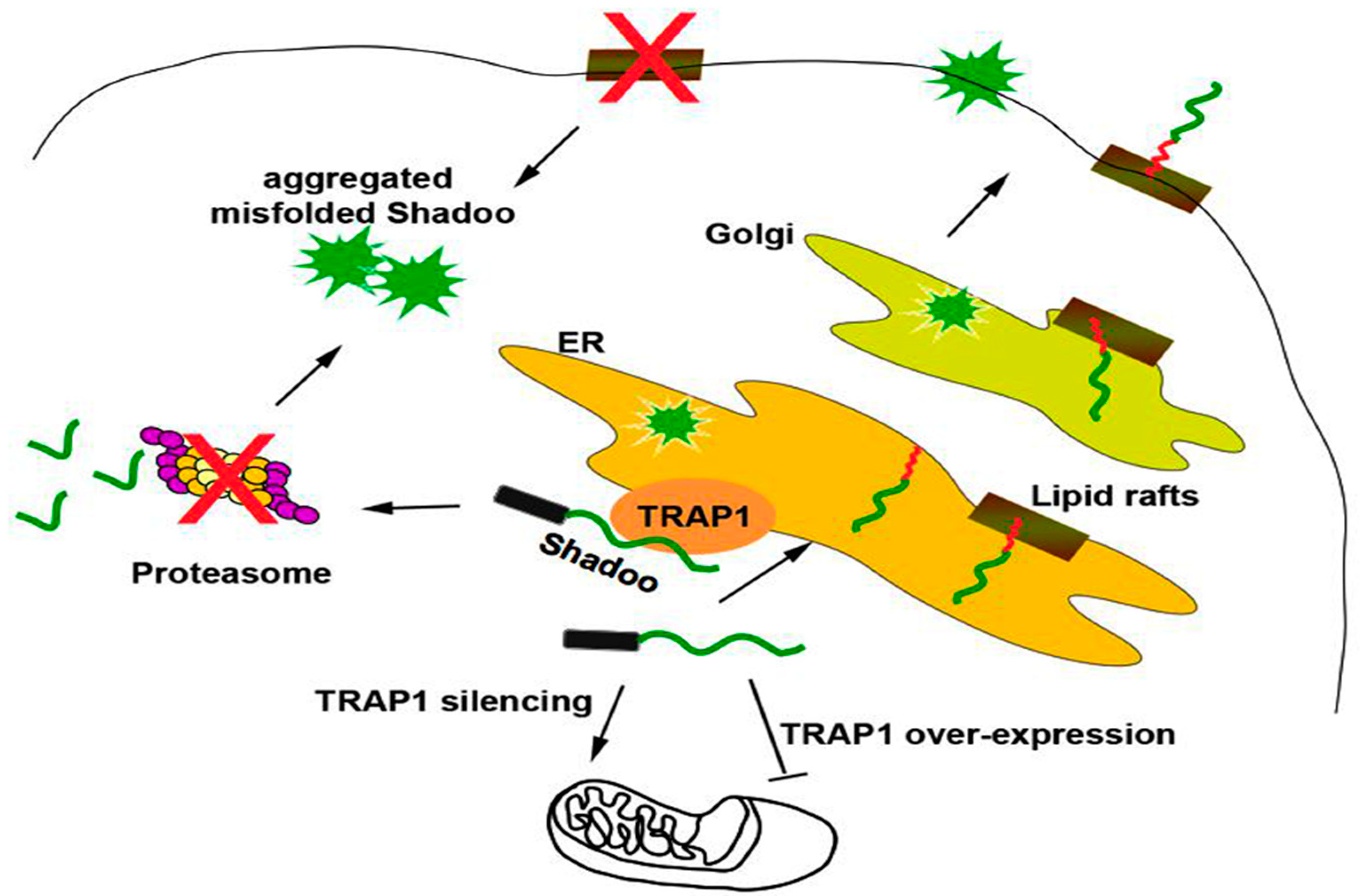
2.1. Quality Control of PrP: The ERAD Pathway
2.2. Quality Control of PrP: The RESET Pathway
2.3. Quality Control of APP
2.4. Quality Control of Tau
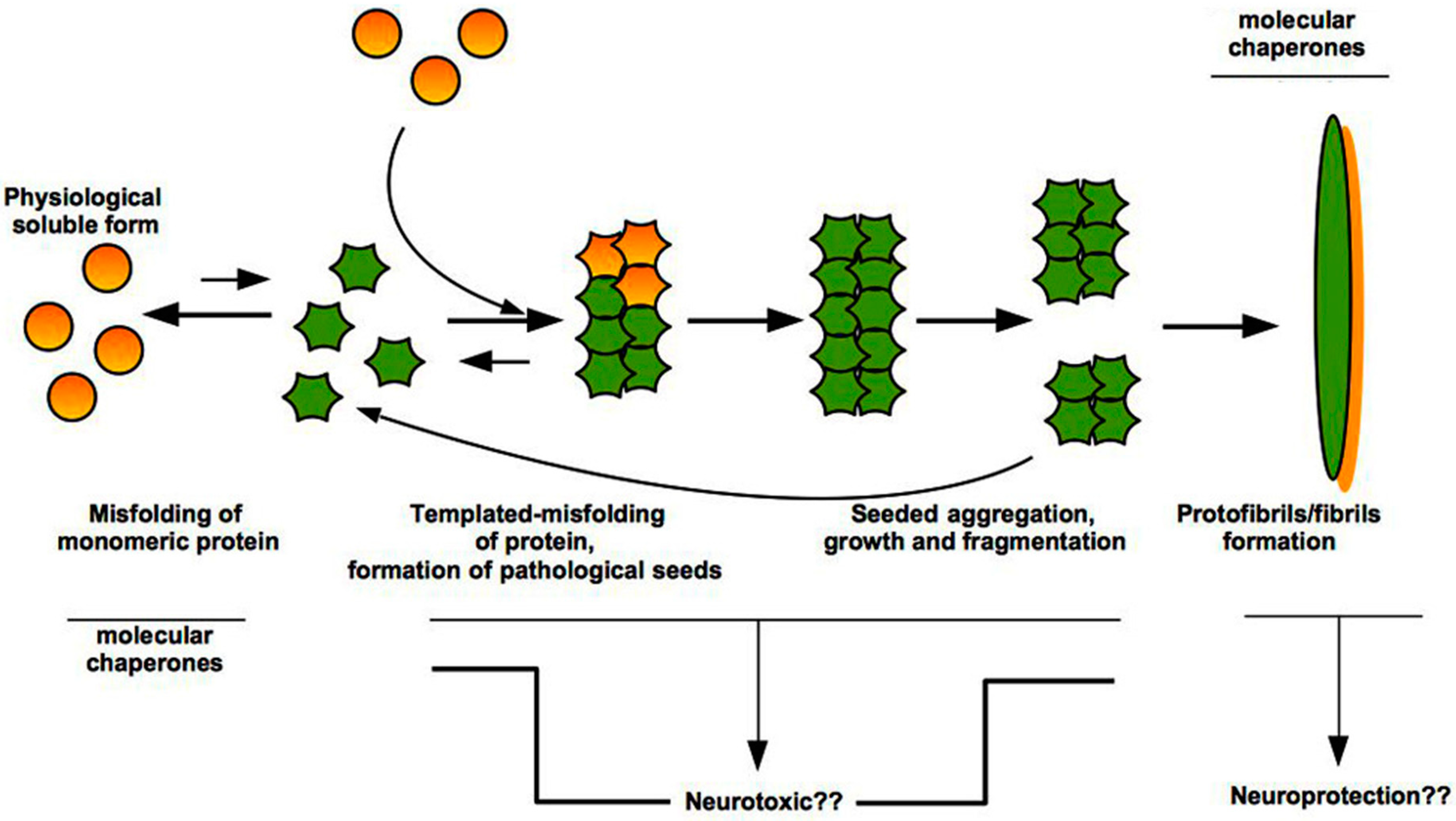
3. “Prion-Like” Misfolding of Aβ and Tau: Implication for Alzheimer’s Disease
Mutations and Polymorphisms That Promote Aβ Misfolding and Aggregation Increase the Risk of AD
4. The Intrinsically Disordered Proteins: A Common Feature of Proteins Associated with Misfolding Diseases
5. Interactions between Misfolded Proteins and Plasma Membrane
5.1. Interaction with ECM Components
5.2. Interaction with Plasma Membrane
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ALS | amyotrophic lateral sclerosis |
| APP | amyloid precursor protein |
| Aβ | amyloid beta |
| BSE | bovine spongiform encephalopathy |
| CJD | Creutzfeldt–Jacob Disease |
| CTFβ | C-terminal fragment beta |
| ECM | extracellular matrix |
| ERAD | ER-associated degradation |
| FFI | Fatal Familial Insomnia |
| GPI | glycosyl-phosphatidyl-inositol |
| GSS | Gerstmann–Sträussler–Scheinker |
| HSPGs | heparan sulfate proteoglycans |
| IDD | intrinsically disordered domain |
| IDP | intrinsically disordered protein |
| MAPT | microtubules-associated tau protein |
| MTBR | microtubules binding region |
| NFTs | neurofibrillary tangles |
| PD | Parkinson’s disease |
| PrPC | cellular prion protein |
| PrPSc | scrapie prion protein |
| RESET | ER stress-induced degradation |
| UPR | unfolding protein response |
References
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.A. Trafficking, turnover and membrane topology of PrP. Br. Med. Bull. 2003, 66, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Mayor, S.; Riezman, H. Sorting GPI-anchored proteins. Nat. Rev. Mol. Cell. Biol. 2004, 5, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Sarnataro, D.; Campana, V.; Paladino, S.; Stornaiuolo, M.; Nitsch, L.; Zurzolo, C. PrPC association with lipid rafts in the early secretory pathway stabilizes its cellular conformation. Mol. Biol. Cell 2004, 15, 4031–4042. [Google Scholar] [CrossRef] [PubMed]
- Campana, V.; Sarnataro, D.; Zurzolo, C. The highways and byways of prion protein trafficking. Trends Cell Biol. 2005, 15, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Campana, V.; Sarnataro, D.; Fasano, C.; Casanova, P.; Paladino, S.; Zurzolo, C. Detergent-resistant membrane domains but not the proteasome are involved in the misfolding of a PrP mutant retained in the endoplasmic reticulum. J. Cell Sci. 2006, 119, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Sarnataro, D.; Pepe, A.; Zurzolo, C. Cell biology of prion protein. Prog. Mol. Biol. Transl. Sci. 2017, 150, 57–82. [Google Scholar] [PubMed]
- Puig, B.; Altmeppen, H.; Glatzel, M. The GPI-anchoring of PrP: Implications in sorting and pathogenesis. Prion 2014, 8, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Victoria, G.S.; Zurzolo, C. Trafficking and degradation pathways in pathogenic conversion of prions and prion-like proteins in neurodegenerative diseases. Virus Res. 2015, 207, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, N.A.; Aperia, A.; Melki, R.; Triller, A. Physico-pathologic mechanisms involved in neurodegeneration: