Receptor-Targeted Glial Brain Tumor Therapies

Brain Tumor Center of Excellence, Department of Cancer Biology, Wake Forest University School of Medicine, Comprehensive Cancer Center of Wake Forest Baptist Medical Center, 1 Medical Center Boulevard, Winston-Salem, NC 27157, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(11), 3326; https://doi.org/10.3390/ijms19113326
Submission received: 25 September 2018
/
Revised: 16 October 2018
/
Accepted: 19 October 2018
/
Published: 25 October 2018
(This article belongs to the Special Issue Receptor-Targeted Cancer Therapy)
Abstract
:Among primary brain tumors, malignant gliomas are notably difficult to manage. The higher-grade tumors represent an unmet need in medicine. There have been extensive efforts to implement receptor-targeted therapeutic approaches directed against gliomas. These approaches include immunotherapies, such as vaccines, adoptive immunotherapy, and passive immunotherapy. Targeted cytotoxic radio energy and pro-drug activation have been designed specifically for brain tumors. The field of targeting through receptors progressed significantly with the discovery of an interleukin 13 receptor alpha 2 (IL-13RA2) as a tumor-associated receptor over-expressed in most patients with glioblastoma (GBM) but not in normal brain. IL-13RA2 has been exploited in novel experimental therapies with very encouraging clinical responses. Other receptors are specifically over-expressed in many patients with GBM, such as EphA2 and EphA3 receptors, among others. These findings are important in view of the heterogeneity of GBM tumors and multiple tumor compartments responsible for tumor progression and resistance to therapies. The combined targeting of multiple receptors in different tumor compartments should be a preferred way to design novel receptor-targeted therapeutic approaches in gliomas.
1. Glial Tumors
In the nineteenth century, glial cells were perceived to hold the central nervous system (CNS) together. Hence, the word ‘glia’ arises from the Greek word, ‘glue’ [1]. Glial cells are supportive cells of the central nervous system. Gliomas arise when these supportive glial cells (or neuronal stem cells) [2,3,4,5] become malignant. The severity of malignancy is categorized in terms of grades. Several pathological features, such as abnormal appearance of the nucleus (nuclear atypia), necrosis, microvascular proliferation, and hemorrhages determine the extent of tumor aggressiveness and prognosis [6,7]. Lower grade (I–II) gliomas (LGG) include ependymomas, plilocytic astocytomas, angiocentric gliomas, papillary glioneural tumors, pituicytomas, choroid plexus carcinomas and rosette-forming glioneural tumors of the fourth ventricle. The latter exhibit more benign features and are associated with a better prognosis [8,9,10].
Unlike higher-grade gliomas, LGG do not usually show contrast enhancement after gadolinium injection on magnetic resonance imaging (MRI) scans; they also have ill-defined margins and appear heterogeneous on radiographic scans [6,8,10,11]. Their various morphologic patterns may not always coincide with specific clinical outcomes because their rate of growth varies. Some defined features of LGG include elongated cells, honeycomb patterns with absence of perinuclear staining, absence of mitotic figures, presence of glial fibrillary acidic protein (GFAP) and calcification, mutations in the isocitrate dehydrogenase gene (IDH1) [12], layers of tumor cells surrounding blood vessels forming pseudo-papillary structures and presence of perivascular pseudo-rosettes [6,8,13]. In over 70% of cases, IDH1 mutations are caused by a single nucleotide substitution at the 132th amino acid from arginine (R) to histidine (H). LGG with IDH1 mutations without co-deletion of 1p and 19q carry a relatively favorable prognosis. However, LGG without IDH1 mutations are associated with the worst prognoses [14].
On the other hand, higher-grade (III-IV) gliomas exhibit more malignant features. Grade III gliomas include anaplastic gliomas [15], whereas Grade IV glioma is represented by glioblastoma (GBM) [16,17]. GBM is characterized by vascular proliferation, necrosis, pseudopalisading features [18], mitoses, and cellular and nuclear atypia [19].
GBM is also characterized by distinct molecular signatures and genetic mutations. Chromosome deletions of 1p and 19q, co-deletions of 1p and 19q [20,21] and mutations in telomerase reverse transcriptase (TERT) [22] are among those present in GBM. While single deletions of 1p and 19q represent an astrocytic lineage, co-deletions of 1p and 19q indicate an oligodendrocytic lineage and respond best to alkylating agent-based radiation therapy or chemotherapy like temozolomide [6]. IDH1 mutations are also present in a small group of tumors called secondary GBM. IDH1 mutations are associated with longer overall survival and progression-free survival among patients with GBM [23].
The Cancer Genome Atlas project first analyzed genomic changes in GBM. There are four major subtypes based on the assessment of mutational changes in 601 genes in GBM: (i) the proneural subtype, which occurs in younger patients with GBM, shows an oligodendrocytic lineage and is characterized by enhanced mutations in tumor protein 53 (TP53) and IDH1 genes. (ii) The neural subtype, which occurs in older patients with GBM, has an astrocytic lineage without particularly high or low rates of mutation within specific genes. (iii) The classical subtype is characterized by lack of TP53 mutations and enhanced expression of epidermal growth factor receptor (EGFR). (iv) The mesenchymal subtype is characterized by mutations in neurofibromin 1, phosphatase and tensin homolog (PTEN) and TP53 genes, and shows an astroglial lineage [23,24,25]. Even though this division was proposed, it appears that nearly every GBM tumor exhibits the features of various subtypes, further indicating high levels of intra- and intertumoral heterogeneity [26].
Proposed in 2010, the Response Assessment in Neuro-Oncology criteria (RANO) is the standard method to assess the response to treatments in GBM. Assessment of treatments is largely based on contrast enhancing computerized axial tomography (CT) or MRI images. The RANO criteria are an improvement over the previously used Macdonald criteria, because they incorporate LGG or smaller tumor components of GBM. The user variability in CT and MRI scans, and evaluation of pseudo-progression after GBM treatment are also lesser concerns with the RANO criteria [27].
GBM accounts for 15% of all primary brain tumors, and represents 56% of all gliomas. About 12,670 new cases of GBM are expected to be diagnosed in 2018 in the U.S. alone [28]. Primary treatment for GBM includes surgical resection of the tumor, chemo therapy and radiation therapy often provided in combination. As a standard therapy, an alkylating agent like temozolomide is given during and after radiation therapy, especially to patients with promoter methylation of the DNA-damage repair protein O6 methylguanine-DNA methyltransferase (MGMT) [29].
Some major challenges associated with GBM treatment include poor penetration of drugs across the blood-brain barrier and the tumor; location of the tumor; infiltrative tumor cells migrating away from the primary tumor into normal brain parenchyma; resistance to chemotherapy and radiation therapy; faulty but extremely well-developed neovasculature; and molecular, genetic and morphological heterogeneity [30,31,32]. While a complete surgical resection of tumors is difficult due to tumor infiltration and proximity to critical motor/sensory nerves, molecular resection via selective targeting of glioma cells might be possible.
Despite relentless research and clinical efforts, over the last eight decades, only one month per decade has been added to survival rates among patients with GBM [32,33]. In particular, the median survival of GBM patients treated with radiation therapy and surgery in 2000–2003 was 12 months, which increased to 14.2 months in 2005–2008 likely due to addition of temozolamide [34]. Today, the median survival of patients with GBM is 15–16 months for those who receive surgery, radiation therapy, chemotherapy, and tumor-treating fields (TTFields) [35]. In this review article (Box 1), we discuss targeted therapies against gliomas with a particular focus on receptors.
Box 1. An overview of the review paper.
- Glial Tumors Overview
- ○
- Glial tumors are malignancies of supportive glial cells.
- ○
- Low Grade Gliomas (LGG) overview: morphological, radiographic, molecular, and genetic characteristics
- ○
- High Grade Gliomas (HGG) overview: morphological, radiographic, molecular, and genetic characteristics
- ○
- Four major subtypes of GBM: proneural, neural, classical, and mesenchymal
- ○
- Response Assessment in Neuro-Oncology (RANO) as a standard method used to assess treatment responses.
- ○
- Major challenges in treatment of GBM: poor penetration of drugs, blood-brain barrier, location of the tumor, infiltrative nature of cells, heterogeneity, and resistance to therapy
- Targeted Therapies for Glioma
- ○
- There is a need to specifically target glioma cells and spare normal cells.
- ○
- Tumor Specific Antigens/ Tumor Associated Antigens (TSA/TAA) are specific to tumor cells and can serve as excellent targets for therapy.
- ○
- Immunotherapy is the fourth pillar of cancer treatment in addition to surgery, chemotherapy and radiation therapy.
- Interleukin 13 Receptor Alpha 2 (IL-13RA2) in Glioma Treatment
- ○
- Overexpressed in up to 75% of patients, IL-13RA2 was discovered as a glioma-specific marker over two decades ago in the Debinski Laboratory.
- ○
- Over time, about 30 variations of targeted therapies have focused on IL-13RA2, including ICT 107 and IL-13-PE38QQR.
- ○
- IL-13RA2 belongs to cancer/testes antigen group.
- ○
- Over 200 cancer/testes antigens belonging to 70 different families have been identified so far.
- ○
- Other cancer/testes antigens found in Glioma: ACTL8, XAGE3, BORIS, OIP5, DDX43, CTAGE1, KNL1, MAGE-A12, BAGE1
- ○
- IL-13RA2 targeting:
- ○
- IL-13 ligand can target IL-13RA2.
- ○
- IL-13 based fusion/conjugated proteins, nanoparticles, liposomes, viruses, IL-13 conjugated radiotherapy agents, antibodies, peptide-pulsed dendritic cells, and CAR T-cells are used to target IL-13RA2.
- ○
- IL-13PE38QQR, or Cintredekin Besudotox (CB) is one of the most potent recombinant cytotoxin in preclinical and clinical trials to treat glioma.
- ○
- Optimizing the ligands for therapies:
- ○
- Mutations in amino acid positions 13, 66, 69 and 105 can enhance the specificity of IL-13 to the IL-13RA2.
- ○
- Addition of Pseudomonas exotoxin domain 2 (D2) and a nuclear localization signal (NLS) to IL-13 can directly transport IL-13 based cytotoxins to the nucleus of the glioma cells.
- ○
- IL-13 based drug delivery:
- ○
- IL-13 directed viruses: modified adeno and lentiviruses expressing IL-13 include LU-13 and MV Hcdelta18-IL-13.
- ○
- IL-13RA2 targeted radiotherapy agents: copper-64 radiolabeled Pep-1L and alpha particle emitter, actinium-225 radiolabeled Pep-1L have been used to target IL-13RA2.
- ○
- Peptide vaccines: multiple TSA/TAA including IL-13RA2, EGFRvIII, AIM-2, gp-100, HER-2, MAGE-1, EphA2, and survivin have been targeted using peptide vaccines.
- ○
- Dendritic cell-based vaccines: multiple TSA/TAA including IL-13RA2, EphA2, Wilms tumor 1 have been targeted using multiple antigen pulsed dendritic cells.
- ○
- CAR T-cell therapy: both first- and second-generation CAR T-cells have successfully targeted IL-13RA2 including a Phase I clinical trial for GBM at The City of Hope.
- Compromising a vastly interconnected tumor ecosystem with a single therapeutic approach is a great challenge. Hence, multiple targeting of various facets of the tumor ecosystem can enhance therapeutic potential and provide significant clinical benefit.
