The Emerging Role of DNA Damage in the Pathogenesis of the C9orf72 Repeat Expansion in Amyotrophic Lateral Sclerosis

1
Centre for MND Research, Department of Biomedical Sciences, Faculty of Medicine & Health Sciences, Macquarie University, Sydney, NSW 2109, Australia
2
La Trobe Institute for Molecular Science, Melbourne, VIC 3086, Australia
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(10), 3137; https://doi.org/10.3390/ijms19103137
Submission received: 20 September 2018
/
Revised: 9 October 2018
/
Accepted: 9 October 2018
/
Published: 12 October 2018
(This article belongs to the Special Issue DNA Replication Stress)
Abstract
:Amyotrophic lateral sclerosis (ALS) is a fatal, rapidly progressing neurodegenerative disease affecting motor neurons, and frontotemporal dementia (FTD) is a behavioural disorder resulting in early-onset dementia. Hexanucleotide (G4C2) repeat expansions in the gene encoding chromosome 9 open reading frame 72 (C9orf72) are the major cause of familial forms of both ALS (~40%) and FTD (~20%) worldwide. The C9orf72 repeat expansion is known to form abnormal nuclei acid structures, such as hairpins, G-quadruplexes, and R-loops, which are increasingly associated with human diseases involving microsatellite repeats. These configurations form during normal cellular processes, but if they persist they also damage DNA, and hence are a serious threat to genome integrity. It is unclear how the repeat expansion in C9orf72 causes ALS, but recent evidence implicates DNA damage in neurodegeneration. This may arise from abnormal nucleic acid structures, the greatly expanded C9orf72 RNA, or by repeat-associated non-ATG (RAN) translation, which generates toxic dipeptide repeat proteins. In this review, we detail recent advances implicating DNA damage in C9orf72-ALS. Furthermore, we also discuss increasing evidence that targeting these aberrant C9orf72 confirmations may have therapeutic value for ALS, thus revealing new avenues for drug discovery for this disorder.
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Maintaining the stability and integrity of the genome is essential for normal cellular viability. Damage to DNA can arise from both endogenous and exogenous sources, and every cell receives numerous DNA injuries per day [1]. These injuries can generate mutations and compromise cellular viability, so safeguarding genetic integrity is of fundamental importance to human health [1]. DNA damage occurs in many forms. Single-stranded breaks (SSBs) involve a cut in the phosphodiester backbone of one DNA strand, whereas both DNA strands are severed in double-stranded breaks (DSBs). Although DSBs are much less common than SSBs, DSBs are difficult to repair and are the most cytotoxic lesion [2]. Alternatively, mismatch or modification of individual bases is another form of DNA damage [2].
Neurons are particularly vulnerable to DNA damage, because they are post-mitotic with high metabolic rates, and they are highly susceptible to oxidative stress, which is a major source of DNA damage [3]. Furthermore, SSBs are predicted to be more detrimental in post-mitotic neurons than in other cell types, because there are fewer options for repairing SSBs compared to proliferating cells. Hence, SSBs in neurons are more likely to be converted to highly cytotoxic DSBs than in other cell types [4]. In addition, DNA damage increases with advancing age, which is a major risk factor for neurodegenerative disorders [5].
DNA damage is now well-documented in neurodegenerative diseases, including ataxia-telangiectasia, Parkinson’s disease, and Alzheimer’s disease [6,7]. Dysfunctional DNA repair and DNA damage is also a growing area of interest in amyotrophic lateral sclerosis (ALS). The greatest proportion of familial cases of ALS are caused by a hexanucleotide repeat expansion in the gene encoding chromosome 9 open reading frame 72 (C9orf72). This review will focus on recent findings revealing a relationship between the formation of abnormal DNA structures, DNA damage, nucleolar stress, and C9orf72-ALS. These studies highlight the importance of genomic integrity in maintaining neuronal viability and they demonstrate a role for DNA damage in the pathogenesis of ALS.
2. Amyotrophic Lateral Sclerosis
ALS is a rapidly progressing and ultimately fatal neurodegenerative disorder affecting both upper motor neurons in the motor cortex and lower motor neurons in the brainstem and spinal cord. [8]. The clinical symptoms are varied, but involve progressive muscle weakness, spasticity, fasciculations, and eventually extensive paralysis [9], resulting in death from respiratory muscle failure usually within 2–5 years of diagnosis [10]. ALS is closely related to frontotemporal dementia (FTD), which affects the frontal lobes of the brain, and is characterised by behavioural changes in personality, emotion, and behaviour. FTD is diagnosed in approximately 20% of ALS cases [11], and an overlap between ALS and FTD exists at the clinical, genetic, and pathological levels. In fact, the discovery of the C9orf72 mutation in both ALS and FTD confirmed that these two disorders represent opposite extremes of the same, continuous clinical disease spectrum [12]. Approximately 10% of ALS cases are caused by dominantly inherited mutations (familial ALS), unlike most cases, which arise sporadically (sALS, 90% of cases). The aetiology of ALS/FTD remains unclear, and the disease is thought to involve both environmental and genetic components.
Hexanucleotide repeat expansions (GGGGCC) in a non-coding region of C9orf72 are the most common genetic abnormality in both ALS and FTD, which is responsible for approximately 40% of familial ALS, 5–10% of sporadic ALS, 40% of familial FTD, and 4–21% of sporadic FTD [13,14,15]. Mutations in the genes encoding the TAR-DNA binding protein (TDP-43) (TARDBP), fused in sarcoma (FUS) (FUS), TANK-binding kinase-1 (TBK-1), Ubiquilin 2 (UBQLN2), optineurin (OPTN), and Cyclin F (CCNF), are also present in both ALS and FTD patients [16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31]. In contrast, mutations in other genes are present in ALS only, including SOD1 and VAPB, encoding superoxide dismutase 1 and VAMP (vesicle-associated membrane protein)-associated protein B and C, respectively [32].
As in other neurodegenerative diseases, the pathological hallmark of ALS is the presence of misfolded protein inclusions in affected tissues [33]. In ALS patients, motor neurons contain these inclusions, and in familial forms of disease, the inclusions contain the specific proteins that are mutated in each case [34]. In sporadic ALS, these inclusions contain several different proteins, including wildtype, misfolded, TDP-43, which is also ubiquitinated, hyper-phosphorylated, and aberrantly mis-localised from the nucleus to the cytoplasm [35]. In fact, this pathological form of TDP-43 is present in motor neurons of almost all cases of ALS/FTD (97%) [36]. Similarly, TDP-43 pathology is present in 45% of FTD cases, implying that TDP-43 is a signature pathological lesion in both ALS and FTD [37]. TDP-43 is also strikingly similar to FUS in terms of its normal cellular functions and its pathological characteristics. Both TDP-43 and FUS are heterogenous nuclear riboproteins (hnRNPs) that perform multiple roles in RNA processing, including alternative splicing and regulation of transcription and translation [38]. The ALS mutations in FUS are primarily found in the nuclear localisation signal (NLS); like TDP-43, FUS mislocalises from the nucleus to the cytoplasm, where it forms stress granules and aggregates [39,40,41].
The accumulation of misfolded proteins in ALS implies that dysfunctional protein homeostasis (proteostasis) mechanisms are central to pathogenesis, and several of these processes are implicated in neurodegeneration, including defects in protein degradation (autophagy and the proteasome), protein trafficking (particularly nucleo-cytoplasmic transport), and protein folding. Similarly, several of the genes mutated in ALS encode proteins that function in proteostasis. However, the growing abundance of RNA binding proteins linked to ALS/FTD has also revealed a role for abnormal RNA metabolism in pathophysiology [42]. Whilst these two mechanisms are often highlighted as being central to ALS, this rather simplistic division does not fully capture the complexity of the range of functions performed by the proteins associated with ALS, nor the cellular signalling pathways that are known to be dysfunctional in this disorder. Recently, DNA damage has been linked to ALS [40,43,44,45,46,47]. Interestingly, many of the signalling pathways associated with DNA damage are also implicated in ALS, including oxidative stress, mitochondrial function, RNA metabolism, autophagy, and proteosomal function [48,49,50,51,52].
3. DNA Damage Signalling
Cells have developed elaborate signalling systems to detect and repair damage to DNA, termed the “DNA damage response” (DDR). Many normal physiological events induce DNA damage, particularly transcription and mitochondrial respiration, which generates reactive oxygen species [53]. It is therefore essential that the cell normally maintains genomic stability. Depending on the extent of damage and risk of mutation, the cell induces DNA repair pathways, or following chronic activation of DDR, induces apoptosis to protect the organism. Various sensor proteins detect DNA damage, and the most widely used sensor experimentally is the phosphorylated histone variant H2AX (γH2AX). The formation of SSBs or DSBs activates phosphorylation of H2AX, hence γH2AX flanks sites of DNA damage. This initiates recruitment of the ataxia-telangiectasia mutated (ATM) and Rad3-related (ATR) protein kinases, which trigger the DDR. Poly (ADP-ribose) polymerase (PARP) and p53 binding protein (53-BP1) are other important sensors that signal DNA damage during DDR. Depending on the type of DNA damage, the DDR can activate several different DNA repair pathways. SSBs are repaired primarily by excision repair mechanisms, whereas DSBs are repaired by either homologous recombination (HR) or non-homologous end-joining (NHEJ). However, in neurons, NHEJ is the primary mechanism, because HR requires active progression through the cell cycle [54,55]
The nucleolus is a prominent cellular compartment located within the nucleus, and it is implicated in the DDR because it contains over 160 DNA repair proteins [56]. However, it is unclear whether the nucleolus is simply a storage facility for DDR proteins, or if these proteins have specific roles in DNA repair in the nucleolus. The nucleolus is also responsible for the biogenesis of ribosomes and the regulation of cellular stress responses.
4. DNA Damage and Neurodegeneration
Nucleotide repeat elements are common in the eukaryotic genome. Microsatellite (short-repeat) expansions are responsible for almost 40 different diseases, including many neurological disorders [57,58]. These repeat expansions lead to instability of the repeat and the formation of abnormal DNA structures. Interestingly, mutations in DNA repair genes lead to disorders that produce neurological phenotypes: xeroderma pigmentosum and ataxia-telangiectasia [59]. Furthermore, defects in DNA repair can be manifested primarily in neural tissues, leading to neurological conditions [60]. It is therefore not surprising that DNA damage has also been detected in neurodegenerative disorders, including Alzheimer’s, Parkinson’s, and Huntington’s diseases [6,7], as well as ALS. These findings therefore imply a close relationship between DNA damage/repair and neuronal function.
Most previous historical studies on DNA damage in ALS were examined in relation to oxidative stress, which occurs in mitochondrial rather than nuclear DNA; they also preceded the discovery of the relationship between TDP-43, C9orf72, and ALS. DNA damage was present in sporadic ALS patients in regions of the CNS that contain motor neurons, but not in other regions [61]. Apoptosis in spinal motor neurons follows DNA damage [62] and DNA repair enzymes are up-regulated in the brain, indicating increased DNA damage [63]. Similarly, in ALS mouse models based on transgenic expression of mutant SOD1, motor neuron degeneration was associated with DNA damage [64], although these animals do not possess the TDP-43 pathology present in almost all ALS cases. DNA damage has also been detected in neuronal cells expressing G93A mutant SOD1 [65]. DNA repair activity detected by 8-hydroxy-2-deoxyguanosine (OHdG) immunoreactivity is also increased in the motor cortex of sALS patients [66], and elevated levels of 8-OHdG were also identified in familial ALS spinal cords bearing SOD1 mutations [66]. Apurinic/apyrimidinic endonuclease 1 (APE1) is elevated in the brain and spinal cord of sporadic ALS patients [45]. Activation of cellular DNA repair processes has been previously implicated in motor neuron degeneration [61,67], and mice lacking the gene encoding ERCC1, which is essential for SSB nucleotide excision repair and repair of DSBs, show age-related motor dysfunction [68].