Misfolded protein-plasma membrane interactions. Neuron 2017, 95, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Clavaguera, F.; Tolnay, M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosc. 2010, 33, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.; Zurzolo, C. The cell biology of prion-like spread of protein aggregates: Mechanisms and implication in neurodegeneration. Biochem. J. 2013, 452, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Yedidia, Y.; Horonchik, L.; Tzaban, S.; Yanai, A.; Taraboulos, A. Proteasomes and ubiquitin are involved in the turnover of the wild-type prion protein. EMBO J. 2001, 20, 5383–5391. [Google Scholar] [CrossRef] [PubMed]
- Anelli, T.; Sitia, R. Protein quality control in the early secretory pathway. EMBO J. 2008, 27, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Meusser, B.; Hirsch, C.; Jarosch, E.; Sommer, T. ERAD: The long road to destruction. Nat. Cell Biol. 2005, 7, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Vembar, S.S.; Brodsky, J.L. One step at a time: Endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008, 9, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, P.; Goder, V.; Rapoport, T.A. Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 2006, 126, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Zanusso, G.; Petersen, R.B.; Jin, T.; Jing, Y.; Kanoush, R.; Ferrari, S.; Gambetti, P.; Singh, N. Proteasomal degradation and N-terminal protease resistance of the codon 145 mutant prion protein. J. Biol. Chem. 1999, 274, 23396–23404. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Gu, Y.; Zanusso, G.; Sy, M.; Kumar, A.; Cohen, M.; Gambetti, P.; Singh, N. The chaperone protein BiP binds to a mutant protein and mediates its degradation by the proteasome. J. Biol. Chem. 2000, 275, 38699–38704. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, C.; Goold, R.; Andre, R.; Devoy, A.; Ortega, Z.; Moonga, J.; Linehan, J.M.; Brandner, S.; Lucas, J.J.; Collinge, J.; et al. Prion-mediated neurodegeneration is associated with early impairment of the ubiquitin-proteasome system. Acta Neuropathol. 2016, 131, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Lindquist, S. Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proc. Natl. Acad. Sci. USA 2001, 98, 14955–14960. [Google Scholar] [CrossRef] [PubMed]
- Pepe, A.; Avolio, R.; Matassa, D.S.; Esposito, F.; Nitsch, L.; Zurzolo, C.; Paladino, S.; Sarnataro, D. Regulation of subcompartmental targeting and folding properties of the prion-like protein Shadoo. Sci. Rep. 2017, 7, 3731. [Google Scholar] [CrossRef] [PubMed]
- Satpute-Krishnan, P.; Ajinkya, M.; Bhat, S.; Itakura, E.; Hegde, R.S.; Lippincott-Schwartz, J. ER stress-induced clearance of misfolded GPI-anchored proteins via the secretory pathway. Cell 2014, 158, 522–533. [Google Scholar] [CrossRef] [PubMed]
- Fujita, M.; Kinoshita, T. GPI-anchor remodeling: Potential functions of GPI-anchors in intracellular trafficking and membrane dynamics. Biochim. Biophys. Acta 2012, 1821, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- Ashok, A.; Hegde, R.S. Selective processing and metabolism of disease-causing mutant prion proteins. PLoS Pathog. 2009, 5, e1000479. [Google Scholar] [CrossRef] [PubMed]