- Eph/ephrin receptor system in gliomas
- ○
- EphA2, EphA3, and EphB3 are overexpressed in gliomas and have been extensively utilized for targeted therapy.
- EGFR/EGF receptor system in gliomas
- ○
- EGFR/EGF is one of the most studied receptor systems in brain tumors.
- ○
- EGFR is amplified in 40-60% patients and 30-50% EGFR amplifications contain EGFRvIII mutations.
- ○
- EGFR targeted therapies include: siRNA, small molecule inhibitors, antibodies, and CAR T-cells.
- Other receptors/targeted therapies
- ○
- Other receptors used to target glioma include: VEGFR, PDGFR, TGFR, FGFR, c-Met receptor, uPAR, GPCR, LDL-R, LRP1, and FOLR.
- Immune checkpoint inhibitors
- ○
- GBM cells consistently work to maintain and generate an immunosuppressive environment.
- ○
- While the use of immune checkpoint inhibitors shows great promise, its clinical benefits are yet to be established in glioma.
- Viral/genetic therapies
- ○
- Over 20 oncolytic virus candidates including reovirus, oncolytic measles, oncolytic herpes simplex virus, and vesicular stomatitis virus have been tested in clinical trials to treat GBM.
- ○
- While the use of viral therapy in glioma shows great promise, its consistent clinical benefits are yet to be established.
- Conclusions
- ○
- Effective treatment of malignant gliomas is a great challenge in oncology.
- ○
- Notable advancements in antigen-specific targeting, viral and genetic silencing, immune checkpoint inhibitors, peptide and dendritic cell vaccines, CAR T-cell therapy, chemotherapy and radiation therapy, in conjugation with effective drug delivery systems like CED offer promise to tackle this grave challenge in glioma therapy.
- ○
- Targeting multiple facets of the tumor ecosystem, personalized antigen screening, and using state of the art ex vivo technologies can enhance the clinical benefits of current and future treatment options.
2. Targeted Therapies for Gliomas
The discovery of tumor-specific and tumor-associated antigens (TSA and TAA, respectively) have permitted development of a new array of therapeutic methods that selectively target gliomas and spare non-diseased cells of the CNS [36,37]. Many of these antigens are plasma membrane receptors [38,39]. Strategies to target TSA/TAA involve using antibodies to block ligand/receptor interactions, antibodies conjugated with modified versions of proteinaceous toxins or cytotoxic drugs or labels and nanoparticles or liposomes loaded with drugs that selectively target glioma TSA/TAA [40,41]. Immune checkpoint inhibitors that interact with a subset of T cell receptors have resulted in significant clinical effects in almost 10 cancers [42,43], to date, but not in brain tumors. Lastly, advances in immunotherapy treatments that involve peptide vaccines [44], antigen pulsed dendritic cells [45], tumor-infiltrating lymphocytes, and chimeric antigen receptor T-cells (CAR T-cells) have shown promise, but their clinical benefits in gliomas remain to be seen [46]. The U.S. Food and Drug Administration (FDA) recently approved CAR T-cell therapy, which targets cluster of differentiation (CD) 19+ cells in acute lymphoblastic leukemia (ALL) [47]. This is seen as one of the most powerful treatment options in oncology and potentially also for gliomas (Figure 1). This approach is being tested extensively for the treatment of GBM, as discussed below.
Immunotherapy has been recognized as the fourth pillar of cancer treatment after surgery, chemotherapy and radiation therapy [48]. The FDA’s approval of ipilimumab (a monoclonal antibody that targets cytotoxic T-lymphocyte-associated protein 4 (CTL-4) in melanoma) and sipuleucel-T (a personalized immunotherapy treatment for prostate cancers) encourages further immunotherapy research in glioma. While the benefit of immunotherapy has been well established in melanoma and prostate cancers [14,49], it is still being evaluated for the treatment of GBM and other astrocytic tumors. For example, a peptide-based vaccine plus poly-ICLC for low-grade gliomas using four targets (IL-13RA2, EphA2 receptor, Wilms tumor 1 and survivin) was well tolerated in patients with grade II glioma [50]. Patients exhibited significant increases in interferon γ (IFN γ) following vaccination. The median survival of patients was 17 and 12 months, respectively, for two cohorts, and one patient had progression-free survival for 67 months.
Traditionally considered an immune privileged system, recent research has shown that immune cells can cross the blood-brain barrier (BBB) and reach the brain parenchyma [51]. In addition, brain cells can perform the function of immune cells like microglial cells [52,53]. This line of research has opened novel avenues for new immunotherapeutic treatment opportunities for GBM [54,55,56,57,58,59]. This approach is gaining wider acceptance as reflected in the recent National Institute of Health (NIH) Cancer Moonshot Blue Ribbon Panel. The Panel had 10 transformative research recommendations, including ‘Create a translational science network devoted exclusively to immunotherapy’ [60]. One of the major challenges of these therapies is to selectively target antigens specific to gliomas. Henceforth, we will elaborate on TSA/TAA targeted at gliomas, and specifically membrane receptors that have been exploited to target therapies in laboratory, preclinical, and clinical settings.
3. Interleukin 13 Receptor Alpha 2 (IL-13RA2) in Glioma Treatment
IL-13RA2 is one of the most thoroughly studied TAA in glioblastoma research. It was revealed as a glioma-specific marker over two decades ago in the Debinski laboratory, when a cytotoxic ligand-toxin fusion protein based on interleukin 13 (IL-13) demonstrated extreme potency against glioma cells (Figure 2) [61,62]. It is overexpressed in up to 75% of glioma patients [63]. Over the years, overexpression of IL-13RA2 in GBM has become a subject of intense interest by multiple laboratories, confirming IL-13RA2 as a powerful target for anti-glioma therapies. Over 30 variations of treatments focused on IL-13RA2 have been used to target and kill glioma cells in vitro, preclinical, and clinical settings [64]. CAR T-cells targeting IL-13RA2 are now being tested in a Phase I clinical trial [56].
One promising example is ICT-107, which is an autologous dendritic cell-based vaccine pulsed with six different antigens: IL-13RA2, melanoma associated antigen 1 (MAGE-1), tyrosine-related protein 2 (TRP-2), glycoprotein 100 (gp100), epidermal growth factor receptor (HER-2), and absent in melanoma 2 (AIM-2). However, a Phase III clinical trial of ICT-107 (NCT02546102) was suspended by ImmunoCellular due to the lack of funding to complete the trial. Nonetheless, progression-free survival was significantly improved in the Phase II trial.
The completion of a Phase III trial for IL-13-PE38QQR, the first generation of IL-13-based cytotoxins, showed good tolerance of the drug candidate with promising effects [65]. Unfortunately, the trial sought to extend the survival of patients with recurrent GBM by 50% compared to historical controls. Virtually no drug in oncology has shown such activity. Hence, not surprisingly, the trial did not achieve the company’s chosen clinical endpoint. Nevertheless, the survival rate achieved in patients with recurrent GBM in this trial has not been improved or matched since.
IL-13RA2 is coded by the X chromosome, and is composed of 380 amino acids with a 17 amino acid intracellular domain and a 26 amino acid signaling sequence [64]. Gene expression of IL-13RA2 in normal tissues is low to undetectable, but high in testis. Protein expression of IL-13RA2 is high in testis and placenta, but very low or undetectable in other organs. Male reproductive cells show characteristics that are advantageous to tumor cells, such as proliferation, migration, and evading immune responses. Hence, a number of antigens are commonly expressed in cancer cells and testes. Like IL-13RA2, an array of antigens described as the cancer/testis antigen group are commonly found in cancer and testis [66]. They are broadly classified into two groups, one coded by the X chromosome and the other coded by autosomes. MAGE-1 was the first of this group to be discovered, over 25 years ago [67]. Other cancer/testis antigens coded by the X chromosome include synovial sarcoma X (SSX), cancer/testis antigen 45 (CT45), and G-antigen (GAGE), whereas those coded by autosomes include B melanoma antigen (BAGE), sperm protein (SP17) and helicase antigen (HAGE). To date, over 200 cancer/testis antigens have been identified among 70 different families [66,68].
Some cancer/testis antigens overexpressed in glioma include actin like protein 8 (ACTL8), X-antigen family member 3 (XAGE3), brother of the regulator of imprinted sites (BORIS), and opa-interacting protein 5 (OIP5) [69]. Glioma cell lines derived from patients with glioma also exhibit overexpression of cancer/testis antigens, especially IL-13RA2, dead-box helicase 43 (DDX43), cutaneous T-cell lymphoma associated antigen 1 (CTAGE1), kinetochore scaffold 1 (KNL1), MAGE-A12 and BAGE l [70]. Higher expression of these antigens was associated with worse prognosis in patients. Some of the hypotheses that possibly explain the advantage of amplified cancer/testis genes in tumor cells are discussed below.
One hypothesis is that glioma cells evade immunosuppression via the overexpression of IL-13RA2. Interleukin 13 (IL-13) is primarily produced by T helper 2 (TH2) cells, and also by CD8+ T cells after activation, mast cells, basophils, and eosinophils [71]. It is believed to inhibit pro-inflammatory cytokines and play a role in anti-inflammatory responses. Overexpression of IL-13 has also been associated with immunosuppression [72] and low serum levels of IL-13 have been associated with poor overall survival in colorectal cancer [73]. IL-13RA2 is highly overexpressed in GBM, with up to 30,000 binding sites per cultured GBM cells [61]. The GBM cell line G48a expresses about 4,000,000 binding sites for IL-13RA2 per cell [74,75,76,77,78,79,80]. IL-13 binds to IL-13 receptor A1 (IL-13RA1), forms a more stable heterodimer with IL-4 receptor A (IL-4RA), and then activates downstream signaling pathways [62,74,81]. IL-13 binds more readily to IL-13RA2 than to IL-13RA1 to form a heterodimer with IL-4RA, making IL-13RA2 an IL-13 sequestering receptor in the GBM microenvironment. Hence, by sequestering the IL-13 available in the tumor microenvironment, GBM cells could be evading immunosuppression.
Another hypothesis is that by sequestering IL-13, GBM cells can escape apoptosis that ordinarily would have been activated by the signal transducer and activator of transcription 6 (STAT6) pathway as an aftermath of IL-13 binding to IL-13RA1/IL-4RA, which is also expressed on tumor cells to some extent. Hence, by taking IL-13 away from the system, GBM tumor cells could avoid apoptosis [82]. IL-13RA2 expression is also correlated with higher glioma grades and cells exhibiting a mesenchymal phenotype in glioma [83]. Eliminating IL-13RA2 positive cells can make tumors less tumorigenic and invasive [84]. Hence, the advantages of targeting IL-13RA2 seem to be threefold: (a) effective and selective targeting of glioma cells sparing normal brain tissue, as normal brain cells do not express IL-13RA2; (b) making IL-13 available for activating the heterodimeric receptor shared with IL-4 by avoiding sequestration; and (c) eliminating the more invasive mesenchymal components within the tumor population.