Further evidence linking DNA damage to ALS is the increasing number of proteins mutated in this disorder that possess normal cellular functions in DNA repair, particularly FUS. DNA damage is present in transgenic mice expressing ALS-associated mutant FUS-R521C in cortical neurons and spinal motor neurons [69]. DSBs trigger FUS phosphorylation by ATM and DNA-dependent protein kinase (DNA-PK), both of which are involved in the DDR [70,71]. FUS binds to histone deacetylase 1 (HDAC1), and therefore indirectly regulates HR and NHEJ in primary mouse neurons [72]. ALS-associated FUS mutations in the nuclear localization sequence NLS cause impartment of PARP-dependent DDR, which leads to neurodegeneration and the formation of FUS aggregates [40]. FUS mutations are also responsible for defects in DNA nick ligation and oxidative damage repair in ALS patients [43]. Similarly, increased expression of γH2AX has been found in ALS patients carrying FUS-R521C or FUS-P525L mutations [72]. Moreover, FUS and TDP-43 function in the prevention or repair of transcription-associated DNA damage [73]. These findings therefore indicate that FUS is a DDR protein that functions in DNA repair, whereas in ALS, DNA repair is defective. However, despite the marked functional and pathological similarities between TDP-43 and FUS, a convincing role for TDP-43 in DNA repair has not yet been demonstrated.
Similarly, senataxin is a helicase that can resolve R-loops [74], and which is mutated in juvenile forms of ALS [75]. Mutations in other DNA damage/repair proteins are also present in more typical forms of ALS, such as valosin-containing protein (VCP) [76] and cyclin F [31]. VCP is implicated in the repair of DSBs [77], and cyclin F controls genome stability through ubiquitin-mediated proteolysis [78]. NIMA-related kinase 1 (NEK1), which is mutated in familial and sporadic ALS [22,79], is necessary for cellular checkpoint control in the DDR, independent of ATM or ATR [80], where it functions in replication fork stability in the completion of HR [81]. ALS-associated NEK1 mutations were shown to induce DNA damage in induced pluripotent stem cells (iPSC)-derived motor neurons [44]. C21ORF2 interacts with NEK1 and functions in HR, but not in NHEJ-mediated DSB repair, and it is also mutated in sporadic ALS [82].
5. Chromosome 9 Open Reading Frame 72 and Amyotrophic Lateral Sclerosis
Hexanucleotide repeat expansions in C9orf72 are central to both ALS and FTD. Whilst the normal population bears fewer than eight GGGGCC repeats, and 50% of these individuals possesses only two repeats [83], in ALS and FTD this region is expanded up to several thousand times [84]. C9orf72 uses alternative splicing to produce at least three different transcript variants. V2 and V3 encode a long isoform, whereas V1 encodes a short isoform. The repeat expansion is located either in intron 1 for transcripts V1 and V3 or within the promoter sequence for V2 [13]. Despite a common genetic cause, however, C9orf72 repeat expansion carriers exhibit remarkably heterogeneous clinical and pathological characteristics. There also appears to be no unambiguous clinical correlation between the length of the repeat and disease onset or progression [85]. Interestingly, genetic analysis of the C9orf72 repeat expansion has identified a common haplotype, but the lengths of the repeat vary among carriers. This implies that the repeats are either unstable and result from a single founder [86], or alternatively, that the repeat sequence is inherently prone to instability and results from different founders [87]. The C9orf72 repeat expansion is linked to other neurological conditions including Alzheimer’s disease [88], multiple system atrophy [89], Huntington’s disease [90], cerebellar ataxia [91], multiple sclerosis [92], Parkinson’s disease [93], bipolar disorder [94,95], and schizophrenia [96].
The mechanism of how the C9orf72 repeat expansion induces motor neuron death is unclear, but this may reflect the intronic nature of the repeat expansion. Three major mechanisms have been proposed, and their relative contributions to pathogenicity are consistently debated. Haploinsufficiency was initially proposed, given that C9orf72 carriers express reduced levels of the C9orf72 transcript compared to individuals without the repeat expansion [97]. However, mice with C9orf72 deficiency, or those expressing loss-of-function mutations, develop immune defects, increased expression of inflammatory cytokines, and autoimmunity [98,99], but no neurodegenerative phenotype, arguing against haploinsufficiency as a single causative factor in ALS/FTD. However, these findings may be reflective of the strong expression of C9orf72 in myeloid cells. Furthermore, a recent study concluded that both loss- and gain-of-function mechanisms cooperate and lead to neurodegeneration in ALS/FTD by a process involving the impairment of vesicle trafficking [100]. In contrast, there is more evidence in favour of gain of a toxic function in C9orf72-ALS/FTD. One possible mechanism is the induction of toxicity by transcription of the C9orf72 repeat expansion, producing greatly expanded RNA, which forms predominately nuclear RNA foci in affected tissues [101]. These RNA foci are thought to sequester important RNA-binding proteins (RBPs), such as those involved in alternative splicing, leading to impairment of RNA processing [102,103,104]. Furthermore, many studies report widespread transcriptome changes in ALS carrying the C9orf72 repeat expansion [102,105,106,107,108]. One report also highlighted the splicing factor hnRNP H as a major C9orf72 binding protein, which was linked to the formation of abnormal nucleic acid structures [109].
Another possible process associated with a gain of toxic function of the C9orf72 repeat expansion is repeat-associated non-ATG dependent (RAN) translation [101], whereby expanded repeat sequences are translated in the absence of an ATG initiation codon. RAN translation has now been described for several non-coding repeat expansions, including C9orf72-ALS/FTD [110]. Recent studies have revealed that translation of the C9orf72 repeat is initiated from a CUG codon upstream from the repeat sequence, which is induced in response to stress stimuli and depends on phosphorylation of the α-subunit of eukaryotic initiation factor-2 (eIF2α) [111,112,113]. In ALS/FTD, RAN translation produces dipeptide repeat proteins (DRPs), which result from translation on the both sense and antisense strands. This results in expression of five DPRs: poly GA, poly GR, poly PA, poly PR, and poly GP (which is produced on both sense and anti-sense strands). The biochemical properties of each DPR are quite distinct, and the arginine-containing peptides (poly GR and poly PR) appear to be the most toxic, at least in disease models [114,115]. Furthermore, these peptides display features associated with neurodegeneration, including liquid–liquid phase separation, perturbation of nucleocytoplasmic transport, and stress granule formation [116,117].
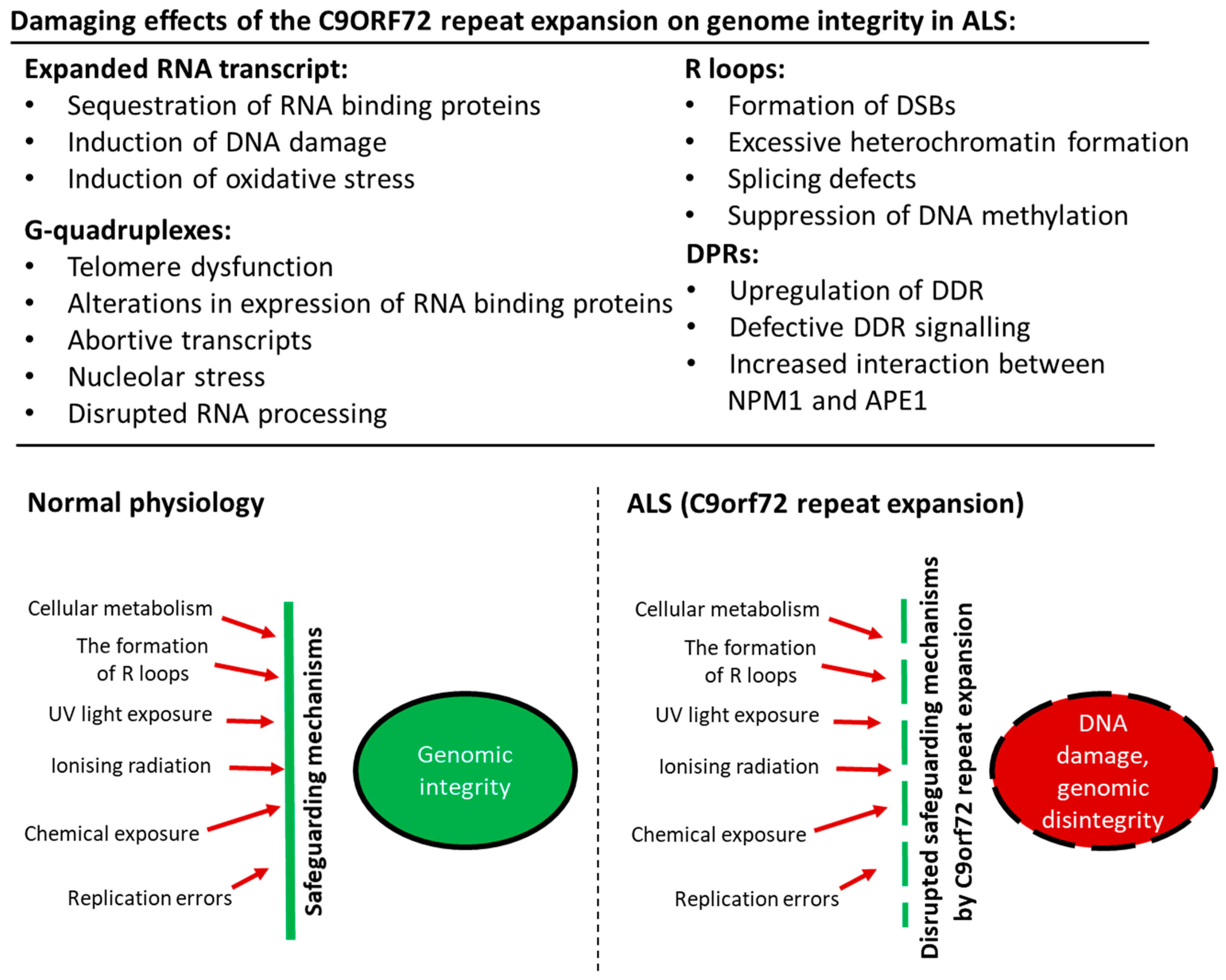
The cause of the selective neurodegeneration of motor neurons in ALS associated with C9orf72 repeat expansions is unknown. However, there are several hypotheses, based on the unique characteristics of motor neurons. As explained above, neurons themselves are particularly susceptible to DNA damage. However, in addition, motor neurons are extremely large cells with high levels of cellular respiration, and thus they are especially prone to oxidative stress. This may render them particularly susceptible to DNA damage, even compared to other types of neurons. There are also other possibilities to explain the selective neurodegeneration of motor neurons in ALS. It has been shown that the excitabilities of corticospinal tract pathways are abnormally increased in ALS, especially in the early stages of disease [118], and an imbalance in excitatory to inhibitory synaptic input precedes motor neuron degeneration in animal models [119]. Interestingly, it has also been demonstrated that physiological neuronal activity causes DNA damage [120,121]. Whilst this may be effectively repaired in normal physiological conditions, the presence of the C9orf72 repeat expansion may disrupt the natural cellular safeguarding mechanisms, thus contributing to neurodegeneration in ALS.