- Campana, V.; Caputo, A.; Sarnataro, D.; Paladino, S.; Tivodar, S.; Zurzolo, C. Characterization of the properties and trafficking of an anchorless form of the prion protein. J. Biol. Chem. 2007, 282, 22747–22756. [Google Scholar] [CrossRef] [PubMed]
- Ashok, A.; Hegde, R.S. Retrotranslocation of prion proteins from the endoplasmic reticulum by preventing GPI signal transamidation. Mol. Biol. Cell 2008, 19, 3463–3476. [Google Scholar] [CrossRef] [PubMed]
- Sikorska, N.; Lemus, L.; Aguilera-Romero, A.; Manzano-Lopez, J.; Riezman, H.; Muñiz, M.; Goder, V. Limited ER quality control for GPI-anchored proteins. J. Cell Biol. 2016, 213, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Puig, B.; Altmeppen, H.C.; Ulbrich, S.; Linsenmeier, L.; Krasemann, S.; Chakroun, K.; Acevedo-Morantes, C.Y.; Wille, H.; Tatzelt, J.; Glatzel, M. Secretory pathway retention of mutant prion protein induces p38-MAPK activation and lethal disease in mice. Sci. Rep. 2016, 6, 24970. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Venditti, R.; Rega, L.R.; Colanzi, A.; D’Angelo, G.; de Matteis, M.A. The Golgi apparatus: An organelle with multiple complex functions. Biochem. J. 2011, 433, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Arvan, P.; Zhao, X.; Ramos-Castaneda, J.; Chang, A. Secretory pathway quality control operating in Golgi, plasmalemmal, and endosomal system. Traffic 2002, 3, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Paladino, S.; Lebreton, S.; Zurzolo, C. Trafficking and membrane organization of GPI-anchored proteins in health and diseases. Curr. Top. Membr. 2015, 75, 269–303. [Google Scholar] [PubMed]
- Uchiyama, K.; Muramatsu, N.; Yano, M.; Usui, T.; Miyata, H.; Sakaguchi, S. Prions disturb post-Golgi trafficking of membrane proteins. Nat. Commun. 2013, 4, 1846. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.S.; Hong, H.; Kim, C.; Mook-Jung, I. Acute ER stress regulates amyloid precursor protein processing through ubiquitin-dependent degradation. Sci. Rep. 2015, 5, 8805–8813. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Okuma, Y.; Nomura, Y. Molecular approaches to the treatment, prophylaxis, and diagnosis of Alzheimer’s disease: Possible involvement of HRD1, a novel molecule related to endoplasmic reticulum stress, in Alzheimer’s disease. J. Pharmacol. Sci. 2012, 118, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Koike, H.; Saito, R.; Kitamura, Y.; Okuma, Y.; Nomura, Y. Loss of HRD1-mediated protein degradation causes amyloid precursor protein accumulation and amyloid-β generation. J. Neurosci. 2010, 30, 3924–3932. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, H.A.; Rivera-Dictter, A.; Cavieres, V.A.; Munoz, V.C.; Gonzalez, A.; Yimo, L.; Mardones, G.A.; Burgos, P.V. Turnover of C99 is controlled by a crosstalk between ERAD and ubiquitin-independent lysosomal degradation in human neuroglioma cells. PLoS ONE 2013, 8, e83096. [Google Scholar] [CrossRef] [PubMed]
- Meier, S.; Bell, M.; Lyons, D.N.; Ingram, A.; Chen, J.; Gensel, J.C.; Zhu, H.; Nelson, P.T.; Abisambra, J.F. Identification of novel tau interactions with endoplasmic reticulum proteins in Alzheimer’s disease brain. J. Alzheimers Dis. 2015, 48, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Markesbery, W.R.; Chen, Q.; Li, F.; Keller, J.N. Ribosome dysfunction is an early event in Alzheimer’s disease. J. Neurosci. 2005, 25, 9171–9175. [Google Scholar] [CrossRef] [PubMed]
