De Novo Transcriptome Sequencing Analysis of Goose (Anser anser) Embryonic Skin and the Identification of Genes Related to Feather Follicle Morphogenesis at Three Stages of Development

Abstract
:
1. Introduction
2. Results
2.1. Micro Anatomic Observation of Feather Follicle Characters
2.2. Transcriptome Sequencing, De Novo Assembly, and Functional Annotation
2.3. Differential Gene Expression at Embryonic Day 13, 18, and 28 (E13, E18, and E28)
2.4. Gene Ontology (GO) Enrichment Analysis of Differentially Expressed Genes (DEGs)
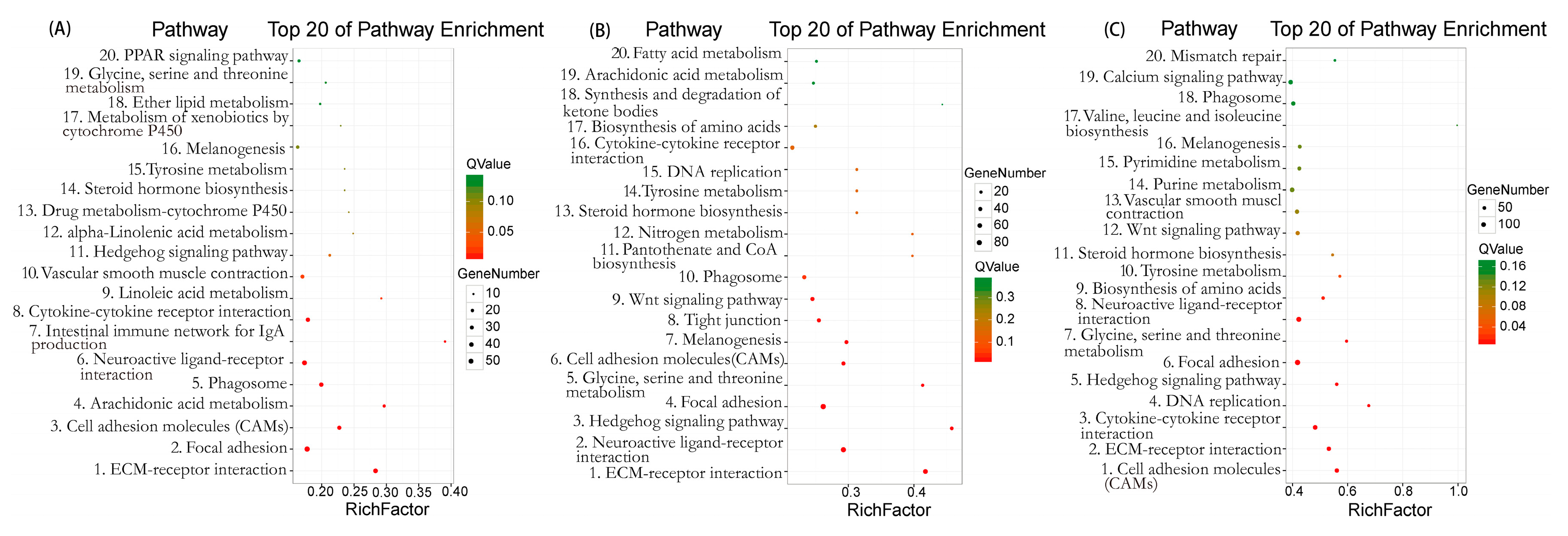
2.5. Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Enrichment Analysis of DEGs
2.6. Cluster Analysis of DEGs among the Three Developmental Stages
2.7. Validation of RNA-Sequencing (RNA-Seq) Data Using Quantitative Real-Time PCR (qRT-PCR)
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Animal Sample Preparation
4.3. Histological Observation
4.4. RNA Extraction, Library Preparation, and Sequencing
4.5. De Novo Assembly and Gene Annotation
4.6. Expression Annotation
4.7. Validation of RNA-Seq Data by qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| E13 | embryonic day 13 |
| E18 | embryonic day 18 |
| E28 | embryonic day 28 |
| DEGs | differentially expressed genes |
| H&E | Hematoxylin and Eosin |
| RPKM | reads per kb per million reads |
| RNA-Seq | RNA-Sequencing |
| FDR | False Discovery Rate |
| WNT3A | Wnt family member 3A |
| IGFBP3 | insulin-like growth factor binding protein 3 |
| BMP7 | bone morphogenetic protein 7 |
| SFRP2 | secreted-frizzled related protein 2 |
| ILs | interleukins |
| IL-20-RI | Interleukin-20-receptor I complex |
| IL6R | Interleukin-6 receptor |
| IL-1R1 | Interleukin-1 type I receptor |
| DBI | diazepam binding inhibitor |
References
- Reddy, N.; Yang, Y. Structure and properties of chicken feather barbs as natural protein fibers. J. Polym. Environ. 2007, 15, 81–87. [Google Scholar] [CrossRef]
- Adejumo, I.O.; Adetunji, C.O.; Ogundipe, K.; Osademe, S.N. Chemical composition and amino acid profile of differently processed feather meal. J. Agric. Sci. 2016, 61, 237–246. [Google Scholar] [CrossRef]
- Wu, W.; Xu, R.F.; Guo, X.; Li, C.H.; Wu, C.X. Characterization of embryonic feather follicle development in the Chinese indigenous Jilin white goose. Asian Australas. J. Anim. 2008, 21, 346–352. [Google Scholar] [CrossRef]
- Chuong, C.M.; Bhat, R.; Widelitz, R.B.; Bissell, M.J. SnapShot: Branching Morphogenesis. Cell 2014, 158, 1212. [Google Scholar] [CrossRef] [PubMed]
- Chuong, C.M.; Randall, V.A.; Widelitz, R.B.; Wu, P.; Jiang, T.X. Physiological regeneration of skin appendages and implications for regenerative medicine. Physiology (Bethesda) 2012, 27, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.F.; Wu, W.; Xu, H. Investigation of feather follicle development in embryonic geese. Poult. Sci. 2007, 86, 2000–2007. [Google Scholar] [CrossRef] [PubMed]
- Andl, T.; Reddy, S.T.; Gaddapara, T.; Millar, S.E. WNT signals are required for the initiation of hair follicle development. Dev. Cell 2002, 2, 643–653. [Google Scholar] [CrossRef]