6. Abnormal Nucleotide Structures: R-Loops, G-Quadruplexes, and Hairpins
Nucleic acids are structurally polymorphic. Whilst the double-stranded, right-handed helix is the regular conformation employed by both DNA and RNA, non-canonical alternative structures, such as hairpins, branched junctions, and quadruplexes, also exist [122]. Normal cellular processes leading to transient separation of nucleic acid strands, such as DNA replication, recombination, repair and transcription, can lead to instability in their sequences. Not all non-canonical conformations are stable under physiological conditions, but increasing evidence links the formation of these structures with different biological functions and pathological conditions. Importantly, aberrant nucleic acid structures are increasingly acknowledged to be an important contributor to human disease [123,124]. They are also major sources of DNA damage and are thus recognised to be a serious threat to genomic integrity [125]. The ability of nucleic acids to form unusual secondary structures is also related to the instability of repeat sequences [126]. Below we discuss two important nucleic acid structures that are formed by the C9orf72 repeat sequence.
G-quadruplex structures are formed in nucleic acids by G-rich sequences, which is not surprising because G-rich DNA is prone to forming stable secondary structures. G-quadruplexes contain G tetrad structures that are stacked on top of one another. G tetrads consist of four guanine bases Hoogsteen hydrogen-bonded to each other and a cation. They possess normal physiological roles, such as in immunoglobulin heavy chain switching [127], and they are often found at important positions in the genome, such as telomeres [128]. However, aberrant, detrimental roles have also been described in disease. G-quadruplexes are gaining increasing interest because of their involvement in signalling pathways that are relevant to cancer and neurodegeneration. In neurological disorders, G-quadruplexes have been implicated in pathogenesis through two main mechanisms. The first is by expansions of G-repeats, which lead to the formation of G-quadruplexes that induce toxicity, such as in C9orf72-ALS. The second mechanism is through mutations that affect the expression of G-quadruplex binding proteins, as in the fragile X mental retardation 1 (FMR1) gene and Fragile X syndrome [129].
R-loops are naturally occurring hybrids between DNA and RNA. They form when an RNA strand displaces a strand of the original DNA double helix, because the stability of the RNA–DNA interaction is greater than DNA–DNA interactions. Hence, the resulting R-loops can be extremely stable. The formation of R loops often occurs in G-rich sequences, again reflecting the propensity of single-stranded G-rich sequences to form stable secondary structures. R-loops occur normally during many cellular processes, including DNA replication, transcription (including reverse transcription), and telomere function, but in these situations, they normally form transiently and do not persist [130,131]. However, the persistence of R-loops can have deleterious effects, resulting in genome instability and DNA damage. This is mediated by at least two distinct mechanisms. Firstly, ssDNA that is exposed via an R-loop is chemically labile, and hence more prone to damage. Secondly, R-loops can block replication fork progression, leading to replication stress and error-prone repair mechanisms [130]. R loops can also lead to reduced protein expression by transcriptional stalling, or by negatively regulating RNA polymerases, thus inhibiting transcription [132]. Furthermore, R-loops can mediate other mechanisms of transcriptional repression, such as the methylation of histones [133,134]. R loops are therefore closely associated with RNA metabolism, which is implicated as a major pathogenic mechanism in ALS [135]. Therefore, it is not surprising that R-loops are linked to various diseases, including multiple cancers and neurodegenerative disorders. Normal cellular mechanisms exist to prevent the formation of R-loops, including senataxin [74]. Interestingly, SETX, the gene encoding senataxin, is mutated in juvenile ALS [58], as is ataxin with oculomotor apraxia type 2 (AOA2) [136].
7. The Chromosome 9 Open Reading Frame 72 Repeat Expansion Induces DNA Damage
The properties of the G-rich GGGGCC repeat render the C9orf72 repeat expansion highly favorable for forming abnormal DNA structures, such as G-quadruplexes and R-loops [137,138,139]. Circular dichroism (CD) and nuclear magnetic resonance spectroscopy (NMR) studies [140,141] have revealed that the C9orf72 repeat expansion forms a heterogenous mixture of G quadruplex conformations, involving both parallel and anti-parallel G structures. Importantly, this interferes with the function of RNA polymerase at repeat sites, leading to abortive transcripts and less full-length transcripts [140]. Treatment with RNase A and RNase H to remove RNA alters the mobility of in vitro transcription products, also providing evidence for the presence of R-loops [140]. In addition, more R-loops were detected by immunohistochemistry in spinal cord motor neurons from C9orf72 ALS patients, compared to controls [46]. Similarly, expression of DPRs in cell culture resulted in the production of more R-loops by immunocytochemistry, which could be reduced by expression of senataxin [46]. R-loops are often found at cytosine–phosphate–guanine (CpG) islands and are proposed to suppress DNA methylation [142]. Interestingly, there are two CpG islands flanking the C9orf72 repeat expansion that are differentially methylated [143].
The formation of G-quadruplexes and R-loops by the C9orf72 repeat expansion implies that these structures could damage DNA. Consistent with this notion, elevated levels of DNA damage markers γH2AX, ATR, GADD45, and p53 were present in motor neurons differentiated from iPSC lines from C9orf72 ALS patients in response to oxidative stress, which could be reduced by pharmacological or genetic reduction of oxidative stress [144]. Similarly, we demonstrated that markers of the DDR, including γH2AX, phosphorylated-ATM, cleaved PARP-1, and 53-BP1, were up-regulated in C9orf72 ALS patient spinal cord motor neurons [47]. This was confirmed using constructs expressing poly (GR)100 and poly (PR)100, but not the native GGGGCC RNA, revealing that DNA damage is activated by the DPRs produced by RAN translation of the C9orf72 repeat expansion in ALS. A subsequent study also found that the DPRs induce DNA damage, and that in addition, the C9orf72 RNA is capable of inducing damage [30]. Expression of the C9orf72 DPRs resulted in suppression of the recruitment of 53BP1 to DSBs. This led to defective ATM signalling and hence DNA repair, which appeared to be driven by the accumulation of p62, and subsequently, defective H2A ubiquitylation. However, a second mechanism was also implicated; the persistent accumulation of R-loops resulting in the formation of DSBs, increased heterochromatin, and splicing defects [46].
8. The Nucleolus and the Chromosome 9 Open Reading Frame 72 Repeat Expansion
The main function of the nucleolus is the rapid production of ribosomal subunits, a process that must be highly regulated to achieve proper cellular proliferation and cell growth [145]. This involves three main events: pre-rRNA transcription, processing, and ribosomal RNP assembly. These functions are concentrated in three distinct sub-nucleolar compartments, the fibrillar center (FC), the dense fibrillar component (DFC), and the granular component (GC). The varied effects on ribosome subunit production and cell growth induced by cellular stress are often accompanied by dramatic changes in the organization and composition of the nucleolus, and the nucleolus is recognised to be a central hub in cellular stress responses. During DNA damage, the nucleolus segregates, resulting in condensation and separation of FC and GC, as well as the formation of ‘‘nucleolar caps’’ around the nucleolar remnant (also called central body) [146].
Dysfunction in the nucleolus is now implicated as an important mechanism related to toxicity of the C9orf72 repeat expansion. The C9orf72 repeat RNA binds nucleolar proteins in vitro [104,140]. Over-expression of poly GR or poly PR repeats in cell culture leads to their localisation in the nucleolus, resulting in abnormal nucleoli, altered ribosomal RNA processing, nucleolar stress, and cell death [140,147,148]. Additionally, in yeast several nucleolar proteins modify poly PR toxicity [116]. Furthermore, nucleolin, an important nucleolar protein involved in the synthesis and maturation of ribosomes, binds specifically to G-quadruplexes formed by the C9orf72 repeat expansion [140]. Whilst the C9orf72 DPRs do not localise to the nucleolus in C9orf72-ALS brains, their neuronal nucleoli display abnormalities [149]. Disrupted nucleocytoplasmic transport is emerging as a central pathogenic mechanism in ALS that is also closely associated with nucleolar stress. The C9orf72 DPRs inhibit nuclear import and export, and enhancement of nuclear import or suppression of nuclear export, suppressed neurodegeneration in yeast and Drosophila [116,150].
Nucleophosmin (NPM1, also known as B23) is a nucleolar-localised DDR protein that regulates nucleolar function and contributes to genomic integrity and stability [151,152]. During DNA damage, NPM1 localises to DSBs, where it mediates the stability, activity, and accumulation of proteins involved in base excision DNA repair (BER) [153]. BER corrects small base lesions, typically resulting from deamination, oxidation, or methylation, which do not significantly distort the DNA helix structure. NPM1 also interacts with APE1, which is central to BER [154]. The NPM1-APE1 interaction regulates multiple cellular functions, including genomic stability and ribosome biogenesis [155]. APE1 is also a growth factor, which is protective against apoptosis induced by DNA damage [156]. Under normal conditions, NPM1 enhances the activity of APE1, thus enhancing BER [157,158,159]. However, during nucleolar stress, NPM1 inhibits the activity of APE1, leading to impairment of BER [157,159,160]. Up-regulation of APE1 was previously reported in sporadic ALS patients [66], and missense mutations in APE1 were found in sporadic and familial SOD1-ALS patients [67]. NPM1 co-localises with both poly GR and poly PR [148]. In C9orf72 ALS patients, we showed that the interaction between NPM1 and APE1 was enhanced compared to control subjects, which may impair the function of both, and in turn disturb RNA processing [47].