- Kundel, F.; De, S.; Flagmeier, P.; Horrocks, M.H.; Kjaergaard, M.; Shammas, S.L.; Jackson, S.E.; Dobson, C.M.; Klenerman, D. Hsp70 inhibits the nucleation and elongation of tau and sequesters tau aggregates with high affinity. ACS Chem. Biol. 2018, 13, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Flach, K.; Hilbrich, I.; Schiffmann, A.; Gärtner, U.; Krüger, M.; Leonhardt, M.; Waschipky, H.; Wick, L.; Arendt, T.; Holzer, M. Tau oligomers impair artificial membrane integrity and cellular viability. J. Biol. Chem. 2012, 287, 43223–43233. [Google Scholar] [CrossRef] [PubMed]
- Duyckaerts, C.; Delatour, B.; Potier, M.C. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009, 118, 5–36. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr71. [Google Scholar] [CrossRef] [PubMed]
- Klein, W.L. Synaptotoxic amyloid-β oligomers: A molecular basis for the cause, diagnosis, and treatment of Alzheimer’s disease? J. Alzheimers Dis. 2013, 33, S49–S65. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C.; Schelle, J.; Jucker, M. The prion-like properties of amyloid-β assemblies: The implication for Alzheimer’s disease. Cold Spring Harb. Perspect. Med. 2016, 6, a024398. [Google Scholar] [CrossRef] [PubMed]
- Mannini, B.; Mulvihill, E.; Sgromo, C.; Cascella, R.; Khodarahmi, R.; Ramazzotti, M.; Dobson, C.M.; Cecchi, C.; Chiti, F. Toxicity of protein oligomers is rationalized by a function combining size and surface hydrophobicity. ACS Chem. Biol. 2014, 9, 2309–2317. [Google Scholar] [CrossRef] [PubMed]
- Binger, K.I.; Ecroyd, H.; Yang, S.; Carver, J.; Howlett, G.J.; Griffin, M.D.W. Avoiding the oligomeric state: αβ-crystallin inhibits fragmentation and induces dissociation of apolipoprotein C-II amyloid fibrils. FASEB J. 2013, 27, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; del Tredici, K.; et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Eisele, Y.S.; Fritschi, S.K.; Hamaguchi, T.; Obermüller, U.; Füger, P.; Skodras, A.; Schäfer, C.; Odenthal, J.; Heikenwalder, M.; Staufenbiel, M.; Jucker, M. Multiple factors contribute to the peripheral induction of cerebral β-amyloiosis. J. Neurosci. 2014, 34, 10264–10273. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Di Fede, G.; Catania, M.; Morbin, M.; Rossi, G.; Suardi, S.; Mazzoleni, G.; Merlin, M.; Giovagnoli, A.R.; Prioni, S.; Erbetta, A.; et al. A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science 2009, 323, 1473–1477. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef] [PubMed]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Rudge, P.; Jaunmuktane, Z.; Adlard, P.; Bjurstrom, N.; Caine, D.; Lowe, J.; Norsworthy, P.; Hummerich, H.; Druyeh, R.; Wadsworth, J.D.; et al. Iatrogenic CJD due to pituitary-derived growth hormone with genetically determined incubation times of up to 40 years. Brain 2015, 138, 3386–3399. [Google Scholar] [CrossRef] [PubMed]
- Frontzek, K.; Lutz, M.I.; Aguzzi, A.; Kovacs, G.G.; Budka, H. Amyloid-β pathology and cerebral amyloid angiopathy are frequent in iatrogenic Creutzfeldt Jakob disease after dural grafting. Swiss Med. Wkly. 2016, 146, w14287. [Google Scholar] [CrossRef] [PubMed]