3.1. IL-13RA2 Targeting
IL-13RA2 targeting agents are represented by IL-13-based fusion/conjugated proteins (e.g., to bacterial toxins), nanoparticles and liposomes, viruses, IL-13 conjugated radiotherapy agents, antibodies (e.g., conjugated to cytotoxic drugs, and unconjugated), peptide pulsed dendritic cells or receptor peptides, and CAR T-cells against IL-13RA2. These can be broadly categorized as types of active, passive, or adoptive immunotherapy. Active immunotherapy entails the arming of immune system to initiate long-term memory by allowing the immune system to fight tumor cells long after the administration of the therapy [85,86]. Passive immunotherapy on the other hand, is the administration of immunotherapeutic agents that are based on the elements of the immune system, temporary, but still could instill a long term memory under certain conditions [87]. Adoptive immunotherapy or adoptive cell transfer (ACT) involves the isolation of T-cells from patients. These T-cells are genetically modified to express TSA or TAA protein-binding moieties. Chemotherapy can then be administered to weaken the existing tumor environment. Finally, the genetically modified T-cells are introduced into a patient so that the engineered T-cells can selectively home and destroy the tumor cells of interest by recognizing TSA/TAA [88].
Recombinant cytotoxins are targeted to GBM as fusions/conjugates to an antibody or a ligand specific to a receptor. Upon ligand/receptor interaction, the toxin/drug is internalized only by tumor cells or elements of the tumor microenvironment expressing the given receptor. The first use of bacterial toxin to treat cancer was reported by Dr. William B. Coley in the 1890s to treat sarcoma [89]. However, due to inconsistent results, it was overtaken by more reliable therapies like chemo- and radiation therapies. One of the most potent recombinant cytotoxins in preclinical and clinical trials to treat glioma has been IL-13PE38QQR. This cytotoxin also led to the discovery of the IL-13RA2 overexpression in gliomas. This recombinant cytotoxin is based on wild-type IL-13 ligand, which binds both IL-13RA1/IL-4A and IL-13RA2 receptors, fused to a form of modified Pseudomonas exotoxin A (PE), PE38QQR [90]. The anti-tumor activity of IL-13-PE38QQR was further confirmed with intratumoral injections in animal models of GBM [91].
In commercial clinical development, IL-13-PE38QQR was called cintredekin besudotox (CB) [92]. CB was delivered by convection-enhanced delivery (CED) and was well tolerated by patients in Phase I/II clinical trials (maximum tolerated concentration 0.5 µg/mL). CB was infused at 0.75 mL/h divided by the number of catheters (up to four) for 96 h. Patients were followed for 30 days after treatment before the next set of patients was enrolled in the study. Some side effects included seizures, numbness, headaches, confusion, fatigue, repeated and uncontrolled eye movements, and gait changes. One of the severe side effects included hemiparesis [93].
In another study of 22 patients with newly diagnosed high-grade glioma, CB was also well tolerated. Only one patient exhibited a dose-limiting grade 4 seizure, and another experienced dose-limiting grade 3 confusion and aphasia, but no patients had grade 3 or 4 hematologic toxicities [94]. Overall, the best results were obtained when catheter positioning was monitored during CB infusion by MRI [93]. When the drug was delivered via more than two catheters in a MRI-guided setting, outcomes were improved [65,93,95]. The median survival of patients with more than two optimally placed catheters was 55.6 weeks, whereas that of patients with less than two optimally placed catheters was 37.4 weeks. Furthermore, suboptimally placed catheters were associated with leakage in the cerebrospinal fluid compartment [93].
A Phase III clinical trial (called the PRECISE study) was performed to compare overall survival and assess toxicity and safety of CB versus Gliadel wafers after the first recurrence of GBM [96]. Using 2 to 4 MRI-guided catheters, drug was delivered to targeted brain areas. The PRECISE study was performed in >50 centers across North America and Europe and involved 296 patients. Patients were randomized to receive CB or Gliadel wafers in a 2:1 ratio. Gliadel wafers were placed immediately after tumor resection. Among patients randomized to CB, 2 to 4 catheters were placed in areas most vulnerable to infiltrating tumors 2–7 days after tumor resection. After 24 h, CB was infused for 96 h at 0.750 mL/h at 0.5 µg/mL; CT scans were used to verify catheter placement. Participants were assessed for radiographic and clinical responses every 8 weeks after treatment. Patients treated at centers more experienced with catheter positioning and neurosurgery had better outcomes than those at centers where less than 2 patients were enrolled [97]. Similarly, most side effects were psychiatric or neurologic in nature and included headache, speech and gait disorder, nausea, sensory disturbance, facial paresis, and memory impairment. These could have been an aftermath of IL-13-PE38QQR binding to IL-13RA1 receptors in normal brain tissues. Patients were not screened for IL-13RA2 in their tumors as an eligibility criterion, and drug distribution was not monitored during prolonged infusion. Gene array studies involving 959 GBM samples have shown that IL-13RA2 is over-expressed in 58% [83] and the protein is present up to 75% of GBM. Therefore, screening of patients for IL-13RA2 positivity could have helped to obtain better responses to CB.
Future considerations to make the targeted cytotoxin therapy more effective could be divided into clinical (methods of drug delivery improvement) and laboratory (ligand/drug improvement) settings. Clinical considerations could include real-time monitoring via MRI of the drug infusion using CED, proper positioning of the catheter through pre-infusion planning, potentially restricting the treatment to more experienced centers, and screening patients for targets before treatment. Overall, optimizing the binding of IL-13 to IL-13RA2 while minimizing its binding to the normal tissue/tumor IL-13RA1/IL-4RA complex could improve outcomes of IL-13RA2 targeting in GBM patients.
3.2. Optimizing the Ligand for Therapies
Of the four alpha helices of the IL-13 ligand, helix D is primarily responsible for its interaction with IL-13RA2 [98]. Mutations such as replacing the 13th amino acid from glutamic acid (E) to lysine (K) to produce IL-13.E13K—or changing from glutamic acid (E) to tyrosine (Y) to produce IL-13.E13Y —could make the ligand more specific to IL-13RA2 by considerably decreasing its association with IL-4A and formation of the IL-13RA1/IL-4 receptor [61,99,100]. Additional mutations in arginine to aspartic acid, serine to aspartic acid, and lysine to arginine at positions 66, 69 and 105 (in α-helices C and D, respectively) enhance specificity of binding of the IL-13 ligand to IL-13RA2 [100,101].
The IL-13.E13K ligand has been incorporated in a single-chain protein with domain 2 (D2) of PE, and a nuclear localization signal (NLS) used to precisely deliver to nuclei the portion of the engineered protein produced by a proteolytic cleavage in an endocytic compartment [78,79,102]. A doxorubicin variant was also conjugated to the C-terminal end of the engineered protein and was toxic to glioma cells. Studies from the Debinski lab have shown that the protein was encapsulated by glioma cells in which IL-13RA2 was overexpressed, and the protein was transferred to the nucleus to deliver the cytotoxic load as planned [79,102].
Several antibodies for IL-13RA2 have been generated with high affinity to glioma cells in vitro [77,103]. Furthermore, even without conjugation to a bacterial toxin, the antibody enhanced survival of mice injected with glioma cells [103]. Thus, there is a possibility that conjugating antibody fragments to toxins may further enhance the cytotoxic effects of therapy on glioma cells.
3.3. IL-13-Based Drug Delivery
Drug delivery systems armed with modified IL-13 to specifically target IL-13RA2 include (but are not limited to) liposomes, nanoparticles, and nanosheets. Liposomes range from 20 nm–3 µm in diameter and are composed of phospholipid lipid bilayers that allow simultaneous loading of hydrophobic and hydrophilic drugs. Liposomes offer many advantages for drug delivery. While modifications of the liposomal surface could allow decoration of various ligands to target different antigens in glioma, systemic delivery of liposomes is also possible. Liposomes directed by IL-13 significantly reduced tumors in glioma xenograft-bearing mice versus liposomes that were not guided by IL-13 [104,105]. Similarly, the liposomes directed by IL-13 and loaded with doxorubicin reduced tumor volume 5-fold, and mice survived more than 200 days; those that received non-targeted liposomal delivery of doxorubicin survived less than 35 days. Huang et al., also designed IL-13 peptide enabled mesoporous silica nanoparticles to deliver doxorubicin to glioma cells. In U-251 GBM cells that overexpress IL-13RA2, uptake of targeted nanoparticles was significantly higher compared to control nanoparticles [106,107].
3.3.1. IL-13-Directed Viruses
Oncolytic virus-based treatment of glioma uses viruses engineered to induce tumor cell lysis, amplify therapeutic genes within the tumor cells, divide selectively within tumor cells, and/or also initiate immune response to themselves and dying tumor cells. Some oncolytic viruses used to treat GBM include herpes simplex virus, adeno- and lentivirus, measles virus, and polio virus. By modifying viruses to express ligands that more selectively target glioma cells, their side effects and toxicity are expected to decrease. For example, a Phase I clinical trial of 37 patients tested dose escalation and biologic endpoints of DNX-2401, an adenovirus that targets the αvβ3 and αvβ5 integrins in glioma cells via the arginylglycylaspartic acid peptide [108]. Twenty percent of patients survived >3 years, 2 patients had stable disease and 3 patients showed a complete response with >95% tumor reduction when 25 patients were treated with DHX-2401. Since DNX-2401 was well tolerated by patients, no dose-limiting toxicity was established. The maximum dose used (3 × 1010 virus particles (vp) in 1 mL) was formulated by manufacturing limitations, not by toxicity.
Modified adeno- and lentiviruses that express the IL-13 (LU-13) and (MV Hcdelta18-IL-13) target glioma cells more than the control viruses [109,110]. Similarly, R5111, a herpes simplex virus that expresses IL-13, selectively targets glioma cells in vitro [111]. An attenuated measles virus targeting the HER2 receptor significantly prolonged survival of severe combined immunodeficiency (SCID) mice that contained ~35 mm3 tumors from HER2 positive SK-OV-3 xenografts. While tumor volumes for mice in the control group increased significantly, mice that received designed ankyrin repeat proteins (DARPins) measles virus showed complete remission [112]. Furthermore, adenovirus Ad.mhIL4.TRE.mhIL13PE co-expressed mutated ligand IL-4 (IL-4.Y124D) along with IL-13 conjugated to PE (mhIL13PE) [113] have also been designed. This strategy was introduced to abrogate the binding of IL-13 to the IL-13RA1/IL-4A receptor in normal cells, thus minimizing toxicity of the viral construct to normal brain and maximizing therapeutic efficacy. This approach helped to maintain strong anti-tumor activity of the targeted cytotoxin, with significantly decreased side effects compared to IL-13PE38QQR (CB) given alone [113].
3.3.2. IL-13RA2-Targeted Radiotherapy Agents
Radioactive agents have been conjugated with peptides targeting the IL-13RA2 [78]. Pep-1L is a peptide ligand that binds to IL-13RA2 with high specificity. Using a phage display library and bioplanning schemes in glioma cells that overexpressed or did not express IL-13RA2, three peptide ligands were identified. Among them, Pep-1L showed the highest affinity to IL-13RA2, so it was used to home IL-13RA2 [78]. Copper-64 radiolabeled Pep-1L selectively bound to IL-13RA2-expressing cells in vitro [63]. Furthermore, labeled Pep-1L targeted GBM both in vitro and in an orthotropic model of GBM, monitored in real time using positron emission tomography (PET). This study also tested conjugation of Pep-1L with an alpha particle emitter, actinium-225. Using CED, [225Act] Pep-1L increased survival of mice compared to saline control. Treated tumor cells also showed lower amounts of Ki67 staining (proliferation marker) and higher internalization of propidium iodide in glioma cells, all of which indicate induction of apoptosis.