- Sun, L.; Bai, M.; Xiang, L.; Zhang, G.; Ma, W.; Jiang, H. Comparative transcriptome profiling of longissimus muscle tissues from Qianhua Mutton Merino and Small Tail Han sheep. Sci. Rep. 2016, 6, 33586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobert, O. Gene regulation by transcription factors and microRNAs. Science 2008, 319, 1785–1786. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.S.; Chen, C.K.; Fan, W.L.; Wu, P.; Wu, S.M.; Chen, J.J.; Lai, Y.T.; Mao, C.T.; Lu, M.Y.J.; Chen, D.R.; et al. Transcriptomic analyses of regenerating adult feathers in chicken. BMC Genom. 2015, 16, 756. [Google Scholar] [CrossRef] [PubMed]
- Chuong, C.M.; Chodankar, R.; Widelitz, R.B.; Jiang, T.X. Evo-devo of feathers and scales: Building complex epithelial appendages. Curr. Opin. Genet. Dev. 2000, 10, 449–456. [Google Scholar] [CrossRef]
- Dhouailly, D. A new scenario for the evolutionary origin of hair, feather, and avian scales. J. Anat. 2009, 214, 587–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prin, F.; Dhouailly, D. How and when the regional competence of chick epidermis is established: Feathers vs. scutate and reticulate scales, a problem en route to a solution. Int. J. Dev. Biol. 2004, 48, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Qu, Y.; Huang, Y.; Lei, F. Dynamic transcriptome profiling towards understanding the morphogenesis and development of diverse feather in domestic duck. BMC Genom. 2018, 19, 391. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Theurkauf, W.E.; Weng, Z.; Zamore, P.D. Strand-specific libraries for high throughput RNA sequencing (RNA-Seq) prepared without poly (A) selection. Silence 2012, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Góngora-Castillo, E.; Buell, C.R. Bioinformatics challenges in de novo transcriptome assembly using short read sequences in the absence of a reference genome sequence. Nat. Prod. Rep. 2013, 30, 490–500. [Google Scholar] [CrossRef] [PubMed]
- Szpecht, D.; Wiak, K.; Braszak, A.; Szymankiewicz, M.; Gadzinowski, J. Role of selected cytokines in the etiopathogenesis of intraventricular hemorrhage in preterm newborns. Childs Nerv. Syst. 2016, 32, 2097–2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fietta, P.; Costa, E.; Delsante, G. Interleukins (ILs), a fascinating family of cytokines. Part I: ILs from IL-1 to IL-19. Theor. Biol. Forum 2014, 107, 13–45. [Google Scholar] [PubMed]
- Chen, C.F.; Foley, J.; Tang, P.C.; Li, A.; Jiang, T.X.; Wu, P.; Widelitz, R.B.; Chuong, C.M. Development, regeneration, and evolution of feathers. Annu. Rev. Anim. Biosci. 2015, 3, 169–195. [Google Scholar] [CrossRef] [PubMed]
- Wake, K.; Sasaki, K.; Watanabe, S. Conductivities of epidermis, dermis, and subcutaneous tissue at intermediate frequencies. Phys. Med. Biol. 2016, 61, 4376. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Coleman, D.J.; Bajaj, G.; Liang, X.; Ganguli-Indra, G.; Indra, A.K. RXRα ablation in epidermal keratinocytes enhances UVR-induced DNA damage, apoptosis, and proliferation of keratinocytes and melanocytes. J. Investig. Dermatol. 2011, 131, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Kandhare, A.D.; Alam, J.; Patil, M.V.K.; Sinha, A.; Bodhankar, S.L. Wound healing potential of naringin ointment formulation via regulating the expression of inflammatory, apoptotic and growth mediators in experimental rats. Pharm. Biol. 2016, 54, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Blumberg, H.; Conklin, D.; Xu, W.F.; Grossmann, A.; Brender, T.; Carollo, S.; Eagan, M.; Foster, D.; Haldeman, B.A.; Hammond, A. Interleukin 20: Discovery, receptor identification, and role in epidermal function. Cell 2001, 104, 9–19. [Google Scholar] [CrossRef]