- Pradines, E.; Hernandez-Rapp, J.; Villa-Diaz, A.; Dakowski, C.; Ardila-Osorio, H.; Haik, S.; Schneider, B.; Launay, J.M.; Kellermann, O.; Torres, J.M.; et al. Pathogenic prions deviate PrPC signalling in neuronal cells and impair A-β clearance. Cell Death Dis. 2013, 4, e456. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2015, 17, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.X. Tau and neurodegenerative disease: The story so far. Nat. Rev. Neurol. 2016, 12, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.Y.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative Tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Eisenberg, D.S.; Crowther, R.A. Propagation of Tau Aggregates and Neurodegeneration. Annu. Rev. Neurosci. 2017, 40, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed]
- Strang, K.H.; Croft, C.L.; Sorrentino, Z.A.; Chakrabarty, P.; Golde, T.E.; Giasson, B.I. Distinct differences in prion-like seeding and aggregation between tau protein variants provide mechanistic insights into tauopathies. J. Biol. Chem. 2018, 293, 2408–2421. [Google Scholar] [CrossRef] [PubMed]
- Ayers, J.I.; Giasson, B.I.; Borchelt, D.R. Prion-like spreading in tauopathies. Biol. Psychiatry. 2018, 83, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Ollesch, J.; Wille, H.; Diamond, M.I. Conformational diversity of wild-type tau fibril specified by template conformation change. J. Biol. Chem. 2009, 284, 3546–3551. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Tolnay, M.; Clavaguera, F. Argyrophilic grain disease: A late-onset dementia with distinctive features among tauopathies. Neuropathology 2004, 24, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G. A century of Alzheimer’s disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the D2 concept. Ann. Rev. Bioph. 2008, 37, 215–246. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsic disorder, protein–protein interactions, and disease. Adv. Protein Chem. Struct. Biol. 2018, 110, 85–121. [Google Scholar] [PubMed]
- Uversky, A.V.; Uversky, V.N. Amino acid code for protein folding, misfolding, and non-folding. In Amino Acids, Peptides, and Proteins; Farkas & M. Ryadnov, RCS Publishing: London, UK, 2014; Volume 39, pp. 192–236. [Google Scholar]
- Dunker, A.K.; Silman, I.; Uversky, V.N.; Sussman, J.L. Function and structure of inherently disordered proteins. Curr. Opin. Struct. Biol. 2008, 18, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Pal, U.; Das, S.; Bagga, K.; Roy, A.; Mrigwani, A.; Maiti, N.C. Sequence complexity of amyloidogenic regions in intrinsically disordered human proteins. PLoS ONE 2014, 9, e89781. [Google Scholar] [CrossRef] [PubMed]
- Heske, J.; Heller, U.; Winklhofer, K.F.; Tatzelt, J. The C-terminal globular domain of the prion protein is necessary and sufficient for import into the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 5435–5443. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, N.V.; Dirndorfer, D.; Lang, S.; Resenberger, U.K.; Restelli, L.M.; Hemion, C.; Miesbauer, M.; Frank, S.; Neutzner, A.; Zimmermann, R.; et al. Structural features within the nascent chain regulate alternative targeting of secretory proteins to mitochondria. EMBO J. 2013, 32, 1036–1051. [Google Scholar] [CrossRef] [PubMed]