3.4. Peptide Vaccines
Peptide vaccines stimulate the immune response to selectively target antigens expressed in tumors. Neoantigens arise in tumors from somatic mutations and are absent or nearly absent in normal cells. Peptide vaccines could be composed of single or multiple peptides targeting single or multiple neoantigens or TAAs. TAAs specific to glioma that have been used to devise peptide vaccines include EGFRvIII, IL-13RA2, AIM-2, TRP-2, gp-100, HER-2 and MAGE-1 [114]. Although heterogeneous and sporadic, EGFRvIII has been proposed as a potential tumor-initiating cell and cancer stem cell marker [115]. The effects of an EGFRvIII-specific vaccine, Rindopepimut/CDX-110 (a keyhole limpet menocyanin conjugated with a 14 amino acid peptide that targets the deletional junction in EGFRvIII) were examined in 18 patients with GBM who had EGFRvIII mutations [116]. This vaccine was tested in conjunction with surgery and radiation therapy; treated patients survived 26 months on average. However, when tumors recurred, 82% of tumors did not expresses the EGFRvIII mutation likely due to immunoediting. A Phase III clinical trial was performed following earlier phases with negative final result, and did not show overall significant benefit [117].
Immunoediting occurs when the immune system continuously monitors tumor and eliminates cells that exhibit neoplastic mutations. Over time, single-peptide vaccines cause overgrowth of cells that do not express the targeted antigen. Hence, targeting multiple antigens at once should allow more robust treatment against glioma. A peptide vaccine targeting EphA2, IL-13RA2, and survivin was administered with tetanus toxoid and poly I:C in 26 children with low-grade gliomas (NCT01130077) and was well tolerated in children [118]. This peptide vaccine showed encouraging clinical outcomes, and no dose-limiting non-CNS toxicity was established. Nineteen patients showed stable disease, and temporary pseudo-progression was seen in 5 patients, along with edema (indicative of immune cell infiltration within the tumor) [119].
3.5. Dendritic Cell-Based Vaccines
Because dendritic cells can activate T cells, they have been used to make vaccines against tumor cells. Dendritic cells are isolated from blood of patients, pulsed with TAAs, and introduced back into the patients to try to activate CD4+ and CD8+ T cells and thus selectively target tumor cells expressing the antigens of interest. Dendritic cells pulsed with IL-13RA2 peptides increased immune response among glioma patients [120]. Patients with LGGs who were treated with multiple antigen-presenting dendritic cells, including EphA2, IL-13RA2, Wilms tumor 1 and survivin, also had increased immune responses [50]. Another Phase II multicenter study (NCT01280552) tested the use of dendritic cells (ICT-107) pulsed with 6 different antigens, including IL-13RA2 in patients with glioma. Progression-free response was increased with treatment, and among the 22 patients who received the vaccine, 58% had a positive response [114].
3.6. CAR T-Cell Therapy
The idea of using immune cells to fight tumor cells was introduced in the 1980s by Dr. Steven Rosenberg’s group at the NCI, who pioneered the extraction of tumor- infiltrating lymphocytes to treat melanoma [121]. Later, they designed T cell receptors to target melanoma-associated antigen recognized by T cells (MART-1) in patients with malignant melanoma [122]. This treatment caused regression of tumor growth in patients after ACT, but there were difficulties in extraction and few tumor-infiltrating lymphocytes were isolated from tumor sites. In addition, T-cell recognition depended on major histocompatibility complex (MHC), which limited its use. However, CAR T-cells recognized TSAs independent of MHC [123,124]. CAR T-cell therapy involves isolation of white blood cells of the patient, activation of T-cells using artificial dendritic cells, transduction with the CAR gene using a viral vector, expansion, and finally delivery back into patients. Before receiving CAR T-cells, patients can undergo chemotherapy to suppress normal white blood cells that could interfere with efficacy of the CAR T-cells [125].
The first FDA-approved CAR T-cell therapy, Kymriah, was developed to target CD19+ cells for patients with ALL [126]. Kymriah is a second generation CAR T-cell therapy that contains anti-CD19 single chain variable fragment (scFv) fragment, a transmembrane domain, and co-stimulatory domains CD3ζ and CD137. It was reported that 63% of patients showed complete remission, with no traces of blood markers that indicate a relapse 3 months after therapy. Very recently, the FDA also approved a second CAR T therapy (Yescarta) for adult patients with relapsed B-cell lymphoma [127]. Similar to Kymriah, Yescarta also targets CD-19. Currently, many CAR T-cell based therapies are being evaluated for various types of cancers. This approach offers further promise for effective treatment of cancer using the body’s own immune cells.
Both first and second-generation CAR T-cell therapies have been successfully used to target IL-13RA2. These CAR T-cells expressed the mutated IL-13 ligand (IL-13. E13Y) to enhance binding to IL-13RA2 [56,128,129,130,131,132]. IL-13. E13Y CD8+ T cells, which are an example of the first-generation of CAR T-cells, significantly affected cell viability in U-251 GBM cells in vitro and in vivo [129]. The anti-tumor response was also extended to glioma stem-like cells. Also, elevated secretion of immunostimulatory cytokines, including tumor necrosis factor α (TNFα) and INF γ was observed when the IL-13 zetakine+ CD8+ T-cells were co-cultured with U251 cells [133]. The City of Hope group conducted a Phase I clinical trial for GBM, using CAR T-cells against IL-13RA2 [46,56]. A patient with recurrent multifocal GBM who had failed all available therapies received CAR T-cells against IL-13RA2; complete regression was observed of all spinal and intracranial lesions, with no exhibit toxic effects greater than grade 3. This clinical response was coupled with an increase in immune cells and cytokines and lasted for at least 7.5 months after initial therapy [56]. In another study, 3 patients with recurrent glioma received first-generation CAR T-cells against IL-13RA2, which also included herpes simplex virus type 1-thymidine kinase and the PET reporter gene to non-invasively detect CAR T-cells on PET imaging [134,135]. IL-13. E13Y-zetakine/HyTk+ CD8+ T cells were present at the sites of injection and recurrence [135,136]. In another Phase I clinical trial (NCT01082926), IL-13-zetakine/HyTK CAR first-generation CAR CD8+ T-cells were expressed in CD8+ T cells from a healthy donor. These cells were co-administered with interleukin 2 (IL-2) and showed anti-tumor effects in patients with GBM expressing IL-13RA2. These promising results indicate that CAR T-mediated therapy holds potential as a treatment for glioma [64,137].
Nonetheless, one issue with CAR T cell therapy is that the survival and persistence of targeted T-cells in the glioma environment is limited. Using second-generation CAR T-cells with IL-13.E13K.R109K and a co-stimulatory signal CD28 and CD3ζ, Kong et al. reported increased survival using in vivo xenograft models [138]. Along with longer survival, CAR T-cells displayed higher affinity to IL-13RA2 compared to IL-13RA1 in normal tissue, along with significant increased immune responses, including secretion of immunostimulatory cytokines such as IL-2 and INF γ. Ways to improve the efficacy of CAR T-cell therapy could include enhancing the affinity of IL-13 ligand to IL-13RA2, and introduction of the IL-15 gene [139,140,141], IL-12, and nuclear factor of activated T-cells (NFAT), to boost survival and proliferation of T-cells within tumors.
While CAR T-cell therapy has shown encouraging anti-tumor results, challenges remain to improve its clinical efficacy. For example, the Phase II ROCKET trial was terminated prematurely after 5 of 38 patients receiving the treatment died after developing neurotoxicity and brain swelling following treatment [142]. Additionally, CAR T-cell activation usually accompanies elevated levels immunostimulatory cytokines, leading to the cytokine release syndrome [143]. An inhibitor of interleukin 6 (IL-6), Actemra (tocilizumab), is used to address this issue [144]. However, a better alternative would be a potential genetic switch within the CAR T-cell system that could turn off when excess cytokines are being released. Companies like Cellectis and Bellicum Pharmaceuticals are experimenting with UCART 19 [145] and GoCAR T-cells that are activated in the presence of rapamycin and rimiducid, respectively [146]. Similarly, efforts are being made to house caspase-9 based switches in CAR T-cells to target CD20+ cells in lymphoma [147].
Another significant challenge for successful CAR T-cell therapy, or immunotherapy in general, is the loss of a targeted antigen with treatment. Studies of CAR T-cell therapy have suggested that decreased expression of IL-13RA2 could be a factor in tumor recurrence [56]. Loss of EGFRvIII has also been described after CAR T-cell therapy [148]. This has been called ‘antigen-negative immune escape’ [149] and might be caused by unintentional selection of antigen negative cells that proliferate faster than the antigen-positive cells within the tumor. It could also be caused by loss of antigen within tumor cells that previously expressed the antigen. Identifying the mechanism for immunoediting could help design improved CAR T-cells to minimize this unintended effect and eventually reduce therapeutic resistance.
Furthermore, simultaneously targeting of different antigens could enhance CAR T-cell therapy by making loss of antigens less likely [150]. T and CAR T-cells that target HER2 and IL-13RA2 have been tested in a murine model of GBM, and was associated with less antigen escape and significantly improved survival [150]. But depriving tumors of IL-13RA2-expressing cells might have its own beneficial effects, because these cells took the longest to show tumor recurrence [151]. This result suggests that tumor cells lacking IL-13RA2 might be less tumorigenic, which was actually seen in cells selected for IL-13RA2 down-regulation [152].
Resistance to immunotherapy can be inherited, as when a cancer does not respond to therapy in the first place; or it can be acquired, where a tumor initially responds to the therapy, but over time becomes resistant [153]. This resistance can result from various intrinsic factors such as phosphatase and tensin homolog deleted on chromosome 10 (PTEN) loss [154], the mitogen-activated protein kinase pathway (MAPK) [155], and extrinsic factors such as recruitment of immunosuppressive cells like regulatory T cells, myeloid-derived stem cells, and loss of T cells over time specific to certain tumor antigens [153,156]. Given the failure of many monotherapies in Phase III clinical trials, tumors should always be viewed as an ecosystem with multiple interconnected heterogeneous layers that feed off each other by sustaining a supportive biochemical and biophysical microenvironment along with immune system and tumor cells [26,157,158,159].
In the tumor ecosystem, identification of ‘keystone species’ that essentially hold the ecosystem together is extremely difficult, and has not been identified in GBM. It is therefore not surprising to observe tumor recurrence, just as an ecosystem would recover after the loss of one species. Hence, therapeutic approaches to GBM should also acknowledge the complexity of the tumor ecosystem to gain significant clinical benefit. This can be done, as proposed by Debinski’s group more than 10 years ago [32] by using a cocktail of combined therapies that can effectively bring the tumor ecosystem down. One such approach is by targeting multiple receptors in GBM, which will be described in more detail below.
4. Eph/Ephrin Receptor System in Gliomas
Isolated from a hepatocellular carcinoma cell line, erythropoietin-producing-human (Eph)s are the largest family of receptor tyrosine kinases that bind to ephrins (eph receptor interacting ligands) [160]. They are important in cell adhesion, migration, guidance, and embryogenesis. Given their critical role in cell migration, they are also expressed in tumors and are active in wound healing and injuries [161,162,163,164,165]. Eph receptors bind to ephrin As and Bs and are internalized after ligand/receptor interaction and clustering in a matter of minutes. Since the ligands ephrin A and B are attached to the cell membrane, matrix metalloproteins are necessary to detach ligands from the cell surface and thereby establish a ligand/receptor interaction in addition to cell-cell interaction [166]. Overexpression of Eph receptors is quite ubiquitous among malignancies including gliomas, pancreatic, breast, ovarian, and other cancers [167].