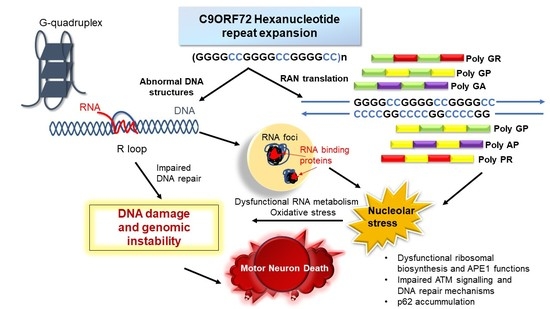
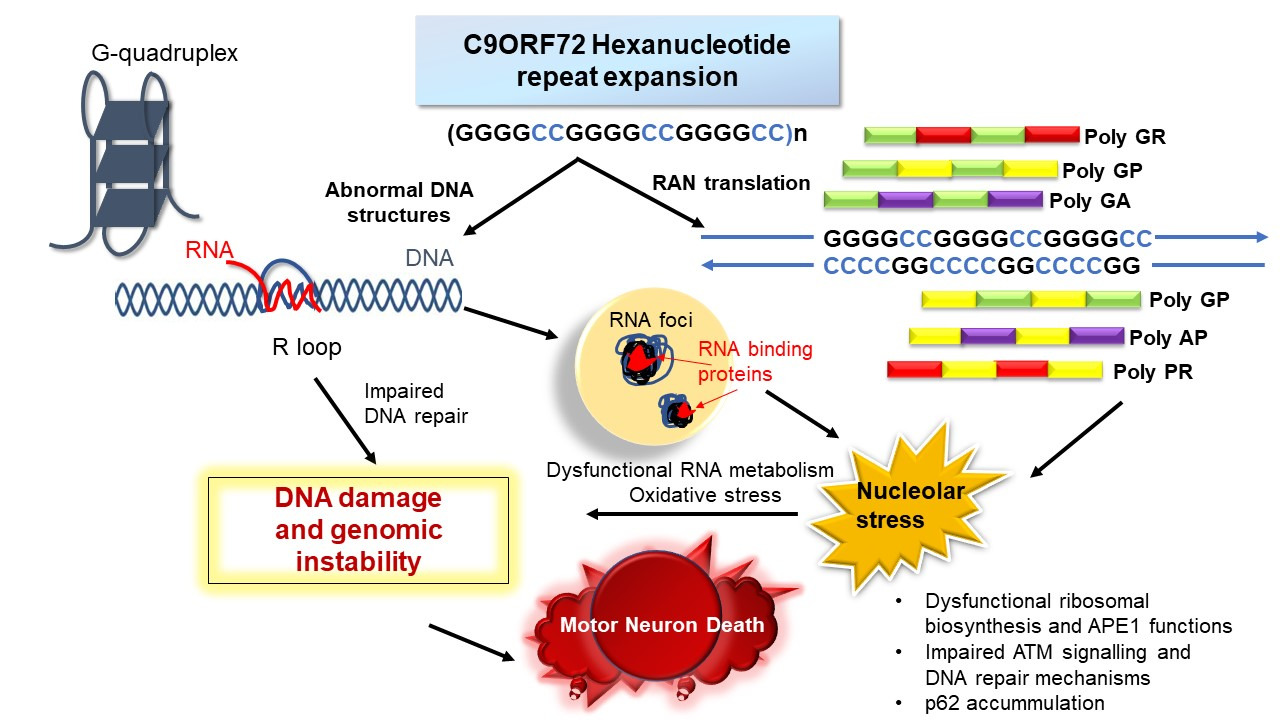
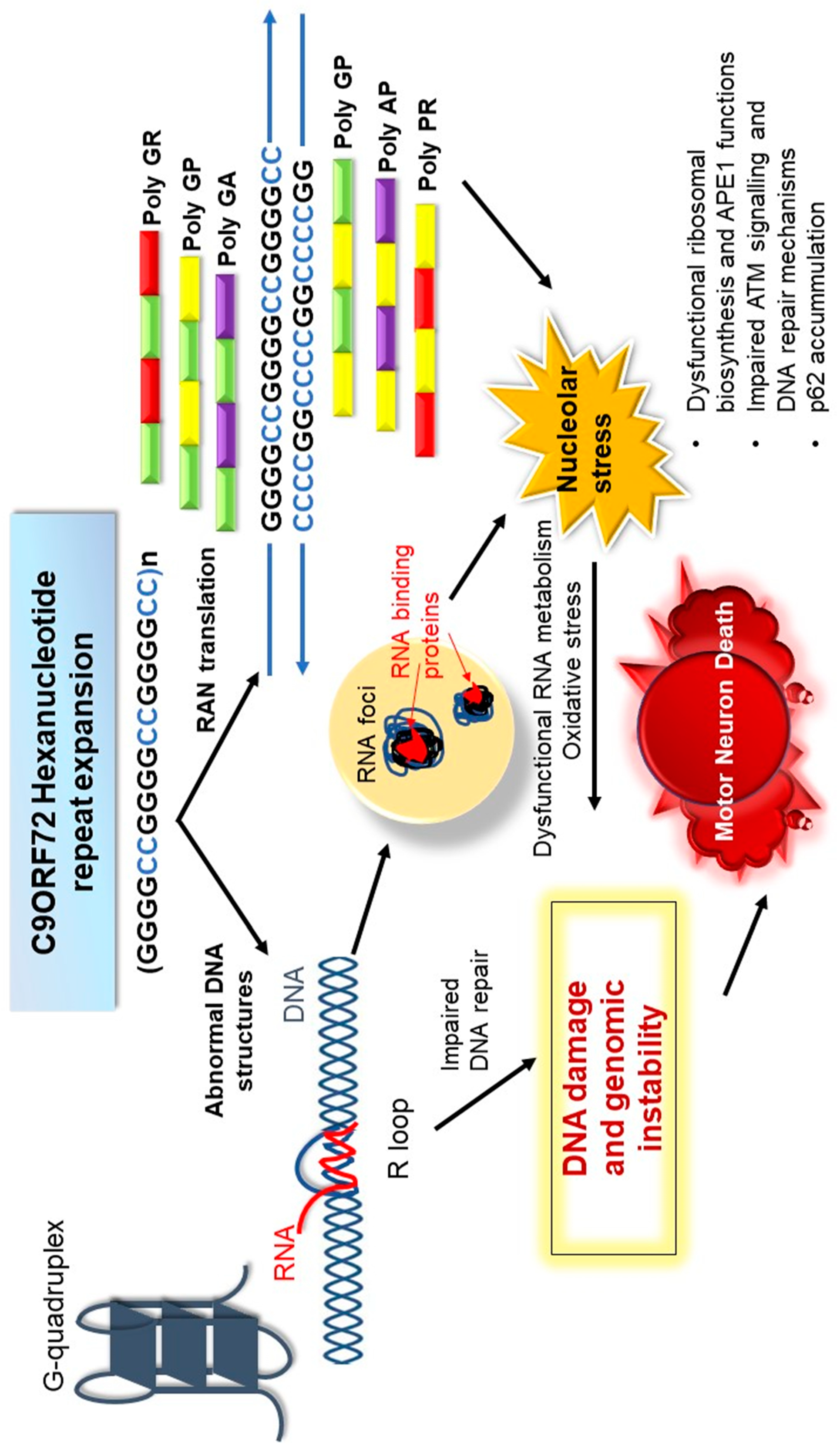
Figure 1 summarises possible mechanisms by which genomic integrity is disrupted by the C9orf72 repeat expansion in ALS, as discussed in the sections above.
9. Novel Therapeutic Strategies for Amyotrophic Lateral Sclerosis Based on the Inhibition of DNA Damage and Abnormal DNA Structures.
The increasing evidence that both the repeat RNA and DPRs contribute to toxicity implies that therapeutic strategies based on targeting both factors may be effective in ALS. In addition, there is evidence that the C9orf72 repeat expansion is upstream of the TDP-43 pathology which is present in almost all ALS cases [161,162], further implying that targeting this region could be an effective strategy in ALS. Abnormal nucleic acid structures, such as G-quadruplexes and R-loops, are increasingly recognised as promising drug targets, and several small molecules are being developed to target these sites.
There are several lines of evidence implying that the targeting the G-quadruplexes formed by the C9orf72 repeat expansion could be an effective therapeutic strategy for C9orf72-ALS and FTD. Targeting the hairpin conformation of the C9orf72 repeat expansion with small chemical lead compounds was protective against the formation of RNA foci and RAN translation [163]. Recently, three small molecules were isolated from chemical libraries that bind and stabilise the C9orf72 G-quadruplex structure [164]. Moreover, these compounds reduced the formation of both C9orf72 RNA foci and DPRs in iPSC-derived motor neurons, and they improved survival in fly models expressing poly GR [164]. A porphyrin compound, TMPyP4, also stabilized C9orf72 G-quadruplexes, reduced the affinity of RanGAP1, a key regulator of nucleocytoplasmic transport, and suppressed nuclear import deficits in fly models [150].
Recent studies indicating that DNA repair pathways are dysregulated by the C9orf72 repeat expansion implies that modulation of the DDR or enhancement of DNA repair processing are also promising therapeutic strategies for ALS. Indeed, there are already many compounds in development that modulate the DDR for cancer therapy. Hence, similar approaches could be examined for ALS and other neurodegenerative disorders. An important tumour suppressor, p53, protects the genome by regulating a variety of DDR mechanisms and controls the induction of apoptosis in genomically compromised cells. Not surprisingly, a broad range of approaches for modulating or inhibiting p53 activity are already underway in studies of cancer therapy [165,166,167]. Interestingly, partial inhibition of the p53 pathway partially suppressed poly GR80 induced toxicity in iPSC-derived patient motor neurons [144]. Another approach is based on the impairment of ATM activation and the suppression of 53BP1 recruitment to DSBs [46] by the C9orf72 repeat expansion. This implies that inhibition of negative regulators of DNA repair, such as the PI3K/AKT mammalian target of rapamycin (mTOR) pathway, which negatively controls ATM [168], may be beneficial for ALS. Consistent with this notion, we also demonstrated the down-regulation of PI3K and p-eIF4G in C9orf72 patient tissues compared to controls [47], in concordance with previous studies demonstrating dysregulation of the AKT/PI3K pathway in ALS motor neurons [169,170,171]. Alternatively, D52 is another recently recognised negative regulator of ATM signalling [172]. Similarly, nucleolar stress also triggers down-regulation of the mTOR pathway [173], and the evidence that the C9orf72 repeat expansion induces nucleolar stress implies that targeting this mechanism could be an important therapeutic strategy in ALS. We also previously showed that overexpression of NPM1 inhibited apoptosis in neuronal cells expressing poly (PR)100 or poly (GR)100, suggesting that depletion of NPM1 is linked to cell death in ALS [47]. These findings suggest that inhibition of nucleolar stress should be investigated in more detail in relation to C9orf72-ALS.
Ultimately however, a potentially more effective way to reduce toxicity of the C9orf72 repeat expansion is by inhibiting its production in the first place. Antisense oligonucleotides (ASOs) are short, single-stranded oligonucleotides that can be designed to hybridize to specific RNAs and thus modulate gene expression [174]. Importantly, ASOs targeting the C9orf72 repeat expansion are currently showing promise for ALS. ASO treatment targeting poly GP reduced both repeat-containing RNA foci and poly GP concentrations in C9orf72 ALS iPSC-derived neurons, although poly GP is particularly stable and required 10 days of ASO treatment to be significantly reduced [175]. In ASO-treated mice, concentrations of C9orf72 repeat-containing RNA were reduced approximately 50%, without affecting endogenous C9orf72 mRNA. Similarly, poly GP concentrations in cerebrospinal fluid (CSF) and the brain, as well as RNA foci and poly GP-containing inclusions, were reduced significantly in the motor cortex of the mouse model of C9orf72 ALS [175]. Furthermore, ASOs targeting C9orf72 RNA prevented nuclear import impairment by the C9orf72 repeat expansion in fly models, as well as in C9orf72 iPSC-derived neurons, and suppressed neurodegeneration [150]. In addition, ASOs selectively reduced the accumulation of C9orf72 GGGGCC sense strand-containing RNA foci, without significantly affecting the level of RNAs encoding C9orf72 itself. Similarly, ASOs targeting the C9orf72 transcript suppressed GGGGCC repeat-containing RNA foci formation, and reversed membrane excitability defects in C9orf72-ALS motor neurons differentiated from iPSCs [106]. Hence these studies reveal that ASOs are capable of reducing pathogenic, expansion-containing RNAs without inducing C9orf72 protein loss [176].
10. Conclusions
There is now convincing evidence that diseases resulting from the expansion of abnormal repeat sequences are nucleic acid diseases, and in C9orf72-ALS, DNA damage and loss of genome integrity are implicated in pathophysiology. In Figure 2, we illustrate the possible mechanisms by which the C9orf72 mutation induces toxicity and genomic instability in ALS. The production of aberrant, toxic nucleic acid conformations formed by the C9orf72 hexanucleotide (GGGGCC) repeat expansion (particularly R-loops) induces DNA damage. This results from impairment of DDR signalling and dysfunction in RNA processing, which compromises genetic integrity and triggers neurodegeneration. Furthermore, the DPRs produced by RAN translation of the C9orf72 repeat expansion, particularly the arginine-containing peptides poly GR and poly PR, are also capable of triggering DNA damage by inducing stress in the nucleolus. They also perturb DNA repair by p62 accumulation and impairing NPM1–APE1 functions. In addition, oxidative stress involving APE1 also further compounds the damage. Hence, expression of the C9orf72 repeat expansion results in a “double whammy” for the cell: both RNA- and DPR-driven mechanisms conspire to produce extensive DNA damage and genomic instability, leading to motor neuron death. Further studies are now warranted to determine exactly how DNA repair mechanisms are compromised in ALS, the extent to which the DDR is induced, and which specific pathways are compromised. Modulation of the DDR, enhancement of DNA repair processes, and the targeting of G-quadruplexes and R-loops, may therefore be novel neuroprotective strategies for ALS that should be harnessed in future studies.
Author Contributions
Both authors, J.D.A. and A.K., conceived, co-wrote, and edited the article throughout for content and style consistency; and prepared the figures.