- Ciric, D.; Richard, C.A.; Moudjou, M.; Chapius, J.; Sibille, P.; Daude, N.; Westaway, D.; Adrover, M.; Beringue, V.; Martin, D.; et al. Interaction between Shadoo and PrP affects PrP-folding pathway. J. Virol. 2015, 89, 6287–6293. [Google Scholar] [CrossRef] [PubMed]
- Batlle, C.; de Groot, N.S.; Iglesias, V.; Navarro, S.; Ventura, S. Characterization of soft amyloid cores in human prion-like proteins. Sci. Rep. 2017, 7, 12134. [Google Scholar] [CrossRef] [PubMed]
- Melo, A.M.; Coraor, J.; Alpha-Cobb, G.; Elbaum-Garfinkle, S.; Nath, A.; Rhoades, E. A functional role for intrinsic disorder in the tau–tubulin complex. Proc. Natl. Acad. Sci. USA 2016, 113, 14336–14341. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Shala, A.; Bezginov, A.; Sljoka, A.; Audette, G.; Wilson, D.J. Hyperphosphorylation of intrinsically disordered tau protein induces an amyloidogenic shift in its conformational ensemble. PLoS ONE 2015, 10, e0120416. [Google Scholar] [CrossRef] [PubMed]
- Elbaum-Garfinkle, S.; Cobb, G.; Compton, J.T.; Li, X.H.; Rhoades, E. Tau mutants bind tubulin heterodimers with enhanced affinity. Proc. Natl. Acad. Sci. USA 2014, 111, 6311–6316. [Google Scholar] [CrossRef] [PubMed]
- Bunker, J.M.; Kamath, K.; Wilson, L.; Jordan, M.A.; Feinstein, S.C. FTDP-17 mutations compromise the ability of tau to regulate microtubule dynamics in cells. J. Biol. Chem. 2006, 281, 11856–11863. [Google Scholar] [CrossRef] [PubMed]
- Mandelkow, E.M.; Mandelkow, E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb. Perspect. Med. 2012, 2, a006247. [Google Scholar] [CrossRef] [PubMed]
- Abisambra, J.F.; Jinwal, U.K.; Blair, L.J.; O’Leary, J.C.; Li, Q.; Brady, S.; Wang, L.; Guidi, C.E.; Zhang, B.; Nordhues, B.A.; et al. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J. Neurosci. 2013, 33, 9498–9507. [Google Scholar] [CrossRef] [PubMed]
- Melki, R. Role of different alpha-synuclein strains in synucleinopathies, similarities with other neurodegenerative diseases. J. Parkinsons Dis. 2015, 5, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Clarke, A.R. A general model of prion strains and their pathogenicity. Science 2007, 318, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, S.K.; Sanders, D.W.; Thomas, T.L.; Ruchinskas, A.J.; Vaquer-Alicea, J.; Sharma, A.M.; Miller, T.M.; Diamond, M.I. Tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo. Neuron 2016, 92, 796–812. [Google Scholar] [CrossRef] [PubMed]
- Stöhr, J.; Condello, C.; Watts, J.C.; Bloch, L.; Oehler, A.; Nick, M.; DeArmond, S.J.; Giles, K.; DeGrado, W.F.; Prusiner, S.B. Distinct synthetic Aβ prion strains producing different amyloid deposits in bigenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10329–10334. [Google Scholar] [CrossRef] [PubMed]
- Abounit, S.; Wu, J.W.; Duff, K.; Victoria, G.S.; Zurzolo, C. Tunneling nanotubes: A possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion 2016, 10, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Suttkus, A.; Holzer, M.; Morawski, M.; Arendt, T. The neuronal extracellular matrix restricts distribution and internalization of aggregated Tau-protein. Neuroscience 2016, 313, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Morawski, M.; Brückner, G.; Jäger, C.; Seeger, G.; Arendt, T. Neurons associated with aggrecan-based perineuronal nets are protected against tau pathology in subcortical regions in Alzheimer’s disease. Neuroscience 2010, 169, 1347–1363. [Google Scholar] [CrossRef] [PubMed]