The EphA2 receptor is over-expressed in ~60% of GBM tumors, and it is associated with grade increases in higher-grade gliomas, and with poor prognosis and survival [168,169,170]. Ephrin-B2 is expressed in astrocytomas; this expression along with expression of EphB4 increases with glioma grade [171]. The EphA7 receptor is present in tumor cells and endothelial cells of the vasculature surrounding the tumor [172]. Also, EphB3, B2, and B4 expression is increased in GBM compared to normal brain [173]. This makes Eph receptors attractive for novel targeted drug/therapy avenues.
Another Eph receptor, EphA3, is overexpressed in almost 60% of GBM patient samples [174] with increased presence in tumor initiating and infiltrating cells both in glioma [174] and leukemia [175], invasive rings, and scattered areas within the tumor and niches close to the blood vessels [174]. EphA3 is overexpressed in mesenchymal GBM, with EphA3 knockdown tumors exhibiting decreased tumorigenic potential [176]. Ephrin-A5 binds to the EphA2, EphB2, and EphA3 receptors [174]. Ephrin-A1 conjugated to a modified PE has been devised in the Debinski lab to target EphA2-overexpressing cells. The conjugate showed significant killing of GBM cells both in vitro (IC50 of 10−11 mol/L) and in vivo [177]. A conjugate of ephrinA5 and modified PE also showed high potency and specificity of tumor cells killing [174].
A study involving a cocktail of ephrin-A1 and IL-13 based cytotoxins has been extended to Phase I trials for glioma in dogs, and is showing extremely encouraging clinical effects [178]. Beta-emitting radionucleotide lutetium (177Lu) conjugated to monoclonal antibodies (mAb IIIA4) that target EphA3 receptors in mice implanted with U-251 MG GBM cells have been shown to prolong animal survival [176]. Similarly, CAR T-cell therapy that selectively targets EphA2 in an orthotropic xenograft SCID mouse model has shown promising results [179]. Combined expression of EphA2, EphA3, EphB2, and IL-13RA2 is observed in almost 100% of patients with glioblastoma patients. Furthermore, expression of these receptors is also present in tumor-infiltrating cells, tumor-initiating cells or GSCs, and neovasculature [180]. Therefore, a cytotoxic agent that simultaneously targets these four receptors could be powerful in destroying the tumor ecosystem of GBM without allowing it to recover and cause recurrence. Ferluga et al. has recently demonstrated such a multi-targeted approach in treating GBM by targeting multiple Eph receptors at once [174].
Desatinib is a multi-targeted kinase inhibitor that inhibits EphA2 and EphB2 receptors and has been tested against breast and ovarian cancer cells [181,182]. Doxazosin is a small molecule inhibitor of EphA2 and EphA4 that suppresses cell migration in glioma, breast cancer, and prostate cancer cells after internalization of receptors [183,184]. While it remains unclear how doxazosin inhibits migration, it is hypothesized that it activates receptor tyrosine kinases involved in inhibition of migration [185]. Eph receptors affect the migration signaling pathway by balancing the Rho versus Rac and Cdc42 pathways [186]. Enhancing the Rho pathway increases acto-myosin contractility within the cell, which explains the ligand-dependent stimulation of cell rounding upon Eph activation [184,187,188]. Suppression of cell migration of cells following doxazosin-Eph interaction may also stem from potential activation of Rho pathways that enhance cell rounding and suppress cell migration. Inhibitors like desatinib and doxazosin could be used in conjunction with other agents to increase anti-tumor activity.
5. EGFR/EGF Receptor System in Gliomas
One of the most studied receptor systems in brain tumors is the EGFR/EGF receptor system [115,189,190,191]. The human EGFR receptor (HER2) family of receptor includes EGFR, HER2/neu, HER3 and HER4. They all contain similar structures except for the absence of tyrosine kinase in HER3. All bind to HER2 with extracellular ligands except for HER2. EGFR is a 170-kD transmembrane protein receptor with an extracellular domain, a transmembrane domain, and a tyrosine kinase domain. The N-terminus containing the extracellular domain creates a cleft of two regions that allow ligands to bind to the receptor [192]. EGFR is amplified in 40–60% of patients with GBM [191,193], while the protein is overexpressed in 38% of patients. However, EGFR expression is not specific to glioma cells [193]. About 30–50% of EGFR amplifications contain EGFRvIII mutations where the coding region of the extracellular domain is absent (deletion of exons 2–7). The mutated protein is expressed in fewer than 20% of patients with GBM, and is very heterogenous and patchy. The absence of the extracellular domain of the EGFR in EGFRvIII causes constitutively active tyrosine kinase activity, making them more invasive and antiapoptotic [190]. EGFRvIII-containing gliomas have enhanced Ras activity, Akt/PI3k signaling, and increased expression of VEGF and IL-8 [194]. Cells containing EGFRvIII also tend to express B-cell lymphoma extra-large (Bcl-XL) protein, which enhances their apoptotic ability [195].
Multiple groups have used EGFR and EGFRvIII receptors in glioma as targets for therapy [189]. Unfortunately, no clinical trials involving EGFR and EGFRvIII have shown significant clinical effects in patients. siRNA have been targeted towards EGFR that inhibited EGFR-expressing cells in vitro [196]. Treatment arrested glioma cells in the G2/M phase, significantly inhibiting cell growth in vitro. The siRNA also reduced tumor growth in vivo [197]. Erlotinib, an EGFR inhibitor, was not able to show significant effects in multicenter Phase I/II trial [198]. However, there was a strong correlation of PTEN and EGFRvIII co-expression with the outcome of erlotinib treatment. Not surprisingly, tumors that were PTEN-negative did not respond to the EGFR inhibitor [199,200].
Another inhibitor of EGFR, geftinib, was evaluated in Phase I/II clinical trials and showed no significant response in patients with newly diagnosed with GBM [201]. The monoclonal antibody cetuximab, which targets the extracellular domain of the EGFR was investigated in Phase I/II studies [202]. While only a few patients responded to the therapy, combining bevacizumab and irinotecan showed improved responses in patients [203]. The study showed 6-month progression- free survival in 46% of patients; 57% of patients showed a partial response, and 6-month overall survival was 77%. However, the study was terminated because one patient developed severe hemorrhage in the central nervous system, and another developed intestinal perforation and died. Nimotuzumab was evaluated in Phase I/II clinical trials for malignant glioma, and was well tolerated in patients [204]. Similarly, TGF-α conjugated to PE38, also called TP-38, has been evaluated in Phase I/II clinical trials. One patient demonstrated a complete response with 83-week survival, and 3 of 15 patients who received therapy showed radiographic response [205].
EGFRvIII-targeted CAR T-cell therapy is also being evaluated by Rosenberg’s lab at the National Cancer Institute (NCT01454596) and at Novartis and the University of Pennsylvania (NCT02209376) [206,207]. In the phase I study, EGFRvIII CAR T-cells were well tolerated by patients and did not show signs of off-target toxicity. A single intravenous dose of CAR T-cells was used and cytokine release syndrome was not observed. The CAR T-cells homed to tumor cells as shown by immunohistochemistry [206]. HER2-specific CAR T-cells have shown antitumor activity with increased production of stimulatory cytokines like IL-2 and interferon-γ. This antitumor response resulted in tumor regression in mice containing xenograft GBM cells. Similarly, HER2 CAR Cytomegalovirus-specific T therapy was evaluated in a phase I clinical trial with 16 patients. One patient showed partial response with about a 62% decrease in tumor volume over 8 months. The median survival of patients was 11.6 months after infusion of the therapeutic agent (NCT02442297 and NCT0110905) [208,209].
6. Other Receptors/Targeted Therapies
Other receptors used to treat gliomas include vascular endothelial growth factor receptor (VEGFR) [210], platelet-derived growth factor receptor (PDGFR) [211], transforming growth factor receptor (TGFR) [212], fibroblast growth factor receptor (FGFR) [213], c-Met receptor [214], IL-4 receptor [215], urokinase-type plasminogen receptor (uPAR) [216], g-protein coupled receptors (GPCR) [217], transferrin [218], lactoferrin [219], low density lipoprotein receptor (LDL-R) and low density lipoprotein receptor-related protein (LRP1) [220], tenascin, folate receptor (FOLR) [221], and integrin-mediated targeting systems [222]. The VEGFR targeted antibody bevacizumab (Avastin) was FDA-approved to treat recurrent GBM [223], based on durable radiologic responses in a Phase II trial [224]. Patients showed a significantly longer progression-free survival time (median survival of 46.8 months) when treated with bevacizumab, compared to the expected survival of 16.3 months with standard temozolamide and radiation treatments [225]. However, it failed to show efficacy in GBM patients in all phase III trials [223].
A Phase III trial investigated the efficacy of cilengitide, an RGD peptide that targets the integrin combined with standard therapy in patients with newly diagnosed GBM. Results from Phase I/IIA studies had shown that patients with MGMT promoter methylation status responded better to cilengitide [226]. In the Phase III trial, 3471 patients were screened for MGMT status; 926 patients had a MGMT promoter and received the treatment. Overall, cilengitide with temozolomide and radiation therapy did not improve clinical outcomes [227]. The lack of clinical response with therapies targeted at individual receptors reaffirms the notion that glioma treatment should simultaneously address various facets of the tumor to confer significant, long-lasting clinical impact.
Tyrosine kinase inhibitors against VEGFR, including cediranib, sorafenib, sunitinib, and vandetanib, have been tested in Phase I/II trials and their efficacy is now being tested for GBM [228]. In a phase III clinical trial (NCT00777153) with 325 patients, the efficacy of cediranib was tested alone and in combination with lamoustine, but with no significant improvement in progression-free survival occurred in either treatment arm [229]. Sorafenib plus emozoalide was evaluated in 43 patients with GBM in a phase II trial and was safe. Six-month progression-free survival was observed in 26% of patients with a median survival of 7.4 months [230]. A phase II evaluation of sunitinib in children with high-grade glioma and ependymoma (NCT014626695) showed that the treatment was well tolerated, but did not result in significant antitumor activity [231]. Tyrosine kinase inhibitors for PDGFR include imatinib, which showed limited activity against glioma in Phase I and II clinical trials [232]. Desatinib, another inhibitor of PDGFR, showed no clinical benefits for recurrent GBM in a Phase II trial (NCT00423735) [233]. Brivanib and XL184, two other tyrosine kinase inhibitors for FGFR and c-Met receptors, respectively, were tested in preclinical and Phase I/II trials, respectively [234].
Targeting the transferrin receptor with Tf-CRM107 immunotoxin was evaluated in Phase I/II/III trials using CED. This was the first clinical trial to test the efficacy of an immunotoxin in GBM. Diphtheria toxin with a point mutation (CRM107) was conjugated with transferrin by a thioester bond to form a cytotoxin-protein conjugate that targets the transferrin receptor in glioma. In vitro analysis toxin showed that the unconjugated toxin was 1000–100,000 times less toxic to glioblastoma cell lines; its IC50 value ranged from 2.6 × 10−12 to 6.5 × 10−11 mmol/L. In a Phase I trial, 18 patients were treated, 9 out of 15 patients showed >50% decrease in tumor and 2 patients showed complete responses [235]. While 7 of 44 patients with recurrent GBM showed a partial response to Tf-CRM107 immunotoxin, the Phase III trial was stopped due to the lack of significant efficacy [235]. The transferrin receptor is expressed in normal endothelium, which may limit the therapeutic window of the immunotoxin.