- Sa, S.M.; Valdez, P.A.; Wu, J.; Jung, K.; Zhong, F.; Hall, L.; Kasman, I.; Winer, J.; Modrusan, Z.; Danilenko, D.M. The effects of IL-20 subfamily cytokines on reconstituted human epidermis suggest potential roles in cutaneous innate defense and pathogenic adaptive immunity in psoriasis. J. Immunol. 2007, 178, 2229. [Google Scholar] [CrossRef] [PubMed]
- Walker, I.D.; Rogers, G.E. Differentiation in Avian Keratinocytes: The Properties of the Proteins of the Chick Down Feather. Eur. J. Biochem. 1976, 69, 329–339. [Google Scholar] [CrossRef]
- Kingo, K.; Mössner, R.; Rätsep, R.; Raud, K.; Krüger, U.; Silm, H.; Vasar, E.; Reich, K.; Kõks, S. Association analysis of IL20RA and IL20RB genes in psoriasis. Genes Immun. 2008, 9, 445–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, K.C. Successfully mapping novel asthma loci by GWAS. Lancet 2011, 378, 967–968. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Jiang, Q.; Hao, H.; Ju, Z.; Yang, C.; Sun, Y.; Wang, C.; Zhong, J.; Huang, J.; et al. DNA methylation rather than single nucleotide polymorphisms regulates the production of an aberrant splice variant of IL6R in mastitic cows. Cell Stress Chaperones 2018, 23, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.C.; Freitag, D.F.; Cutler, A.J.; Howson, J.M.M.; Rainbow, D.B.; Smyth, D.J.; Kaptoge, S.; Clarke, P.; Boreham, C.; Coulson, R.M.; et al. Functional IL6R 358Ala allele impairs classical IL-6 receptor signaling and influences risk of diverse inflammatory diseases. PLoS Genet. 2013, 9, e1003444. [Google Scholar] [CrossRef] [PubMed]
- Correia-Álvarez, E.; Gómez, E.; Martín, D.; Carrocera, S.; Pérez, S.; Otero, J.; Peynot, N.; Giraud-Delville, C.; Caamaño, J.N.; Sandra, O.; et al. Expression and localization of interleukin 1 beta and interleukin 1 receptor (type I) in the bovine endometrium and embryo. J. Reprod. Immunol. 2015, 110, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Boraschi, D.; Italiani, P.; Weil, S.; Martin, M.U. The family of the interleukin-1 receptors. Immunol. Rev. 2018, 281, 197–232. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.L.; Zhang, G.R.; Zhu, D.M.; Ji, W.; Shi, Z.C.; Jiang, R.; Fan, Q.X.; Wei, K.J. Molecular cloning and expression analysis of interleukin-1β and interleukin-1 receptor type I genes in yellow catfish (Pelteobagrus fulvidraco): Responses to challenge of Edwardsiella ictaluri. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2018, 223, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.; Foitzik, K.; Eichmüller, S.; Handjiski, B.; Czarnetzki, B.M.; Paus, R. IL-1 receptor, TGF-β receptor, and alkaline phosphatase activity are markers of early murine hair follicle development. J. Investig. Dermatol. 1995, 105, 494. [Google Scholar]
- Bai, M.; Sun, L.; Zhao, J.; Xiang, L.; Cheng, X.; Li, J.; Jia, C.; Jiang, H. Histological analysis and identification of spermatogenesis-related genes in 2-, 6-, and 12-month-old sheep testes. Sci. Nat. 2017, 104, 84. [Google Scholar] [CrossRef] [PubMed]