- Pujadas, L.; Rossi, D.; Andrés, R.; Teixeira, C.M.; Serra-Vidal, B.; Parcerisas, A.; Maldonado, R.; Giralt, E.; Carulla, N.; Soriano, E. Reelin delays amyloid-β fibril formation and rescues cognitive deficits in a model of Alzheimer’s disease. Nat. Commun. 2014, 5, 3443. [Google Scholar] [CrossRef] [PubMed]
- Horonchik, L.; Tzaban, S.; Ben-Zaken, O.; Yedidia, Y.; Rouvinski, A.; Papy-Garcia, D.; Barritault, D.; Vlodavsky, I.; Taraboulos, A. Heparan sulfate is a cellular receptor for purified infectious prions. J. Biol. Chem. 2005, 280, 17062–17067. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [PubMed]
- Kanekiyo, T.; Zhang, J.; Liu, Q.; Liu, C.C.; Zhang, L.; Bu, G. Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-beta uptake. J. Neurosci. 2011, 31, 1644–1651. [Google Scholar] [CrossRef] [PubMed]
- Rieger, R.; Lasmezas, C.I.; Weiss, S. Role of the 37 kDa laminin receptor precursor in the life cycle of prions. Transfus. Clin. Biol. 1999, 6, 7–16. [Google Scholar] [CrossRef]
- Gauczynski, S.; Peyrin, J.M.; Haïk, S.; Leucht, C.; Hundt, C.; Rieger, R.; Krasemann, S.; Deslys, J.P.; Dormont, D.; Lasmézas, C.I.; et al. The 37-kDa/67-kDa laminin receptor acts as the cell-surface receptor for the cellular prion protein. EMBO J. 2001, 20, 5863–5875. [Google Scholar] [CrossRef] [PubMed]
- Sarnataro, D.; Pepe, A.; Altamura, G.; De Simone, I.; Pesapane, A.; Nitsch, L.; Montuori, N.; Lavecchia, A.; Zurzolo, C. The 37/67kDa laminin receptor (LR) inhibitor, NSC47924, affects 37/67kDa LR cell surface localization and interaction with the cellular prion protein. Sci. Rep. 2016, 6, 24457. [Google Scholar] [CrossRef] [PubMed]
- Rangachari, V.; Dean, D.N.; Rana, P.; Vaidya, A.; Ghosh, P. Cause and consequences of Aβ–lipid interactions in Alzheimer disease pathogenesis. Biochim. Biophys. Acta 2018, in press. [Google Scholar] [CrossRef] [PubMed]
- Di Scala, C.; Chahinian, H.; Yahi, N.; Garmy, N.; Fantini, J. Interaction of Alzheimer’s β-amyloid peptides with cholesterol: Mechanistic insights into amyloid pore formation. Biochemistry 2014, 53, 4489–4502. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Ostaszewski, B.L.; Yang, T.; O’Malley, T.T.; Jin, M.; Yanagisawa, K.; Li, S.; Bartels, T.; Selkoe, D.J. Soluble Ab oligomers are rapidly sequestered from brain ISF in vivo and bind GM1 ganglioside on cellular membranes. Neuron 2014, 82, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.I.; Yi, J.S.; Ko, Y.G. Amyloid β oligomerization is induced by brain lipid rafts. J. Cell. Biochem. 2006, 99, 878–889. [Google Scholar] [CrossRef] [PubMed]
- Fabelo, N.; Martín, V.; Marín, R.; Moreno, D.; Ferrer, I.; Díaz, M. Altered lipid composition in cortical lipid rafts occurs at early stages of sporadic Alzheimer’s disease and facilitates APP/BACE1 interactions. Neurobiol. Aging 2014, 35, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Sarnataro, D.; Campana, V.; Zurzolo, C. Lipid rafts in trafficking and processing of prion protein and amyloid precursor protein. In Lipid Rafts and Caveolae: From Membrane Biophysics to Cell Biology; Fielding, C.J., Ed.; Cardiovascular Research Institute, Department of Medicine, University of California: San Francisco, CA, USA, 2006; pp. 205–231. ISBN 978-352731261-0. [Google Scholar]