7. Immune Checkpoint Inhibitors
A potential new facet of glioma therapy includes immune checkpoint inhibitors. GBM cells consistently work to maintain and generate an immunosuppressive environment. Expression of receptors that bind to cytotoxic T-cells and inhibit their action is common in GBM. Specifically, expression of galectin-9 that binds to T-cell immunoglobulin and mucin domain 3 (TIM-3) [236]; Fas ligand (FasL) that induces apoptosis of lymphocytes [237], B7 which is immunosuppressive to CTLA-4 [238], and programmed death ligand 1 (PD-L1) which is immunosuppressive to programmed death 1 (PD-1) expressing T-cells are significantly high in glioma cells [239]. The expression of PD-L1 in particular is higher in glioma-infiltrating cells than in glioma cells. GBM also overexpress major histocompatibility complex class I (MHCI) molecule human leukocyte antigen G (HLA-G) which inhibit natural killer cell and cytotoxic T-cell responses. Synthesis of anti-inflammatory cytokines including chemokine (C-C motif) ligand 2 (CCL2), tumor growth factor β (TGFβ), interleukin 10 (IL-10) that recruit regulatory T cells and myeloid derived suppressor cells (MDSC), and tumor-associated macrophages (TAMs) that inhibit T cell responses and promote tumor progression are also very common in GBM [240]. Downregulation of human leukocyte antigen I (HLAI) molecules, which allow tumor cells to evade cytotoxic T cells, has been shown in GBM compared to LGGs [241]. Immunomodulation of the glioma environment may therefore provide significant therapeutic benefits.
T cells express CTLA-4 that would be cytotoxic to tumor cells. However, tumor cells express B7 that bind to CTLA-4 and inhibit these cytotoxic effects. Ipilimumab is a mAb against CTLA-4 that was approved by FDA for melanoma in 2011. It has shown benefits to patients with brain metastasis as a single therapy and combined with radiation therapy [242]. CTLA-4 blocking antibodies improved survival in mice. Similarly, anti-CTLA-4 antibodies in conjunction with IL-12, which promotes survival of T cells, was effective against GL261 glioma cells [243]. Furthermore, the CTLA-4 antibody plus cytokine granulocyte macrophage colony stimulating factor was more effective than the treatments alone [244].
PD-1 is expressed in T cells, TAMs, vascular endothelial cells, and neurons of the tumor microenvironment. It binds to PD-L1 and programmed death ligand 2 (PD-L2), which are overexpressed in GBM cells. Inhibiting PD-L1/PD-1 interaction allows cytotoxic T cells to effectively kill tumor cells [245]. Pembrolizumab and ivolumab are PD-1 inhibitors approved by the FDA for the treatment of melanoma in 2014, with subsequent approvals for treatment in seven more cancers. A Phase III trial of nivolumab and ipilimumab was initiated for GBM in 2014, but failed completely [246]. A Phase I trial of ipilimumab plus nivolumab (a PD-1 inhibitor) is ongoing for patients after resection of recurrent glioblastoma (NCT03233152) [247]. Other PD-1/PD-L1 interaction inhibitors include AMP-224, MPDL32804, MSB0010418C, and MEDI4736 for other hematologic and solid tumors [248]. Anti-CTLA-4, anti-PD-1 and 1-methylthryptophan (1-MT) significantly enhanced survival and lowered the number of regulatory T cells compared to monotherapies [249].
TIM-3 is a glycoprotein expressed in T-cells containing an extracellular IgG and mucin domain that binds to Galectin-9. TIM-3 binds to galectin-9 on tumor cells and suppresses immune response to GBM cells. TIM-3 and LAG-3 are also highly expressed other malignancies, e.g., non-small-cell lung carcinoma and melanoma [250]. Expression of TIM-3 is also elevated in CD4+ and CD8+ T cells of glioma patients compared to healthy counterparts [251]. In in vivo studies, anti-PD-1, anti-TIM-3 and radiation synergistically regressed glioma in mice [252].
Lymphocyte activation gene 3 (LAG-3) binds to MHC class II antigens and works together with PD-L1 to suppress the activation and expansion of T cells. LAG-3 is highly expressed in tumor- infiltrating lymphocytes in patients with fibrosarcoma, colorectal cancer, and melanoma [253]. A Phase I trial in patients with recurrent GBM (NCT02658981) testing combined therapy of an anti-LAG-3 monoclonal antibody and nivolumab is ongoing [55].
Immunostimulatory cytokines have also been used to treat glioma. IL-2 was first tested in 1986 [254]. A Phase I trial of suicide gene therapy of herpes simplex virus type 1-thymidine kinase and retrovirus-producing IL-2 shown partial response with minimal side effects [255]. Subcutaneous IL-4 injections plus herpes simplex virus type 1-thymidine kinase increased interferon γ production in a rat model, and is being tested in a Phase I clinical trial [256]. Similarly, administration of interferon γ enhanced efficacy of temozolomide [257]. Liposomal delivery of interferon γ reduced tumors by half in a Phase I clinical trial, and Ad-hIFNγ has shown safety and tumor cell apoptosis [258].
Given that checkpoint inhibitors target immune responses and are critical in maintenance of the tumor ecosystem, checkpoint inhibitors and immunostimulatory cytokines are appealing as treatments for GBM in conjunction with chemotherapy, radiation therapy and a myriad of receptor-targeted therapies like cytotoxins and viral gene therapy. However, substantial clinical effects to back this notion have not yet been observed. Furthermore, although rare, immune checkpoint inhibitors have been associated with immune-related adverse events. For example, in a single-center review of over 500 patients treated with the immune checkpoint inhibitors ipilimumab and nivolumab in 2005–2017, 5 patients had immune-related adverse events (0.95% incidence rate) [259]. These ranged from sensory neuropathy and transverse myelopathy, to oculomotor nerve palsy and posterior reversible encephalopathy syndrome, with more severe events in patients receiving combination immunotherapy.
8. Viral/Genetic Therapies
Development of vectors that can successfully deliver genes into the tumor has allowed testing of genetic therapy for glioma. Delivery of gene therapy is broadly based on viral vectors (adenovirus, herpes simplex, measles, reovirus, and poliovirus) [260] and non-viral vectors like liposomes and nanoparticles. Virus-based therapies can have multiple therapeutic effects including tumor cell death, increased anti-tumor immune response, and release of damage- associated molecules like high mobility group box 1 (HMGB1) [261,262]. Use of suicide gene therapy, where an inactive prodrug ganciclovir thymidine kinase is converted into a toxic metabolite to cause tumor apoptosis, has been investigated in GBM [263]. While the results from Phase I/II preclinical trials were promising, a large Phase III trial of herpes simplex virus type 1-thymidine kinase/gancyclovir plus surgery and radiation therapy did not significantly improve survival among patients newly diagnosed with GBM [264]. A combined cytotoxic and immune-stimulatory therapy for glioma is underway. Phase I trial (NCT01811992) of adenovirus mediated delivery of FMS like tyrosine kinase 3 ligand (Flt3L, which stimulates the migration of dendritic cells) along with Ad tyrosine kinase and ganciclovir increased survival of mice, and generated tumor-specific T-cell responses and long-term cell memory, along with release of HMGB1 by dying tumor cells [265].
Over 20 oncolytic virus candidates have been tested in clinical trials to treat GBM. Several oncolytic viruses have demonstrated safety in Phase I clinical trials for glioma, including reovirus [266], oncolytic measles [267], conditionally replicating adenoviruses (CAds) [268] and oncolytic herpes simplex virus [269]. ONYX-015 (adenovirus mutant dl1520) has been shown to be safe when injected intracranially to treat glioma. This virus has been engineered to divide in cells lacking p53 [270]. A Phase I/II clinical trial of Ad 5Delta 24-RGD and DNX2401 is underway in patients with recurrent GBM (NCT01582516) [108]. Similarly, DNX2401 (NCT02197169) expressing interferon γ was tested in a Phase I trial for patients with recurrent GBM [271]. Other viruses tested for GBM include the M protein mutant (M51R) and vesicular stomatitis virus, which showed selectivity in killing U87 GBM cells versus normal brain cells [272].
Some viral constructs take advantage of the overexpression of CD46, which can serve as a measles virus receptor; the laminin receptor, which could serve as a sindbis virus receptor; and CD155, which is overexpressed in glioma cells and can serve as a receptor for poliovirus. Furthermore, viruses have been designed to specifically target receptors that are overexpressed in glioma cells including PDGFR, IL-13RA2, and EGFRvIII [273]. Presently, a Phase I clinical trial using carcinoembryonic antigen-expressing measles virus is underway for GBM. The study is designed to administer a starting does of 105 TCID50 to be escalated to 2 × 107 TCID50. Until now, no dose-limiting toxicity has been established [110,267,273,274,275]. These viruses have enhanced efficiency of therapeutic gene transduction compared to non-replicating viruses. However, consistent clinical benefits of these viruses are yet to be established.
9. Conclusions
Treating brain tumors like gliomas is extraordinarily difficult. Their location and infiltrative nature, the presence of the BBB, molecular, genetic and phenotypic heterogeneity, the poorly developed but well vascularized immunosuppressive environment, and resistance to therapy are some of the major challenges facing the treatment of brain tumors. Moreover, brain tumors function as an ecosystem with highly interconnected networks that are able to compensate for each other and bounce back after treatment. Hence, the clinical prognosis for brain tumors has not changed substantively over the last few decades. However, there have been notable advancements in antigen-specific targeting, viral and genetic silencing, immune checkpoint inhibitors, peptide and dendritic cell vaccines, CAR T-cell therapy, chemotherapy and radiation therapy, in conjugation with effective drug delivery systems like CED (Table 1). Combinatorial therapies damage multiple facets of the tumor ecosystem, giving hope to eventual cure of brain tumors. Similarly, neoantigen screening of patients using next-generation sequencing would allow treatments to be individually tailored [276]. Furthermore, the antigen profile of tumors could be altered after exposure to ionizing radiation/surgery. Hence, detailed studies of changes in antigen expression before and after radiation or chemotherapy would help predict therapeutic outcomes and modify treatment plans accordingly for maximum efficacy. Also, deciphering the mechanisms of immunoediting could lead to efficient immunotherapies with superior clinical benefits. In addition, effective and innovative monitoring mechanisms that can critically evaluate the efficacy of single and combinatorial therapies, along with CT and MRI imaging, could help enhance the success of therapeutic strategies for brain tumors. Similarly, technologies like the IsoChip and IsoLight [277,278] that can eventually predict the outcome of potential therapies ex vivo before being introduced into patients could provide powerful tools to better understand and treat this devastating disease.
Funding
This work was supported by National Cancer Institute grants R01 CA74145 and P01 CA207206 and Hearn Fund for Brain Tumor Research. We thank Karen Klein for help with editing the manuscript.
Conflicts of Interest
The authors have no conflicts of interest to declare.