- Chu, Q.; Cai, L.; Fu, Y.; Chen, X.; Yan, Z.; Lin, X.; Zhou, G.; Han, H.; Widelitz, R.B.; Chuong, C.M.; et al. Dkk2/Frzb in the dermal papillae regulates feather regeneration. Dev. Biol. 2014, 387, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuda, Y.; Tanaka, M.; Yagi, T.; Tashiro, K. The defect of SFRP2 modulates an influx of extracellular calcium in B lymphocytes. BMC Res. Notes 2014, 7, 780. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, J.; Burgeson, R.E.; Morgan, B.A. Wnt signaling maintains the hair-inducing activity of the dermal papilla. Genes Dev. 2000, 14, 1181–1185. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Jiang, T.X.; Widelitz, R.B.; Chuong, C.M. Wnt3a gradient converts radial to bilateral feather symmetry via topological arrangement of epithelia. Proc. Natl. Acad. Sci. USA 2006, 103, 951–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.; Xiao, Y.; Wen, R.; Zhang, Y.; Li, X.; Wang, H.; Huang, J.; Liu, J.; Long, T.; Tang, J. Promoting roles of the secreted frizzled-related protein 2 as a Wnt agonist in lung cancer cells. Oncol. Rep. 2015, 34, 2259–2266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Techavichit, P.; Yang, G.; Kurenbekova, L.; Shuck, R.; Donehower, L.A.; Yustein, J.T. Secreted Frizzled-Related Protein 2 (sFRP2) promotes osteosarcoma invasion and metastatic potential. BMC Cancer 2016, 16, 869. [Google Scholar] [CrossRef] [PubMed]
- Kwack, M.H.; Ahn, J.S.; Jang, J.H.; Kim, J.C.; Sung, Y.K.; Kim, M.K. SFRP2 augments Wnt/β-catenin signalling in cultured dermal papilla cells. Exp. Dermatol. 2016, 25, 813–815. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.K.; Kim, Y.S. Expression of sfrp2 is increased in catagen of hair follicles and inhibits keratinocyte proliferation. Ann. Dermatol. 2014, 26, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Fessing, M.Y.; Atoyan, R.; Shander, B.; Mardaryev, A.N.; Botchkarev, V.V.; Poterlowicz, K.; Peng, Y.; Efimova, T.; Botchkarev, V.A. BMP signaling induces cell-type-specific changes in gene expression programs of human keratinocytes and fibroblasts. J. Investig. Dermatol. 2010, 130, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; Yadav, P.S.; Prashar, P. BMP signaling in development and diseases: A pharmacological perspective. Biochem. Pharmacol. 2013, 85, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tang, X.; Wang, W.; Yuan, Z.; Bai, Y. Spatiotemporal expression of BMP7 in the development of anorectal malformations in fetal rats. Int. J. Clin. Exp. Pathol. 2015, 8, 3727–3734. [Google Scholar] [PubMed]
- Li, R.X.; Yiu, W.H.; Wu, H.J.; Wong, W.L.; Chan, L.Y.; Lin, M.; Leung, C.K.; Lai, K.N.; Tang, C.W. BMP7 reduces inflammation and oxidative stress in diabetic tubulopathy. Clin. Sci. 2015, 128, 269–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, M.P.; Linkhart, B.L.; Fallon, J.F. Bmp7 mediates early signaling events during induction of chick epidermal organs. Dev. Dyn. 2004, 231, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, K.; Uemura, K.; Asada, M.; Maesako, M.; Akiyama, H.; Shimohama, S.; Takahashi, R.; Kinoshita, A. The participation of insulin-like growth factor-binding protein 3 released by astrocytes in the pathology of Alzheimer’s disease. Mol. Brain. 