Funding
This research was funded by National Health and Medical Research Council: 10305133, 1086887, 1095215.
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia (NHMRC) Project grants (10305133 and 1086887), the MND Research Institute of Australia, and an NHMRC Dementia Team research grant (1095215).
Conflicts of Interest
The authors have no conflict of interest to declare.
References
- Liu, W.F.; Yu, S.S.; Chen, G.J.; Li, Y.Z. DNA damage checkpoint, damage repair, and genome stability. Yi Chuan Xue Bao 2006, 33, 381–390. [Google Scholar] [CrossRef]
- Martin, L.J. DNA damage and repair: Relevance to mechanisms of neurodegeneration. J. Neuropathol. Exp. Neurol. 2008, 67, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Penney, J.; Tsai, L.H. Chromatin regulation of DNA damage repair and genome integrity in the central nervous system. J. Mol. Biol. 2014, 426, 3376–3388. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.L.; Mantha, A.K.; Hazra, T.K.; Bhakat, K.K.; Mitra, S.; Szczesny, B. Oxidative genome damage and its repair: Implications in aging and neurodegenerative diseases. Mech. Ageing Dev. 2012, 133, 157–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Obulesu, M.; Rao, D.M. DNA damage and impairment of DNA repair in Alzheimer’s disease. Int. J. Neurosci. 2010, 120, 397–403. [Google Scholar] [CrossRef] [PubMed]
- McKinnon, P.J. ATM and the molecular pathogenesis of ataxia telangiectasia. Ann. Rev. Pathol. 2012, 7, 303–321. [Google Scholar] [CrossRef] [PubMed]
- Dorst, J.; Ludolph, A.C.; Huebers, A. Disease-modifying and symptomatic treatment of amyotrophic lateral sclerosis. Ther. Adv. Neurol. Disord. 2018, 11, 1756285617734734. [Google Scholar] [CrossRef] [PubMed]
- Wijesekera, L.C.; Leigh, P.N. Amyotrophic lateral sclerosis Orphanet. J. Rare Dis. 2009, 4, 3. [Google Scholar] [CrossRef]
- Ludolph, A.C.; Brettschneider, J.; Weishaupt, J.H. Amyotrophic lateral sclerosis. Curr. Opin. Neurol. 2012, 25, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Liscic, R.M. Als and Ftd: Insights into the disease mechanisms and therapeutic targets. Eur. J. Pharmacol. 2017, 817, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Shahheydari, H.; Ragagnin, A.; Walker, A.K.; Toth, R.P.; Vidal, M.; Jagaraj, C.J.; Perri, E.R.; Konopka, A.; Sultana, J.M.; Atkin, J.D. Protein Quality Control and the Amyotrophic Lateral Sclerosis/Frontotemporal Dementia Continuum. Front. Mol. Neurosci. 2017, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; Van Swieten, J.C.; Myllykangas, L. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Devenney, E.; Hornberger, M.; Irish, M.; Mioshi, E.; Burrell, J.; Tan, R.; Kiernan, M.C.; Hodges, J.R. Frontotemporal dementia associated with the C9ORF72 mutation: A unique clinical profile. JAMA Neurol. 2014, 71, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, N.J.; Zhang, Y.J.; Baker, M.; Gass, J.M.; Finch, N.A.; Xu, Y.F.; Stewart, H.; Kelley, B.J.; Kuntz, K.; Crook, R.J.; et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008, 4, e1000193. [Google Scholar] [CrossRef] [PubMed]
- Borroni, B.; Alberici, A.; Archetti, S.; Magnani, E.; Di Luca, M.; Padovani, A. New insights into biological markers of frontotemporal lobar degeneration spectrum. Curr. Med. Chem. 2010, 17, 1002–1009. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Rogelj, B.; Hortobagyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Deng, H.X.; Siddique, N.; Fecto, F.; Chen, W.; Yang, Y.; Liu, E.; Donkervoort, S.; Zheng, J.G.; Shi, Y.; et al. Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology 2010, 75, 807–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Muller, K.; Marroquin, N.; Nordin, F.; Hubers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef] [PubMed]
- van der Zee, J.; Gijselinck, I.; Van Mossevelde, S.; Perrone, F.; Dillen, L.; Heeman, B.; Baumer, V.; Engelborghs, S.; De Bleecker, J.; Baets, J.; et al. TBK1 Mutation Spectrum in an Extended European Patient Cohort with Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Hum. Mutat. 2017, 38, 297–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.M.; Gorrie, G.H.; Siddique, N.; Yang, Y.; Fecto, F.; Shi, Y.; Zhai, H.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, K.L.; Warraich, S.T.; Yang, S.; Solski, J.A.; Fernando, R.; Rouleau, G.A.; Nicholson, G.A.; Blair, I.P. UBQLN2/ubiquilin 2 mutation and pathology in familial amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, 2527.e3–2527.e10. [Google Scholar] [CrossRef] [PubMed]
- Gellera, C.; Tiloca, C.; Del Bo, R.; Corrado, L.; Pensato, V.; Agostini, J.; Cereda, C.; Ratti, A.; Castellotti, B.; Corti, S.; et al. Ubiquilin 2 mutations in Italian patients with amyotrophic lateral sclerosis and frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 2013, 84, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Maetzler, W.; Grehl, T.; Prudlo, J.; Vom Hagen, J.M.; Haack, T.; Rebassoo, P.; Munz, M.; Schols, L.; Biskup, S. Screening in ALS and FTD patients reveals 3 novel UBQLN2 mutations outside the PXX domain and a pure FTD phenotype. Neurobiol. Aging 2012, 33, 2949.e13–2949.e17. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Pottier, C.; Bieniek, K.F.; Finch, N.; van de Vorst, M.; Baker, M.; Perkersen, R.; Brown, P.; Ravenscroft, T.; van Blitterswijk, M.; Nicholson, A.M.; et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 2015, 130, 77–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, K.L.; Topp, S.; Yang, S.; Smith, B.; Fifita, J.A.; Warraich, S.T.; Zhang, K.Y.; Farrawell, N.; Vance, C.; Hu, X.; et al. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat. Commun. 2016, 7, 11253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Therrien, M.; Dion, P.A.; Rouleau, G.A. ALS: Recent Developments from Genetics Studies. Curr. Neurol. Neurosci. Rep. 2016, 16, 59. [Google Scholar] [CrossRef] [PubMed]
- Parakh, S.; Atkin, J.D. Protein folding alterations in amyotrophic lateral sclerosis. Brain Res. 2016, 1648, 633–649. [Google Scholar] [CrossRef] [PubMed]
- Blokhuis, A.M.; Groen, E.J.; Koppers, M.; van den Berg, L.H.; Pasterkamp, R.J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 2013, 125, 777–794. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Kwong, L.K.; Lee, E.B.; Kremmer, E.; Flatley, A.; Xu, Y.; Forman, M.S.; Troost, D.; Kretzschmar, H.A.; Trojanowski, J.Q.; et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009, 117, 137–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, I.R.; Neumann, M. FET proteins in frontotemporal dementia and amyotrophic lateral sclerosis. Brain Res. 2012, 1462, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, F.E. The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. 2010, 7, 420–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, D.; Seki, M.; Tsunoda, Y.; Uchiyama, H.; Suzuki, N. Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann. Neurol. 2011, 69, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Gunther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat. Commun. 2018, 9, 335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dormann, D.; Rodde, R.; Edbauer, D.; Bentmann, E.; Fischer, I.; Hruscha, A.; Than, M.E.; Mackenzie, I.R.; Capell, A.; Schmid, B.; et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010, 29, 2841–2857. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Tsai, P.Y.; Chern, Y. Energy Homeostasis and Abnormal RNA Metabolism in Amyotrophic Lateral Sclerosis. Front. Cell Neurosci. 2017, 11, 126. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Vandoorne, T.; Eckelmann, B.J.; Mitra, S.; Tomkinson, A.E.; Van Den Bosch, L.; Hegde, M.L. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat. Commun. 2018, 9, 3683. [Google Scholar] [CrossRef] [PubMed]
- Higelin, J.; Catanese, A.; Semelink-Sedlacek, L.L.; Oeztuerk, S.; Lutz, A.K.; Bausinger, J.; Barbi, G.; Speit, G.; Andersen, P.M.; Ludolph, A.C.; et al. NEK1 loss-of-function mutation induces DNA damage accumulation in ALS patient-derived motoneurons. Stem. Cell Res. 2018, 30, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Huang, M.; Wang, F.; Ma, X.; Liu, H.; Tu, Y.; Xing, L.; Zhu, X.; Zheng, H.; Fang, J.; et al. RBM45 competes with HDAC1 for binding to FUS in response to DNA damage. Nucleic. Acids Res. 2017, 45, 12862–12876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, C.; Herranz-Martin, S.; Karyka, E.; Liao, C.; Lewis, K.; Elsayed, W.; Lukashchuk, V.; Chiang, S.C.; Ray, S.; Mulcahy, P.J.; et al. C9orf72 expansion disrupts ATM-mediated chromosomal break repair. Nat. Neurosci. 2017, 20, 1225–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farg, M.A.; Konopka, A.; Ying Soo, K.; Ito, D.; Atkin, J.D. The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in Amyotrophic Lateral Sclerosis. Hum. Mol. Genet. 2017. [Google Scholar] [CrossRef] [PubMed]
- Muyderman, H.; Chen, T. Mitochondrial dysfunction in amyotrophic lateral sclerosis-a valid pharmacological target? Br. J. Pharmacol. 2014, 171, 2191–2205. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.Y.; Cali, C.P.; Lee, E.B. RNA metabolism in neurodegenerative disease. Dis. Model. Mech. 2017, 10, 509–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramesh, N.; Pandey, U.B. Autophagy Dysregulation in ALS: When Protein Aggregates Get Out of Hand. Front. Mol. Neurosci. 2017, 10, 263. [Google Scholar] [CrossRef] [PubMed]
- Kabashi, E.; Agar, J.N.; Strong, M.J.; Durham, H.D. Impaired proteasome function in sporadic amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2012, 13, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Pollari, E.; Goldsteins, G.; Bart, G.; Koistinaho, J.; Giniatullin, R. The role of oxidative stress in degeneration of the neuromuscular junction in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 131. [Google Scholar] [CrossRef] [PubMed]
- Dutertre, M.; Lambert, S.; Carreira, A.; Amor-Gueret, M.; Vagner, S. DNA damage: RNA-binding proteins protect from near and far. Trends Biochem. Sci. 2014, 39, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S. Age-related nonhomologous end joining activity in rat neurons. Brain Res. Bull. 2007, 73, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, K.; McVey, M. Error-Prone Repair of DNA Double-Strand Breaks. J. Cell Physiol. 2016, 231, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, L.M.; Baserga, S.J. Crosstalk between the nucleolus and the DNA damage response. Mol. Biosyst. 2017, 13, 443–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todd, P.K.; Paulson, H.L. RNA-mediated neurodegeneration in repeat expansion disorders. Ann. Neurol. 2010, 67, 291–300. [Google Scholar] [CrossRef] [PubMed]