Abbreviations
| 1-MT | 1-Methylthryptophan |
| ACT | Adoptive Cell Transfer |
| ACTL8 | Actin Like Protein 8 |
| AIM-2 | Absent In Melanoma 2 |
| ALL | Acute Lymphoblastic Leukemia |
| BAGE | B Melanoma Antigen |
| BBB | Blood-Brain Barrier |
| Bcl-XL | B-cell Lymphoma Extra-Large |
| BORIS | Brother Of The Regulator Of Imprinted Sites |
| CAds | Conditionally Replicating Adenoviruses |
| CAR T | Chimeric Antigen Receptor T-cells |
| CB | Cintredekin Besudotox |
| CCL2 | Cytokines Including Chemokine (C-C motif) Ligand 2 |
| CD | Cluster of Differentiation |
| CED | Convection-Enhanced Delivery |
| CNS | Central Nervous System |
| CT | Computerized Axial Tomography |
| CT 45 | Cancer/Testis Antigen 45 |
| CTAGE1 | Cutaneous T-cell Lymphoma Associated Antigen 1 |
| CTL-4 | Cytotoxic T-lymphocyte-Associated Protein 4 |
| D2 | Domain 2 |
| DARPins | Designed Ankyrin Repeat Proteins |
| DDX43 | Dead-Box Helicase 43 |
| E | Glutamic Acid |
| EGF | Endothelial Growth Factor |
| EGFR | Epidermal Growth Factor Receptor |
| Eph | Erythropoietin-Producing-Human |
| ephrin | Erythropoietin-Producing-Human Receptor Interacting Ligands |
| FasL | Fas ligand |
| FDA | Food and Drug Administration |
| FGFR | Fibroblast Growth Factor Receptor |
| FOLR | Folate Receptor |
| GAGE | G-Antigen |
| GBM | Glioblastoma |
| GFAP | Glial Fibrillary Acidic Protein |
| gp-100 | glycoprotein 100 |
| GPCR | G-Protein Coupled Receptors |
| HAGE | Helicase Antigen |
| HER-2 | Epidermal Growth Factor Receptor |
| HLA-G | Human Leukocyte Antigen G |
| HLAI | Human Leukocyte Antigen I |
| HMGB1 | High Mobility Group Box 1 |
| IDH1 | Isocitrate Dehydrogenase Gene 1 |
| IFN γ | Interferon γ |
| IL | Interleukin |
| IL-13RA1 | Interleukin-13 Receptor A1 |
| IL-13RA2 | Interleukin 13 Receptor Alpha 2 |
| IL-4RA | Interleukin-4 Receptor A |
| K | Lysine |
| KNL1 | Kinetochore Scaffold 1 |
| LAG-3 | Lymphocyte Activation Gene 3 |
| LDL-R | Low Density Lipoprotein Receptor |
| LGG | Lower Grade Glioma |
| LRP1 | Low Density Lipoprotein Receptor-Related Protein |
| MAGE | Melanoma Associated Antigen |
| MAPK | Mitogen-Activated Protein Kinase |
| MART | Melanoma-Associated Antigen Recognized by T cells |
| MDSC | Myeloid Derived Suppressor Cells |
| MGMT | Protein O6 Methylguanine-DNA Methyltransferase |
| MHC | Major Histocompatibility Complex |
| MHCI | Major Histocompatibility Complex Class I |
| MRI | Magnetic resonance Imaging |
| NLS | Nucleus Localizing Signal |
| OIP5 | Opa-Interacting Protein 5 |
| PD-1 | Programmed Death 1 |
| PDGFR | Platelet-Derived Growth Factor Receptor |
| PD-L1 | Programmed Death Ligand 1 |
| PD-L2 | Programmed Death Ligand 2 |
| PTEN | Phosphatase and Tensin Homolog |
| PTEN | Phosphatase And Tensin Homolog |
| RANO | Response Assessment in Neuro-Oncology |
| scFv | Small Chain Variable Fragment |
| SCID | Severe Combined Immunodeficiency |
| SP 17 | Sperm Protein 17 |
| SSX | Synovial Sarcoma X |
| STAT6 | Signal Transducer And Activator Of Transcription 6 |
| TAA | Tumor-Associated Antigens |
| TAM | Tumor-Associated Macrophages |
| TERT | Telomerase Reverse Transcriptase |
| TGFR | Transforming Growth Factor Receptor |
| TGFβ | Tumor Growth Factor β |
| TH2 | T Helper 2 |
| TIM-3 | T-cell Immunoglobulin And Mucin Domain 3 |
| TNFα | Tumor Necrosis Factor α |
| TP53 | Tumor Protein 53 |
| TRP-2 | Tyrosine-Related Protein 2 |
| TSA | Tumor-Specific Antigens |
| TT Fields | Tumor-Treating Fields |
| Upar | Low Density Lipoprotein Receptor |
| VEGFR | Vascular Endothelial Growth Factor Receptor |
| XAGE3 | X-Antigen Family Member 3 |
| Y | Tyrosine |
References
- Allen, N.J.; Barres, B.A. Neuroscience: Glia—More than just brain glue. Nature 2009, 457, 675–677. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanai, N.; Alvarez-Buylla, A.; Berger, M.S. Neural Stem Cells and the Origin of Gliomas. N. Engl. J. Med. 2005, 353, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.A.; Cha, S.; Mayo, M.C.; Chen, M.-H.; Keles, E.; VandenBerg, S.; Berger, M.S. Relationship of glioblastoma multiforme to neural stem cell regions predicts invasive and multifocal tumor phenotype. Neuro Oncol. 2007, 9, 424–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goffart, N.; Kroonen, J.; Rogister, B. Glioblastoma-initiating cells: relationship with neural stem cells and the micro-environment. Cancers 2013, 5, 1049–1071. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, P.; Kros, J.M.; Jeuken, J.W.M. The pathological diagnosis of diffuse gliomas: Towards a smart synthesis of microscopic and molecular information in a multidisciplinary context. Diagn. Histopathol. 2011, 17, 486–494. [Google Scholar] [CrossRef]
- Shaw, E.G.; Wang, M.; Coons, S.W.; Brachman, D.G.; Buckner, J.C.; Stelzer, K.J.; Barger, G.R.; Brown, P.D.; Gilbert, M.R.; Mehta, M.P. Randomized trial of radiation therapy plus procarbazine, lomustine, and vincristine chemotherapy for supratentorial adult low-grade glioma: Initial results of RTOG 9802. J. Clin. Oncol. 2012, 30, 3065–3070. [Google Scholar] [CrossRef] [PubMed]
- Buckner, J.C.; Shaw, E.G.; Pugh, S.L.; Chakravarti, A.; Gilbert, M.R.; Barger, G.R.; Coons, S.; Ricci, P.; Bullard, D.; Brown, P.D.; et al. Radiation plus Procarbazine, CCNU, and Vincristine in Low-Grade Glioma. N. Engl. J. Med. 2016, 374, 1344–1355. [Google Scholar] [CrossRef] [PubMed]
- Shaw, E.G.; Wisoff, J.H. Prospective clinical trials of intracranial low-grade glioma in adults and children. Neuro. Oncol. 2003, 5, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillevin, R.; Herpe, G.; Verdier, M.; Guillevin, C. Low-grade gliomas: The challenges of imaging. Diagn. Interv. Imaging 2014, 95, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.L.; Holmen, S.L.; Colman, H. IDH1 and IDH2 mutations in gliomas. Curr. Neurol. Neurosci. Rep. 2013, 13, 345. [Google Scholar] [CrossRef] [PubMed]
- Forst, D.A.; Nahed, B.V; Loeffler, J.S.; Batchelor, T.T. Low-grade gliomas. Oncologist 2014, 19, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Kamran, N.; Calinescu, A.; Candolfi, M.; Chandran, M.; Mineharu, Y.; Asad, A.S.; Koschmann, C.; Nunez, F.J.; Lowenstein, P.R.; Castro, M.G. Recent advances and future of immunotherapy for glioblastoma. Expert Opin. Biol. Ther. 2016, 16, 1245–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuño, M.; Birch, K.; Mukherjee, D.; Sarmiento, J.M.; Black, K.L.; Patil, C.G. Survival and Prognostic Factors of Anaplastic Gliomas. Neurosurgery 2013, 73, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.K.; Llaguno, S.R.A.; McKay, R.M.; Parada, L.F. Glioblastoma multiforme: A perspective on recent findings in human cancer and mouse models. BMB Rep. 2011, 44, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Durden, D.L.; Van Meir, E.G.; Brat, D.J. “Pseudopalisading” necrosis in glioblastoma: A familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J. Neuropathol. Exp. Neurol. 2006, 65, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Bastien, J.I.L.; Mcneill, K.A.; Fine, H.A. Molecular Characterizations of Glioblastoma, Targeted Therapy, and Clinical Results to Date. Cancer 2014, 121, 502–518. [Google Scholar] [CrossRef] [PubMed]
- von Deimling, A.; Nagel, J.; Bender, B.; Lenartz, D.; Schramm, J.; Louis, D.N.; Wiestler, O.D. Deletion mapping of chromosome 19 in human gliomas. Int. J. Cancer 1994, 57, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Idbaih, A.; Marie, Y.; Pierron, G.; Brennetot, C.; Hoang-Xuan, K.; Kujas, M.; Mokhtari, K.; Sanson, M.; Lejeune, J.; Aurias, A.; et al. Two types of chromosome 1p losses with opposite significance in gliomas. Ann. Neurol. 2005, 58, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Qi, C.; Maling, G.; Xiang, W.; Yanhui, L.; Ruofei, L.; Yunhe, M.; Jiewen, L.; Qing, M. TERT mutation in glioma: Frequency, prognosis and risk. J. Clin. Neurosci. 2016, 26, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Cancer Genome Atlas Research Network Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Cancer Genome Atlas Research Network Identification of a CpG Island Methylator Phenotype that Defines a Distinct Subgroup of Glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellingson, B.M.; Wen, P.Y.; Cloughesy, T.F. Modified Criteria for Radiographic Response Assessment in Glioblastoma Clinical Trials. Neurotherapeutics 2017, 14, 307–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrom, Q.T.; Gittleman, H.; de Blank, P.M.; Finlay, J.L.; Gurney, J.G.; McKean-Cowdin, R.; Stearns, D.S.; Wolff, J.E.; Liu, M.; Wolinsky, Y.; et al. American Brain Tumor Association Adolescent and Young Adult Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro Oncol. 2016, 18, i1–i50. [Google Scholar] [CrossRef] [PubMed]
- Kitange, G.J.; Carlson, B.L.; Schroeder, M.A.; Grogan, P.T.; Lamont, J.D.; Decker, P.A.; Wu, W.; James, C.D.; Sarkaria, J.N. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro Oncol. 2009, 11, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, D.J. Cancer genetics: Initially complex, always heterogeneous. Nat. Rev. Cancer 2011, 11, 153. [Google Scholar] [CrossRef] [PubMed]
- Ellis, H.P.; Greenslade, M.; Powell, B.; Spiteri, I.; Sottoriva, A.; Kurian, K.M. Current Challenges in Glioblastoma: Intratumour Heterogeneity, Residual Disease, and Models to Predict Disease Recurrence. Front. Oncol. 2015, 5, 251. [Google Scholar] [CrossRef] [PubMed]
- Debinski, W. Drug cocktails for effective treatment of glioblastoma multiforme. Expert Rev. Neurother. 2008, 8, 515–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.R.; O’Neill, B.P. Glioblastoma survival in the United States before and during the temozolomide era. J. Neurooncol. 2012, 107, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Bi, W.L.; Beroukhim, R. Beating the odds: Extreme long-term survival with glioblastoma. Neuro Oncol. 2014, 16, 1159–1160. [Google Scholar] [CrossRef] [PubMed]
- Malpass, K. Identification of novel glioblastoma-associated antigens reveals targets for immunotherapy. Nat. Rev. Neurol. 2012, 8, 240. [Google Scholar] [CrossRef] [PubMed]
- Dutoit, V.; Herold-Mende, C.; Hilf, N.; Schoor, O.; Beckhove, P.; Bucher, J.; Dorsch, K.; Flohr, S.; Fritsche, J.; Lewandrowski, P.; et al. Exploiting the glioblastoma peptidome to discover novel tumour-associated antigens for immunotherapy. Brain 2012, 135, 1042–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatano, M.; Eguchi, J.; Tatsumi, T.; Kuwashima, N.; Dusak, J.E.; Kinch, M.S.; Pollack, I.F.; Hamilton, R.L.; Storkus, W.J.; Okada, H. EphA2 as a glioma-associated antigen: A novel target for glioma vaccines. Neoplasia 2005, 7, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Johanns, T.M.; Bowman-Kirigin, J.A.; Liu, C.; Dunn, G.P. Targeting Neoantigens in Glioblastoma: An Overview of Cancer Immunogenomics and Translational Implications. Neurosurgery 2017, 64, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.R.D.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal Transduct. Target. Ther. 2017, 2, 17040. [Google Scholar] [CrossRef] [PubMed]
- Yaghi, N.K.; Wei, J.; Hashimoto, Y.; Kong, L.-Y.; Gabrusiewicz, K.; Nduom, E.K.; Ling, X.; Huang, N.; Zhou, S.; Kerrigan, B.C.P.; et al. Immune modulatory nanoparticle therapeutics for intracerebral glioma. Neuro Oncol. 2017, 19, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Pico De Coañ, Y.; Choudhury, A.; Kiessling, R. Checkpoint blockade for cancer therapy: Revitalizing a suppressed immune system. Trends Mol. Med. 2015, 21, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swartz, A.M.; Batich, K.A.; Fecci, P.E.; Sampson, J.H. Peptide vaccines for the treatment of glioblastoma. J. Neurooncol. 2015, 123, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Müller, I.; Altherr, D.; Eyrich, M.; Flesch, B.; Friedmann, K.S.; Ketter, R.; Oertel, J.; Schwarz, E.C.; Technau, A.; Urbschat, S.; et al. Tumor antigen–specific T cells for immune monitoring of dendritic cell–treated glioblastoma patients. Cytotherapy 2016, 18, 1146–1161. [Google Scholar] [CrossRef] [PubMed]
- Migliorini, D.; Dietrich, P.-Y.; Stupp, R.; Linette, G.P.; Posey, A.D.; June, C.H. CAR T-Cell Therapies in Glioblastoma: A First Look. Clin. Cancer Res. 2018, 24, 535–540. [Google Scholar] [CrossRef] [PubMed]
- FDA Approval Brings First Gene Therapy to the United States. Available online: https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm574058.htm (accessed on 5 September 2018).