2015, 8, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzec, K.A.; Lin, M.Z.; Martin, J.L.; Baxter, R.C. Involvement of p53 in insulin-like growth factor binding protein-3 regulation in the breast cancer cell response to DNA damage. Oncotarget 2015, 6, 26583–26598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weger, N.; Schlake, T. Igfbp3 Modulates Cell Proliferation in the Hair Follicle. J. Investig. Dermatol. 2005, 125, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Rosso, F.; Giordano, A.; Barbarisi, M.; Barbarisi, A. From Cell–ECM interactions to tissue engineering. J. Cell. Physiol. 2004, 199, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.E.; Bundred, N.J.; Landberg, G.; Clarke, R.B.; Farnie, G. Focal adhesion kinase and Wnt signaling regulate human ductal carcinoma in situ stem cell activity and response to radiotherapy. Stem Cells 2015, 33, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Xu, T.; Yuan, J.; Guo, X.; Liu, D. Transcriptome Sequencing Reveals Differences between Primary and Secondary Hair Follicle-derived Dermal Papilla Cells of the Cashmere Goat (Capra hircus). PLoS ONE 2013, 8, e76282. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, X.; Yan, H.; Zeng, J.; Ma, S.; Niu, Y.; Zhou, G.; Jiang, Y.; Chen, Y. Comparative Transcriptome Analysis of Fetal Skin Reveals Key Genes Related to Hair Follicle Morphogenesis in Cashmere Goats. PLoS ONE 2016, 11, e0151118. [Google Scholar] [CrossRef] [PubMed]
- Alho, H.; Vaalasti, A.; Podkletnova, I.; Rechardt, L. Expression of diazepam-binding inhibitor peptide in human skin: An immunohistochemical and ultrastructural study. J. Investig. Dermatol. 1993, 101, 800–803. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Ge, K.; Wang, M.; Zhang, C.; Geng, Z. Integrative analysis of the Pekin duck (Anas anas) MicroRNAome during feather follicle development. BMC Dev. Biol. 2017, 17, 12. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Jiang, X.; Pan, Y.; Chen, L.; Zhang, L.; Zhu, K.; Cai, Y.; Ling, Y.; Chen, F.; Xu, X.; et al. Transcriptomic analysis of hepatic responses to testosterone deficiency in miniature pigs fed a high-cholesterol diet. BMC Genom. 2015, 16, 59. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Kim, H.; Zhong, S.; Chen, H.; Hu, Z.; Zhou, B. De novo transcriptome assembly for rudimentary leaves in Litchi chinesis Sonn. And identification of differentially expressed genes in response to reactive oxygen species. BMC Genom. 2014, 15, 805. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Before Filter Reads Number | After Filter Reads Number (%) | Reads Len | GC | Adapter (%) | Low Quality (%) |
|---|---|---|---|---|---|---|
| E13-1 | 51,704,342 | 50,591,630 (97.85%) | 150 | 57.45% | 237,982 (0.46%) | 869,940 (1.68%) |
| E13-2 | 53,263,218 | 52,209,704 (98.02%) | 150 | 56.54% | 226,048 (0.42%) | 822,340 (1.54%) |
| E13-3 | 52,416,882 | 51,381,802 (98.03%) | 150 | 56.84% | 227,124 (0.43%) | 803,064 (1.53%) |
| E18-1 | 51,066,402 | 50,087,188 (98.08%) | 150 | 56.80% | 212,366 (0.42%) | 762,056 (1.49%) |
| E18-2 | 55,460,086 | 54,329,966 (97.96%) | 150 | 56.78% | 255,134 (0.46%) | 869,672 (1.57%) |
| E18-3 | 50,868,330 | 49,791,358 (97.88%) | 150 | 57.04% | 231,472 (0.46%) | 840,530 (1.65%) |
| E28-1 | 49,794,350 | 48,680,268 (97.76%) | 150 | 56.43% | 221,302 (0.44%) | 888,292 (1.78%) |