- La Spada, A.R.; Taylor, J.P. Repeat expansion disease: Progress and puzzles in disease pathogenesis. Nat. Rev. Genet. 2010, 11, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Sordet, O.; Redon, C.E.; Guirouilh-Barbat, J.; Smith, S.; Solier, S.; Douarre, C.; Conti, C.; Nakamura, A.J.; Das, B.B.; Nicolas, E.; et al. Ataxia telangiectasia mutated activation by transcription- and topoisomerase I-induced DNA double-strand breaks. EMBO Rep. 2009, 10, 887–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosshard, M.; Markkanen, E.; van Loon, B. Base excision repair in physiology and pathology of the central nervous system. Int. J. Mol. Sci. 2012, 13, 16172–16222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaikh, A.Y.; Martin, L.J. DNA base-excision repair enzyme apurinic/apyrimidinic endonuclease/redox factor-1 is increased and competent in the brain and spinal cord of individuals with amyotrophic lateral sclerosis. Neuromol. Med. 2002, 2, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Liu, Z.; Chen, K.; Price, A.C.; Pan, Y.; Swaby, J.A.; Golden, W.C. Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 transgenic mice: Mechanisms of mitochondriopathy and cell death. J. Comp. Neurol. 2007, 500, 20–46. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Engelhardt, J.I.; Henkel, J.S.; Siklos, L.; Soos, J.; Goodman, C.; Appel, S.H. Widespread increased expression of the DNA repair enzyme PARP in brain in ALS. Neurology 2004, 62, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J. Transgenic mice with human mutant genes causing Parkinson’s disease and amyotrophic lateral sclerosis provide common insight into mechanisms of motor neuron selective vulnerability to degeneration. Rev. Neurosci. 2007, 18, 115–136. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, L.F.; Cerqueira, F.M.; Macedo, A.F.; Garcia, C.C.; Angeli, J.P.; Schumacher, R.I.; Sogayar, M.C.; Augusto, O.; Carri, M.T.; Di Mascio, P.; et al. Increased SOD1 association with chromatin, DNA damage, p53 activation, and apoptosis in a cellular model of SOD1-linked ALS. Biochim. Biophys. Acta 2010, 1802, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar] [CrossRef] [PubMed]
- Olkowski, Z.L. Mutant AP endonuclease in patients with amyotrophic lateral sclerosis. Neuroreport 1998, 9, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Selfridge, J.; Song, L.; Brownstein, D.G.; Melton, D.W. Mice with DNA repair gene Ercc1 deficiency in a neural crest lineage are a model for late-onset Hirschsprung disease. DNA Repair 2010, 9, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Lee, S.; Shang, Y.; Wang, W.Y.; Au, K.F.; Kamiya, S.; Barmada, S.J.; Finkbeiner, S.; Lui, H.; Carlton, C.E.; et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J. Clin. Investig. 2014, 124, 981–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Q.; Holler, C.J.; Taylor, G.; Hudson, K.F.; Watkins, W.; Gearing, M.; Ito, D.; Murray, M.E.; Dickson, D.W.; Seyfried, N.T.; et al. FUS is phosphorylated by DNA-PK and accumulates in the cytoplasm after DNA damage. J. Neurosci. 2014, 34, 7802–7813. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, M.; Toth, R.; Vandermoere, F.; Morrice, N.A.; Rouse, J. Identification and characterization of FUS/TLS as a new target of ATM. Biochem. J. 2008, 415, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.; Huang, E.J.; Tsai, L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, S.J.; Mordes, D.A.; Cameron, L.A.; Neuberg, D.S.; Landini, S.; Eggan, K.; Livingston, D.M. Two familial ALS proteins function in prevention/repair of transcription-associated DNA damage. Proc. Natl. Acad. Sci. USA 2016, 113, E7701–E7709. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.Z.; Bennett, C.L.; Huynh, H.M.; Blair, I.P.; Puls, I.; Irobi, J.; Dierick, I.; Abel, A.; Kennerson, M.L.; Rabin, B.A.; et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 2004, 74, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Koppers, M.; van Blitterswijk, M.M.; Vlam, L.; Rowicka, P.A.; van Vught, P.W.; Groen, E.J.; Spliet, W.G.; Engelen-Lee, J.; Schelhaas, H.J.; de Visser, M.; et al. VCP mutations in familial and sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2012, 33, e813–e837. [Google Scholar] [CrossRef] [PubMed]
- Vaz, B.; Halder, S.; Ramadan, K. Role of p97/VCP (Cdc48) in genome stability. Front. Genet. 2013, 4, 60. [Google Scholar] [CrossRef] [PubMed]
- D’Angiolella, V.; Esencay, M.; Pagano, M. A cyclin without cyclin-dependent kinases: Cyclin F controls genome stability through ubiquitin-mediated proteolysis. Trends Cell Biol. 2013, 23, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.; Muller, K.; Wieland, T.; Weydt, P.; Bohm, S.; Lule, D.; Hubers, A.; Neuwirth, C.; Weber, M.; Borck, G.; et al. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain 2016, 139, e28. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, C.F.; Riley, D.J.; Chen, P.L. Nek1 kinase functions in DNA damage response and checkpoint control through a pathway independent of ATM and ATR. Cell Cycle 2011, 10, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spies, J.; Waizenegger, A.; Barton, O.; Surder, M.; Wright, W.D.; Heyer, W.D.; Lobrich, M. Nek1 Regulates Rad54 to Orchestrate Homologous Recombination and Replication Fork Stability. Mol. Cell 2016, 62, 903–917. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Lin, H.; Wang, X.; Zuo, Q.; Qin, J.; Zhang, P. The NEK1 interactor, C21ORF2, is required for efficient DNA damage repair. Acta Biochim. Biophys. Sin. 2015, 47, 834–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutherford, N.J.; Heckman, M.G.; Dejesus-Hernandez, M.; Baker, M.C.; Soto-Ortolaza, A.I.; Rayaprolu, S.; Stewart, H.; Finger, E.; Volkening, K.; Seeley, W.W.; et al. Length of normal alleles of C9ORF72 GGGGCC repeat do not influence disease phenotype. Neurobiol. Aging 2012, 33, 2950.e5–2950.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubers, A.; Marroquin, N.; Schmoll, B.; Vielhaber, S.; Just, M.; Mayer, B.; Hogel, J.; Dorst, J.; Mertens, T.; Just, W.; et al. Polymerase chain reaction and Southern blot-based analysis of the C9orf72 hexanucleotide repeat in different motor neuron diseases. Neurobiol Aging 2014, 35, 1214.e1–1214.e6. [Google Scholar] [CrossRef] [PubMed]
- Van Mossevelde, S.; van der Zee, J.; Cruts, M.; Van Broeckhoven, C. Relationship between C9orf72 repeat size and clinical phenotype. Curr. Opin. Genet. Dev. 2017, 44, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.N.; Newhouse, S.; Shatunov, A.; Vance, C.; Topp, S.; Johnson, L.; Miller, J.; Lee, Y.; Troakes, C.; Scott, K.M.; et al. The C9ORF72 expansion mutation is a common cause of ALS+/-FTD in Europe and has a single founder. Eur. J. Hum. Genet. 2013, 21, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Fratta, P.; Polke, J.M.; Newcombe, J.; Mizielinska, S.; Lashley, T.; Poulter, M.; Beck, J.; Preza, E.; Devoy, A.; Sidle, K.; et al. Screening a UK amyotrophic lateral sclerosis cohort provides evidence of multiple origins of the C9orf72 expansion. Neurobiol. Aging 2015, 36, e541–e547. [Google Scholar] [CrossRef] [PubMed]
- Rollinson, S.; Halliwell, N.; Young, K.; Callister, J.B.; Toulson, G.; Gibbons, L.; Davidson, Y.S.; Robinson, A.C.; Gerhard, A.; Richardson, A.; et al. Analysis of the hexanucleotide repeat in C9ORF72 in Alzheimer’s disease. Neurobiol Aging 2012, 33, e1845–e1846. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.S.; Quinzii, C.; Dunning-Broadbent, J.; Waters, C.; Mitsumoto, H.; Brannagan, T.H., 3rd; Cosentino, S.; Huey, E.D.; Nagy, P.; Kuo, S.H. Multiple system atrophy and amyotrophic lateral sclerosis in a family with hexanucleotide repeat expansions in C9orf72. JAMA Neurol. 2014, 71, 771–774. [Google Scholar] [CrossRef] [PubMed]
- Hensman Moss, D.J.; Poulter, M.; Beck, J.; Hehir, J.; Polke, J.M.; Campbell, T.; Adamson, G.; Mudanohwo, E.; McColgan, P.; Haworth, A.; et al. C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology 2014, 82, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Corcia, P.; Vourc’h, P.; Guennoc, A.M.; Del Mar Amador, M.; Blasco, H.; Andres, C.; Couratier, P.; Gordon, P.H.; Meininger, V. Pure cerebellar ataxia linked to large C9orf72 repeat expansion. Amyotroph. Lateral Scler. Frontotemporal. Degener. 2016, 17, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.; Cooper-Knock, J.; Highley, J.R.; Milano, A.; Kirby, J.; Goodall, E.; Lowe, J.; Scott, I.; Constantinescu, C.S.; Walters, S.J.; et al. Concurrence of multiple sclerosis and amyotrophic lateral sclerosis in patients with hexanucleotide repeat expansions of C9ORF72. J. Neurol. Neurosurg. Psychiatry 2013, 84, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Le Ber, I.; Condroyer, C.; Broussolle, E.; Gabelle, A.; Thobois, S.; Pasquier, F.; Mondon, K.; Dion, P.A.; Rochefort, D.; et al. C9orf72 repeat expansions are a rare genetic cause of parkinsonism. Brain 2013, 136, 385–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floris, G.; Borghero, G.; Cannas, A.; Stefano, F.D.; Murru, M.R.; Corongiu, D.; Cuccu, S.; Tranquilli, S.; Marrosu, M.G.; Chio, A.; et al. Bipolar affective disorder preceding frontotemporal dementia in a patient with C9ORF72 mutation: Is there a genetic link between these two disorders? J. Neurol. 2013, 260, 1155–1157. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, D.; Reif, A.; Dell’Osso, B.; Palazzo, C.; Villa, C.; Fenoglio, C.; Kittel-Schneider, S.; Leonhard, C.; Olmes, D.G.; Serpente, M.; et al. C9ORF72 hexanucleotide repeat expansion as a rare cause of bipolar disorder. Bipolar. Disord. 2014, 16, 448–449. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, D.; Reif, A.; Dell’osso, B.; Kittel-Schneider, S.; Leonhard, C.; Herr, A.; Palazzo, C.; Villa, C.; Fenoglio, C.; Serpente, M.; et al. C9ORF72 hexanucleotide repeat expansion is a rare cause of schizophrenia. Neurobiol. Aging 2014, 35, e1214–e1217. [Google Scholar] [CrossRef] [PubMed]
- Waite, A.J.; Baumer, D.; East, S.; Neal, J.; Morris, H.R.; Ansorge, O.; Blake, D.J. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol. Aging 2014, 35, 1779.e5–1779.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burberry, A.; Suzuki, N.; Wang, J.Y.; Moccia, R.; Mordes, D.A.; Stewart, M.H.; Suzuki-Uematsu, S.; Ghosh, S.; Singh, A.; Merkle, F.T.; et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci. Transl. Med. 2016, 8, 347. [Google Scholar] [CrossRef] [PubMed]
- Atanasio, A.; Decman, V.; White, D.; Ramos, M.; Ikiz, B.; Lee, H.C.; Siao, C.J.; Brydges, S.; LaRosa, E.; Bai, Y.; et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci. Rep. 2016, 6, 23204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Lin, S.; Staats, K.A.; Li, Y.; Chang, W.H.; Hung, S.T.; Hendricks, E.; Linares, G.R.; Wang, Y.; Son, E.Y.; et al. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 2018, 24, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Liu, Y.; Banez-Coronel, M.; Reid, T.; Pletnikova, O.; Lewis, J.; Miller, T.M.; Harms, M.B.; Falchook, A.E.; Subramony, S.H.; et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2013, 110, E4968–E4977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnelly, C.J.; Zhang, P.W.; Pham, J.T.; Haeusler, A.R.; Mistry, N.A.; Vidensky, S.; Daley, E.L.; Poth, E.M.; Hoover, B.; Fines, D.M.; et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013, 80, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.B.; Chen, H.J.; Peres, J.N.; Gomez-Deza, J.; Attig, J.; Stalekar, M.; Troakes, C.; Nishimura, A.L.; Scotter, E.L.; Vance, C.; et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013, 5, 1178–1186. [Google Scholar] [CrossRef] [PubMed]