- Hernandez, P.; Lee, G.; Sjoberg, M.; Maccioni, R.B. Tau phosphorylation by cdk5 and Fyn in response to amyloid peptide Aβ (25–35): Involvement of lipid rafts. J. Alzheimers Dis. 2009, 16, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.W.; Nicoll, A.J.; Zhang, D.; Mably, A.J.; O’Malley, T.; Purro, S.A.; Terry, C.; Collinge, J.; Walsh, D.M.; Rowan, M.J. mGlu5 receptors and cellular prion protein mediate amyloid-b-facilitated synaptic long-term depression in vivo. Nat. Commun. 2014, 5, 3374. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.; Vasefi, M.; Vander Tuin, C.; McQuaid, R.J.; Anisman, H.; Ferguson, S.S.G. Chronic pharmacological mglur5 inhibition prevents cognitive impairment and reduces pathogenesis in an Alzheimer disease mouse model. Cell Rep. 2016, 15, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Küffer, A.; Lakkaraju, A.K.K.; Mogha, A.; Petersen, S.C.; Airich, K.; Doucerain, C.; Marpakwar, R.; Bakirci, P.; Senatore, A.; Monnard, A.; et al. The prion protein is an agonistic ligand of the G protein coupled receptor Adgrg6. Nature 2016, 536, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Falsig, J. Prion propagation, toxicity and degradation. Nat. Neurosci. 2012, 15, 936–939. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Garmy, N.; Mahfoud, R.; Yahi, N. Lipid rafts: Structure, function and role in HIV, Alzheimers and prion diseases. Expert Rev. Mol. Med. 2002, 4, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Baron, G.S.; Caughey, B. Effect of glycosylphosphatidylinositol anchor-dependent and -independent prion protein association with model raft membranes on conversion to the protease-resistant isoform. J. Biol. Chem. 2003, 278, 14883–14892. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.B.M.; Mogk, A.; Bukau, B. Spatially organized aggregation of misfolded proteins as cellular stress defense strategy. J. Mol. Biol. 2015, 427, 1564–1574. [Google Scholar] [CrossRef] [PubMed]
- Mannini, B.; Chiti, F. Chaperones as suppressors of protein misfolded oligomer toxicity. Front. Mol. Neurosci. 2017, 10, 98. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarnataro, D. Attempt to Untangle the Prion-Like Misfolding Mechanism for Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 3081. https://doi.org/10.3390/ijms19103081
Sarnataro D. Attempt to Untangle the Prion-Like Misfolding Mechanism for Neurodegenerative Diseases. International Journal of Molecular Sciences. 2018; 19(10):3081. https://doi.org/10.3390/ijms19103081
Chicago/Turabian StyleSarnataro, Daniela. 2018. "Attempt to Untangle the Prion-Like Misfolding Mechanism for Neurodegenerative Diseases" International Journal of Molecular Sciences 19, no. 10: 3081. https://doi.org/10.3390/ijms19103081
APA StyleSarnataro, D. (2018). Attempt to Untangle the Prion-Like Misfolding Mechanism for Neurodegenerative Diseases. International Journal of Molecular Sciences, 19(10), 3081. https://doi.org/10.3390/ijms19103081