| E28-2 | 48,238,510 | 47,164,092 (97.77%) | 150 | 57.06% | 234,242 (0.49%) | 835,516 (1.73%) |
| E28-3 | 72,214,054 | 69,719,918 (96.55%) | 150 | 56.56% | 295,872 (0.41%) | 2,192,284 (3.04%) |
| Total | 485,026,174 | 473,955,926(97.72%) | 2,141,542 (0.44%) | 8,883,694 (1.83%) |
| Gene_ID | Gene_Symbol | RPKM 1-E13 | RPKM-E18 | RPKM-E28 |
|---|---|---|---|---|
| Unigene0004079 | IL16 | 0.313533 | 0.810267 | 1.96877 |
| Unigene0004463 | IL20RA | 9.75107 | 3.5371 | 1.67913 |
| Unigene0009028 | IL36RN | 0.7632 | 3.4918 | 12.3211 |
| Unigene0029291 | IL17D | 0.470633 | 1.89013 | 2.22343 |
| Unigene0033163 | ILDR2 | 4.27267 | 10.8527 | 10.6716 |
| Unigene0036855 | Il15ra | 1.66583 | 3.69263 | 4.09673 |
| Unigene0008165 | IL7R | 0.001 | 0.0513667 | 1.09823 |
| Unigene0016840 | IL17C | 3.03347 | 1.74543 | 0.0239 |
| Unigene0022008 | IL31RA | 0.230633 | 0.4919 | 1.37237 |
| Unigene0022142 | Il34 | 0.631367 | 0.951633 | 2.5645 |
| Unigene0025806 | IL16 | 0.248633 | 0.353767 | 1.08887 |
| Unigene0028720 | IL22RA1 | 2.47243 | 1.55193 | 0.228967 |
| Unigene0028801 | IL1RAP | 16.0848 | 16.0816 | 6.3824 |
| Unigene0030418 | Il1rl1 | 0.0184333 | 0.132833 | 0.776233 |
| Unigene0030419 | Il1rl1 | 0.2798 | 0.466433 | 0.1425 |
| Unigene0031991 | IL13RA2 | 0.934333 | 0.863233 | 8.49547 |
| Unigene0036009 | IL12RB2 | 1.8168 | 1.71347 | 0.647733 |
| Unigene0038243 | IL1R1 | 1.13113 | 2.188 | 6.5063 |
| Unigene0041147 | Il15ra | 0.0634 | 0.0751667 | 1.1632 |
| Unigene0001585 | IL1RN | 9.347 | 18.6492 | 24.2625 |
| Unigene0008166 | IL7R | 0.001 | 0.145 | 0.9396 |
| Unigene0017728 | IL10RB | 0.511767 | 1.11633 | 1.19017 |
| Unigene0022006 | Il31ra | 0.0695667 | 0.252933 | 1.08303 |
| Unigene0027228 | IL1RL2 | 1.91883 | 1.12357 | 0.711267 |
| Unigene0028870 | IL11RA | 21.1164 | 30.1148 | 50.5107 |
| Unigene0030116 | IL17RC | 14.7142 | 21.909 | 40.9831 |
| Unigene0030288 | IL2RG | 6.4065 | 11.2284 | 16.348 |
| Unigene0031047 | IL6R | 14.2714 | 20.6134 | 39.1989 |
| Unigene0052404 | IL16 | 0.100367 | 0.320633 | 0.8613 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Sello, C.T.; Sun, Y.; Zhou, Y.; Lu, H.; Sui, Y.; Hu, J.; Xu, C.; Sun, Y.; Liu, J.; et al. De Novo Transcriptome Sequencing Analysis of Goose (Anser anser) Embryonic Skin and the Identification of Genes Related to Feather Follicle Morphogenesis at Three Stages of Development. Int. J. Mol. Sci. 2018, 19, 3170. https://doi.org/10.3390/ijms19103170
Liu C, Sello CT, Sun Y, Zhou Y, Lu H, Sui Y, Hu J, Xu C, Sun Y, Liu J, et al. De Novo Transcriptome Sequencing Analysis of Goose (Anser anser) Embryonic Skin and the Identification of Genes Related to Feather Follicle Morphogenesis at Three Stages of Development. International Journal of Molecular Sciences. 2018; 19(10):3170. https://doi.org/10.3390/ijms19103170
Chicago/Turabian StyleLiu, Chang, Cornelius Tlotliso Sello, Yongfeng Sun, Yuxuan Zhou, Hongtao Lu, Yujian Sui, Jingtao Hu, Chenguang Xu, Yue Sun, Jing Liu, and et al. 2018. "De Novo Transcriptome Sequencing Analysis of Goose (Anser anser) Embryonic Skin and the Identification of Genes Related to Feather Follicle Morphogenesis at Three Stages of Development" International Journal of Molecular Sciences 19, no. 10: 3170. https://doi.org/10.3390/ijms19103170