- Cooper-Knock, J.; Walsh, M.J.; Higginbottom, A.; Robin Highley, J.; Dickman, M.J.; Edbauer, D.; Ince, P.G.; Wharton, S.B.; Wilson, S.A.; Kirby, J.; et al. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain 2014, 137, 2040–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prudencio, M.; Belzil, V.V.; Batra, R.; Ross, C.A.; Gendron, T.F.; Pregent, L.J.; Murray, M.E.; Overstreet, K.K.; Piazza-Johnston, A.E.; Desaro, P.; et al. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat. Neurosci. 2015, 18, 1175–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sareen, D.; O’Rourke, J.G.; Meera, P.; Muhammad, A.K.; Grant, S.; Simpkinson, M.; Bell, S.; Carmona, S.; Ornelas, L.; Sahabian, A.; et al. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 2013, 5, 208. [Google Scholar] [CrossRef] [PubMed]
- Cooper-Knock, J.; Bury, J.J.; Heath, P.R.; Wyles, M.; Higginbottom, A.; Gelsthorpe, C.; Highley, J.R.; Hautbergue, G.; Rattray, M.; Kirby, J.; et al. C9ORF72 GGGGCC Expanded Repeats Produce Splicing Dysregulation which Correlates with Disease Severity in Amyotrophic Lateral Sclerosis. PLoS ONE 2015, 10, e0127376. [Google Scholar] [CrossRef] [PubMed]
- Highley, J.R.; Kirby, J.; Jansweijer, J.A.; Webb, P.S.; Hewamadduma, C.A.; Heath, P.R.; Higginbottom, A.; Raman, R.; Ferraiuolo, L.; Cooper-Knock, J.; et al. Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones. Neuropathol. Appl. Neurobiol. 2014, 40, 670–685. [Google Scholar] [CrossRef] [PubMed]
- Conlon, E.G.; Lu, L.; Sharma, A.; Yamazaki, T.; Tang, T.; Shneider, N.A.; Manley, J.L. The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Cleary, J.D.; Ranum, L.P. Repeat-associated non-ATG (RAN) translation in neurological disease. Hum. Mol. Genet. 2013, 22, R45–R51. [Google Scholar] [CrossRef] [PubMed]
- Sonobe, Y.; Ghadge, G.; Masaki, K.; Sendoel, A.; Fuchs, E.; Roos, R.P. Translation of dipeptide repeat proteins from the C9ORF72 expanded repeat is associated with cellular stress. Neurobiol. Dis. 2018, 116, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Wang, S.; Mestre, A.A.; Fu, C.; Makarem, A.; Xian, F.; Hayes, L.R.; Lopez-Gonzalez, R.; Drenner, K.; Jiang, J.; et al. C9ORF72 GGGGCC repeat-associated non-AUG translation is upregulated by stress through eIF2alpha phosphorylation. Nat. Commun. 2018, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Tabet, R.; Schaeffer, L.; Freyermuth, F.; Jambeau, M.; Workman, M.; Lee, C.Z.; Lin, C.C.; Jiang, J.; Jansen-West, K.; Abou-Hamdan, H.; et al. CUG initiation and frameshifting enable production of dipeptide repeat proteins from ALS/FTD C9ORF72 transcripts. Nat. Commun. 2018, 9, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizielinska, S.; Gronke, S.; Niccoli, T.; Ridler, C.E.; Clayton, E.L.; Devoy, A.; Moens, T.; Norona, F.E.; Woollacott, I.O.C.; Pietrzyk, J.; et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 2014, 345, 1192–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moens, T.G.; Mizielinska, S.; Niccoli, T.; Mitchell, J.S.; Thoeng, A.; Ridler, C.E.; Gronke, S.; Esser, J.; Heslegrave, A.; Zetterberg, H.; et al. Sense and antisense RNA are not toxic in Drosophila models of C9orf72-associated ALS/FTD. Acta Neuropathol. 2018, 135, 445–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovicic, A.; Mertens, J.; Boeynaems, S.; Bogaert, E.; Chai, N.; Yamada, S.B.; Paul, J.W., 3rd; Sun, S.; Herdy, J.R.; Bieri, G.; et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci. 2015, 18, 1226–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.H.; Zhang, P.; Kim, H.J.; Mitrea, D.M.; Sarkar, M.; Freibaum, B.D.; Cika, J.; Coughlin, M.; Messing, J.; Molliex, A.; et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 2016, 167, 774–788.e717. [Google Scholar] [CrossRef] [PubMed]
- Kohara, N.; Kaji, R.; Kojima, Y.; Mills, K.R.; Fujii, H.; Hamano, T.; Kimura, J.; Takamatsu, N.; Uchiyama, T. Abnormal excitability of the corticospinal pathway in patients with amyotrophic lateral sclerosis: A single motor unit study using transcranial magnetic stimulation. Electroencephalogr. Clin. Neurophysiol. 1996, 101, 32–41. [Google Scholar] [CrossRef]
- Schutz, B. Imbalanced excitatory to inhibitory synaptic input precedes motor neuron degeneration in an animal model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2005, 20, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Suberbielle, E.; Sanchez, P.E.; Kravitz, A.V.; Wang, X.; Ho, K.; Eilertson, K.; Devidze, N.; Kreitzer, A.C.; Mucke, L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-beta. Nat. Neurosci. 2013, 16, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Gao, F.; Pfenning, A.R.; Pan, L.; Yamakawa, S.; Seo, J.; Rueda, R.; Phan, T.X.; Yamakawa, H.; Pao, P.C.; et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 2015, 161, 1592–1605. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, S.M. Discovery of alternative DNA structures: A heroic decade (1979–1989). Front. Biosci. 2008, 13, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Manley, J.L. R Loops and Links to Human Disease. J. Mol. Biol. 2017, 429, 3168–3180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Brosh, R.M., Jr. G-quadruplex nucleic acids and human disease. FEBS J. 2010, 277, 3470–3488. [Google Scholar] [CrossRef] [PubMed]
- Hamperl, S.; Cimprich, K.A. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair 2014, 19, 84–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, C.E.; Nichol Edamura, K.; Cleary, J.D. Repeat instability: Mechanisms of dynamic mutations. Nat. Rev. Genet. 2005, 6, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Chedin, F.; Hsieh, C.L.; Wilson, T.E.; Lieber, M.R. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B. cells. Nat. Immunol. 2003, 4, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.L.; Balasubramanian, S. G-quadruplexes in promoters throughout the human genome. Nucleic. Acids Res. 2007, 35, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Simone, R.; Fratta, P.; Neidle, S.; Parkinson, G.N.; Isaacs, A.M. G-quadruplexes: Emerging roles in neurodegenerative diseases and the non-coding transcriptome. FEBS Lett. 2015, 589, 1653–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilera, A.; Garcia-Muse, T. R loops: From transcription byproducts to threats to genome stability. Mol. Cell 2012, 46, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Graf, M.; Bonetti, D.; Lockhart, A.; Serhal, K.; Kellner, V.; Maicher, A.; Jolivet, P.; Teixeira, M.T.; Luke, B. Telomere Length Determines TERRA and R-Loop Regulation through the Cell Cycle. Cell 2017, 170, 72–85.e14. [Google Scholar] [CrossRef] [PubMed]
- Belotserkovskii, B.P.; Hanawalt, P.C. PNA binding to the non-template DNA strand interferes with transcription, suggesting a blockage mechanism mediated by R.-loop formation. Mol. Carcinog. 2015, 54, 1508–1512. [Google Scholar] [CrossRef] [PubMed]
- Groh, M.; Lufino, M.M.; Wade-Martins, R.; Gromak, N. R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X. syndrome. PLoS Genet. 2014, 10, e1004318. [Google Scholar] [CrossRef] [PubMed]
- Skourti-Stathaki, K.; Kamieniarz-Gdula, K.; Proudfoot, N.J. R-loops induce repressive chromatin marks over mammalian gene terminators. Nature 2014, 516, 436–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Droppelmann, C.A.; Campos-Melo, D.; Ishtiaq, M.; Volkening, K.; Strong, M.J. RNA metabolism in ALS: When normal processes become pathological. Amyotroph Lateral Scler. 2014, 15, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Groh, M.; Albulescu, L.O.; Cristini, A.; Gromak, N. Senataxin: Genome Guardian at the Interface of Transcription and Neurodegeneration. J. Mol. Biol. 2017, 429, 3181–3195. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.A.; Aristizabal, M.J.; Lu, P.Y.; Luo, Z.; Hamza, A.; Kobor, M.S.; Stirling, P.C.; Hieter, P. Genome-wide profiling of yeast DNA:RNA hybrid prone sites with DRIP-chip. PLoS Genet. 2014, 10, e1004288. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P.; Aguilera, A. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell 2003, 12, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Panigrahi, G.B.; Lau, R.; Montgomery, S.E.; Leonard, M.R.; Pearson, C.E. Slipped (CTG)·(CAG) repeats can be correctly repaired, escape repair or undergo error-prone repair. Nat. Struct. Mol. Biol. 2005, 12, 654–662. [Google Scholar] [CrossRef] [PubMed]
- Haeusler, A.R.; Donnelly, C.J.; Periz, G.; Simko, E.A.; Shaw, P.G.; Kim, M.S.; Maragakis, N.J.; Troncoso, J.C.; Pandey, A.; Sattler, R.; et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 2014, 507, 195–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Liu, C.; Geng, Y.; Zhu, G. Topology of a G-quadruplex DNA formed by C9orf72 hexanucleotide repeats associated with ALS and FTD. Sci. Rep. 2015, 5, 16673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chedin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Zinman, L.; Moreno, D.; Schymick, J.; Liang, Y.; Sato, C.; Zheng, Y.; Ghani, M.; Dib, S.; Keith, J.; et al. Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am. J. Hum. Genet. 2013, 92, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.B. Poly(GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Lempiainen, H.; Shore, D. Growth control and ribosome biogenesis. Curr. Opin. Cell Biol. 2009, 21, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Shav-Tal, Y.; Blechman, J.; Darzacq, X.; Montagna, C.; Dye, B.T.; Patton, J.G.; Singer, R.H.; Zipori, D. Dynamic sorting of nuclear components into distinct nucleolar caps during transcriptional inhibition. Mol. Biol. Cell 2005, 16, 2395–2413. [Google Scholar] [CrossRef] [PubMed]
- Kwon, I.; Xiang, S.; Kato, M.; Wu, L.; Theodoropoulos, P.; Wang, T.; Kim, J.; Yun, J.; Xie, Y.; McKnight, S.L. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 2014, 345, 1139–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Z.; Wang, H.; Xia, Q.; Li, K.; Li, K.; Jiang, X.; Xu, G.; Wang, G.; Ying, Z. Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum. Mol. Genet. 2015, 24, 2426–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizielinska, S.; Ridler, C.E.; Balendra, R.; Thoeng, A.; Woodling, N.S.; Grasser, F.A.; Plagnol, V.; Lashley, T.; Partridge, L.; Isaacs, A.M. Bidirectional nucleolar dysfunction in C9orf72 frontotemporal lobar degeneration. Acta Neuropathol. Commun. 2017, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.; Wang, Y.; Mukhopadhyay, D.; Backlund, P.; Kolli, N.; Yergey, A.; Wilkinson, K.D.; Dasso, M. Nucleolar protein B23/nucleophosmin regulates the vertebrate SUMO pathway through SENP3 and SENP5 proteases. J. Cell Biol. 2008, 183, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haindl, M.; Harasim, T.; Eick, D.; Muller, S. The nucleolar SUMO-specific protease SENP3 reverses SUMO modification of nucleophosmin and is required for rRNA processing. EMBO Rep. 2008, 9, 273–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koike, A.; Nishikawa, H.; Wu, W.; Okada, Y.; Venkitaraman, A.R.; Ohta, T. Recruitment of phosphorylated NPM1 to sites of DNA damage through RNF8-dependent ubiquitin conjugates. Cancer Res. 2010, 70, 6746–6756. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Lai, Y.; Torner, J.; Zhang, Y.; Zhang, Z.; Liu, Y. Base excision repair of oxidative DNA damage coupled with removal of a CAG repeat hairpin attenuates trinucleotide repeat expansion. Nucleic Acids Res. 2014, 42, 3675–3691. [Google Scholar] [CrossRef] [PubMed]
- Vascotto, C.; Fantini, D.; Romanello, M.; Cesaratto, L.; Deganuto, M.; Leonardi, A.; Radicella, J.P.; Kelley, M.R.; D’Ambrosio, C.; Scaloni, A.; et al. APE1/Ref-1 Interacts with NPM1 within Nucleoli and Plays a Role in the rRNA Quality Control Process. Mol. Cell Biol. 2009, 29, 1834–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, K.; Motoda, Y.; Ito, F. Role of transcription factor activator protein 1 (AP1) in epidermal growth factor-mediated protection against apoptosis induced by a DNA-damaging agent. FEBS J. 2006, 273, 3743–3755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vascotto, C.; Lirussi, L.; Poletto, M.; Tiribelli, M.; Damiani, D.; Fabbro, D.; Damante, G.; Demple, B.; Colombo, E.; Tell, G. Functional regulation of the apurinic/apyrimidinic endonuclease 1 by nucleophosmin: Impact on tumor biology. Oncogene 2014, 33, 2876–2887. [Google Scholar] [CrossRef] [PubMed]
- Londero, A.P.; Orsaria, M.; Tell, G.; Marzinotto, S.; Capodicasa, V.; Poletto, M.; Vascotto, C.; Sacco, C.; Mariuzzi, L. Expression and prognostic significance of APE1/Ref-1 and NPM1 proteins in high-grade ovarian serous cancer. Am. J. Clin. Pathol. 2014, 141, 404–414. [Google Scholar] [CrossRef] [PubMed]
- Poletto, M.; Lirussi, L.; Wilson, D.M., 3rd; Tell, G. Nucleophosmin modulates stability, activity, and nucleolar accumulation of base excision repair proteins. Mol. Biol. Cell 2014, 25, 1641–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindstrom, M.S. NPM1/B23: A Multifunctional Chaperone in Ribosome Biogenesis and Chromatin Remodeling. Biochem. Res. Int. 2011, 2011, 195209. [Google Scholar] [CrossRef] [PubMed]
- Baborie, A.; Griffiths, T.D.; Jaros, E.; Perry, R.; McKeith, I.G.; Burn, D.J.; Masuda-Suzukake, M.; Hasegawa, M.; Rollinson, S.; Pickering-Brown, S.; et al. Accumulation of dipeptide repeat proteins predates that of TDP-43 in frontotemporal lobar degeneration associated with hexanucleotide repeat expansions in C9ORF72 gene. Neuropathol. Appl. Neurobiol. 2015, 41, 601–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nonaka, T.; Masuda-Suzukake, M.; Hosokawa, M.; Shimozawa, A.; Hirai, S.; Okado, H.; Hasegawa, M. C9ORF72 dipeptide repeat poly-GA inclusions promote: Intracellular aggregation of phosphorylated TDP-43. Hum. Mol. Genet. 2018. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Zhang, Y.; Gendron, T.F.; Bauer, P.O.; Chew, J.; Yang, W.Y.; Fostvedt, E.; Jansen-West, K.; Belzil, V.V.; Desaro, P.; et al. Discovery of a Biomarker and Lead Small Molecules to Target r(GGGGCC)-Associated Defects in c9FTD/ALS. Neuron 2014, 84, 239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simone, R.; Balendra, R.; Moens, T.G.; Preza, E.; Wilson, K.M.; Heslegrave, A.; Woodling, N.S.; Niccoli, T.; Gilbert-Jaramillo, J.; Abdelkarim, S.; et al. G-quadruplex-binding small molecules ameliorate C9orf72 FTD/ALS pathology in vitro and in vivo. EMBO Mol. Med. 2018, 10, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P.; Cheok, C.F.; Lain, S. p53-based cancer therapy. Cold Spring Harb. Perspect. Biol. 2010, 2, a001222. [Google Scholar] [CrossRef] [PubMed]
- Komarov, P.G.; Komarova, E.A.; Kondratov, R.V.; Christov-Tselkov, K.; Coon, J.S.; Chernov, M.V.; Gudkov, A.V. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999, 285, 1733–1737. [Google Scholar] [CrossRef] [PubMed]
- Komarova, E.A.; Gudkov, A.V. Suppression of p53: A new approach to overcome side effects of antitumor therapy. Biochemistry (Mosc) 2000, 65, 41–48. [Google Scholar] [PubMed]
- Shen, C.; Houghton, P.J. The mTOR pathway negatively controls ATM by up-regulating miRNAs. Proc. Natl. Acad. Sci. USA 2013, 110, 11869–11874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leger, B.; Vergani, L.; Soraru, G.; Hespel, P.; Derave, W.; Gobelet, C.; D’Ascenzio, C.; Angelini, C.; Russell, A.P. Human skeletal muscle atrophy in amyotrophic lateral sclerosis reveals a reduction in Akt and an increase in atrogin-1. FASEB J. 2006, 20, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Peviani, M.; Salvaneschi, E.; Bontempi, L.; Petese, A.; Manzo, A.; Rossi, D.; Salmona, M.; Collina, S.; Bigini, P.; Curti, D. Neuroprotective effects of the Sigma-1 receptor (S1R) agonist PRE-084, in a mouse model of motor neuron disease not linked to SOD1 mutation. Neurobiol. Dis. 2014, 62, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Peviani, M.; Cheroni, C.; Troglio, F.; Quarto, M.; Pelicci, G.; Bendotti, C. Lack of changes in the PI3K/AKT survival pathway in the spinal cord motor neurons of a mouse model of familial amyotrophic lateral sclerosis. Mol. Cell Neurosci. 2007, 34, 592–602. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kamili, A.; Hardy, J.R.; Groblewski, G.E.; Khanna, K.K.; Byrne, J.A. Tumor protein D52 represents a negative regulator of ATM protein levels. Cell Cycle 2013, 12, 3083–3097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieker, C.; Engblom, D.; Kreiner, G.; Domanskyi, A.; Schober, A.; Stotz, S.; Neumann, M.; Yuan, X.; Grummt, I.; Schutz, G.; et al. Nucleolar disruption in dopaminergic neurons leads to oxidative damage and parkinsonism through repression of mammalian target of rapamycin signaling. J. Neurosci. 2011, 31, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, C.; Wood, M.J.A. Antisense oligonucleotides: The next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2018, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Gendron, T.F.; Chew, J.; Stankowski, J.N.; Hayes, L.R.; Zhang, Y.J.; Prudencio, M.; Carlomagno, Y.; Daughrity, L.M.; Jansen-West, K.; Perkerson, E.A.; et al. Poly(GP) proteins are a useful pharmacodynamic marker for C9ORF72-associated amyotrophic lateral sclerosis. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Baughn, M.; Rigo, F.; Sun, S.; Liu, P.; Li, H.R.; Jiang, J.; Watt, A.T.; Chun, S.; Katz, M.; et al. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. USA 2013, 110, E4530–4539. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Scheme illustrating mechanisms by which genomic integrity is disrupted by the C9orf72 repeat expansion in ALS. Cells are exposed to exogenous and endogenous sources of DNA damage, such as normal cellular metabolism, the formation of R-loops, UV light exposure, ionising radiation, chemical exposure, and replication errors. In normal physiological conditions, the integrity of the genome is preserved by safeguarding mechanisms: the DDR and the nucleolus. However, in ALS, transcription of the C9orf72 repeat expansion leads to the production of expanded RNA transcripts. Furthermore, DPRs and abnormal DNA structures, such as R-loops, hairpins, and G-quadruplexes, are formed. These conformations compromise the normal cellular protective mechanisms, leading to persistent DNA damage and loss of genomic integrity.
Figure 1.
Scheme illustrating mechanisms by which genomic integrity is disrupted by the C9orf72 repeat expansion in ALS. Cells are exposed to exogenous and endogenous sources of DNA damage, such as normal cellular metabolism, the formation of R-loops, UV light exposure, ionising radiation, chemical exposure, and replication errors. In normal physiological conditions, the integrity of the genome is preserved by safeguarding mechanisms: the DDR and the nucleolus. However, in ALS, transcription of the C9orf72 repeat expansion leads to the production of expanded RNA transcripts. Furthermore, DPRs and abnormal DNA structures, such as R-loops, hairpins, and G-quadruplexes, are formed. These conformations compromise the normal cellular protective mechanisms, leading to persistent DNA damage and loss of genomic integrity.

Figure 2.
Mechanisms of amyotrophic lateral sclerosis (ALS) pathogenesis induced by the chromosome 9 open reading frame 72 (C9orf72) repeat expansion. The C9orf72 repeat expansion forms abnormal nucleic acid structures, including R-loops, which perturb DNA repair processes involving ATM, and probably other mechanisms. The expanded RNA forms foci and sequesters RNA binding proteins, leading to dysfunction in RNA processing. The C9orf72 DPRs, produced by RAN translation, also accumulate in the nucleolus, leading to perturbations in nucleolar function, including DNA repair processes, ribosomal biogenesis, and APE-dependent mechanisms (including oxidative stress). They also impair DNA repair by p62-dependent mechanisms. Together, these events result in the accumulation of DNA damage, genome instability, and motor neuron death.
Figure 2.
Mechanisms of amyotrophic lateral sclerosis (ALS) pathogenesis induced by the chromosome 9 open reading frame 72 (C9orf72) repeat expansion. The C9orf72 repeat expansion forms abnormal nucleic acid structures, including R-loops, which perturb DNA repair processes involving ATM, and probably other mechanisms. The expanded RNA forms foci and sequesters RNA binding proteins, leading to dysfunction in RNA processing. The C9orf72 DPRs, produced by RAN translation, also accumulate in the nucleolus, leading to perturbations in nucleolar function, including DNA repair processes, ribosomal biogenesis, and APE-dependent mechanisms (including oxidative stress). They also impair DNA repair by p62-dependent mechanisms. Together, these events result in the accumulation of DNA damage, genome instability, and motor neuron death.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Konopka, A.; Atkin, J.D. The Emerging Role of DNA Damage in the Pathogenesis of the C9orf72 Repeat Expansion in Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2018, 19, 3137. https://doi.org/10.3390/ijms19103137
AMA Style
Konopka A, Atkin JD. The Emerging Role of DNA Damage in the Pathogenesis of the C9orf72 Repeat Expansion in Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2018; 19(10):3137. https://doi.org/10.3390/ijms19103137
Chicago/Turabian StyleKonopka, Anna, and Julie D Atkin. 2018. "The Emerging Role of DNA Damage in the Pathogenesis of the C9orf72 Repeat Expansion in Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 19, no. 10: 3137. https://doi.org/10.3390/ijms19103137
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.