- Hunter, P. The fourth pillar. EMBO Rep. 2017, 18, 1889–1892. [Google Scholar] [CrossRef] [PubMed]
- Janiczek, M.; Szylberg, Ł.; Kasperska, A.; Kowalewski, A.; Parol, M.; Antosik, P.; Radecka, B.; Marszałek, A. Immunotherapy as a Promising Treatment for Prostate Cancer: A Systematic Review. J. Immunol. Res. 2017, 2017, 4861570. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Butterfield, L.H.; Hamilton, R.L.; Hoji, A.; Sakaki, M.; Ahn, B.J.; Kohanbash, G.; Drappatz, J.; Engh, J.; Amankulor, N.; et al. Induction of robust type-I CD8+ T-cell responses in WHO grade 2 low-grade glioma patients receiving peptide-based vaccines in combination with poly-ICLC. Clin. Cancer Res. 2015, 21, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.H.; Weninger, W.; Hunter, C.A. Trafficking of immune cells in the central nervous system. J. Clin. Investig. 2010, 120, 1368–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, I.; Han, S.J.; Kaur, G.; Crane, C.; Parsa, A.T. The role of microglia in central nervous system immunity and glioma immunology. J. Clin. Neurosci. 2010, 17, 6–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendeln, A.-C.; Degenhardt, K.; Kaurani, L.; Gertig, M.; Ulas, T.; Jain, G.; Wagner, J.; Häsler, L.M.; Wild, K.; Skodras, A.; et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature 2018, 556, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Filley, A.C.; Henriquez, M.; Dey, M. Recurrent glioma clinical trial, CheckMate-143: The game is not over yet. Oncotarget 2017, 8, 91779–91794. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Kang, W.Y.; Yoon, Y.; Jin, J.Y.; Song, H.J.; Her, J.H.; Kang, S.M.; Hwang, Y.K.; Kang, K.J.; Joo, K.M.; et al. Natural killer (NK) cells inhibit systemic metastasis of glioblastoma cells and have therapeutic effects against glioblastomas in the brain. BMC Cancer 2015, 15, 1011. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genßler, S.; Schönfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. JNCI J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Kmiecik, J.; Zimmer, J.; Chekenya, M. Natural killer cells in intracranial neoplasms: Presence and therapeutic efficacy against brain tumours. J. Neurooncol. 2014, 116, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Cancer Institute, Blue Ribbon Panel Report 2016. Available online: https://www.cancer.gov/research/key-initiatives/moonshot-cancer-initiative/blue-ribbon-panel/blue-ribbon-panel-report-2016.pdf (accessed on 14 September 2018).
- Debinski, W.; Obiri, N.I.; Powers, S.K.; Pastan, I.; Puri, R.K. Human glioma cells overexpress receptors for interleukin 13 and are extremely sensitive to a novel chimeric protein composed of interleukin 13 and pseudomonas exotoxin. Clin. Cancer Res. 1995, 1, 1253–1258. [Google Scholar] [PubMed]
- Debinski, W.; Gibo, D.M.; Hulet, S.W.; Connor, J.R.; Gillespie, G.Y. Receptor for interleukin 13 is a marker and therapeutic target for human high-grade gliomas. Clin. Cancer Res. 1999, 5, 985–990. [Google Scholar] [PubMed]
- Sattiraju, A.; Sai, K.K.S.; Xuan, A.; Pandya, D.N.; Almaguel, F.G.; Wadas, T.J.; Herpai, D.M.; Debinski, W.; Mintz, A. IL13RA2 targeted alpha particle therapy against glioblastomas. Oncotarget 2017, 8, 42997–43007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaci, B.; Brown, C.E.; Binello, E.; Werbaneth, K.; Sampath, P.; Sengupta, S. Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro Oncol. 2014, 16, 1304–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunwar, S. Convection enhanced delivery of IL13-PE38QQR for treatment of recurrent malignant glioma: Presentation of interim findings from ongoing phase 1 studies. Acta Neurochir. Suppl. 2003, 88, 105–111. [Google Scholar] [PubMed]
- Scanlan, M.J.; Gure, A.O.; Jungbluth, A.A.; Old, L.J.; Chen, Y.-T. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol. Rev. 2002, 188, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van den Eynde, B.; Knuth, A.; Boon, T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.-H.; Wong, E.W.; Cheng, C.Y. Cancer/testis (CT) antigens, carcinogenesis and spermatogenesis. Spermatogenesis 2011, 1, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freitas, M.R.P.; Malheiros, S.M.F.; Stávale, J.N.; Biassi, T.P.; Zamunér, F.T.; Begnami, M.D.F.S.; Soares, F.A.; Vettore, A.L.; Freitas, M.R.P.; Malheiros, S.M.F.; et al. Expression of Cancer/Testis Antigens is Correlated with Improved Survival in Glioblastoma. Oncotarget 2013, 4, 636–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, Y.; Komiyama, M.; Miyata, H.; Yagoto, M.; Ashizawa, T.; Iizuka, A.; Oshita, C.; Kume, A.; Nogami, M.; Ito, I.; et al. Novel cancer-testis antigen expression on glioma cell lines derived from high-grade glioma patients. Oncol. Rep. 2014, 31, 1683–1690. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, G.J.; Bancroft, A.; Grencis, R.K.; McKenzie, A.N. A distinct role for interleukin-13 in Th2-cell-mediated immune responses. Curr. Biol. 1998, 8, 339–342. [Google Scholar] [CrossRef] [Green Version]
- Deepak, P.; Kumar, S.; Acharya, A. Overexpression of Interleukin-13 in a Murine T-Cell Lymphoma: A Possible Factor of DL-Induced Immunosuppression and Tumor Progression. Cancer Invest. 2009, 27, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, S.; Tanaka, K.; Inoue, Y.; Toiyama, Y.; Okugawa, Y.; Iwata, T.; Mohri, Y.; Kusunoki, M. Low serum interleukin-13 levels correlate with poorer prognoses for colorectal cancer patients. Int. Surg. 2014, 99, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Sai, K.K.S.; Sattiraju, A.; Almaguel, F.G.; Xuan, A.; Rideout, S.; Krishnaswamy, R.S.; Zhang, J.; Herpai, D.M.; Debinski, W.; Mintz, A. Peptide-based PET imaging of the tumor restricted IL13RA2 biomarker. Oncotarget 2017, 8, 50997–51007. [Google Scholar] [CrossRef] [PubMed]
- Mintz, A.; Gibo, D.M.; Slagle-Webb, B.; Christensen, N.D.; Debinski, W. IL-13Ralpha2 is a glioma-restricted receptor for interleukin-13. Neoplasia 2002, 4, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Mintz, A.; Gibo, D.M.; MadhanKumar, A.B.; Cladel, N.M.; Christensen, N.D.; Debinski, W. Protein- and DNA-Based Active Immunotherapy Targeting Interleukin-13 Receptor Alpha2. Cancer Biother. Radiopharm. 2008, 23, 581–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debinski, W.; Dickinson, P.; Rossmeisl, J.H.; Robertson, J.; Gibo, D.M. New Agents for Targeting of IL-13RA2 Expressed in Primary Human and Canine Brain Tumors. PLoS ONE 2013, 8, e77719. [Google Scholar] [CrossRef] [PubMed]
- Pandya, H.; Gibo, D.M.; Garg, S.; Kridel, S.; Debinski, W. An interleukin 13 receptor α 2–specific peptide homes to human Glioblastoma multiforme xenografts. Neuro. Oncol. 2012, 14, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Pandya, H.; Gibo, D.M.; Debinski, W. Molecular targeting of intracellular compartments specifically in cancer cells. Genes Cancer 2010, 1, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Mintz, A.; Gibo, D.M.; Madhankumar, A.B.; Debinski, W. Molecular targeting with recombinant cytotoxins of interleukin-13 receptor alpha2-expressing glioma. J. Neurooncol. 2003, 64, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Arima, K.; Sato, K.; Tanaka, G.; Kanaji, S.; Terada, T.; Honjo, E.; Kuroki, R.; Matsuo, Y.; Izuhara, K. Characterization of the Interaction between Interleukin-13 and Interleukin-13 Receptors. J. Biol. Chem. 2005, 280, 24915–24922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsi, L.C.; Kundu, S.; Palomo, J.; Xu, B.; Ficco, R.; Vogelbaum, M.A.; Cathcart, M.K. Silencing IL-13Rα2 promotes glioblastoma cell death via endogenous signaling. Mol. Cancer Ther. 2011, 10, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Warden, C.D.; Starr, R.; Deng, X.; Badie, B.; Yuan, Y.-C.; Forman, S.J.; Barish, M.E. Glioma IL13Rα2 is associated with mesenchymal signature gene expression and poor patient prognosis. PLoS ONE 2013, 8, e77769. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.; Conyers, J.M.; Zhu, D.; Gibo, D.M.; Dorsey, J.F.; Debinski, W.; Mintz, A. IL-13Rα2-Targeted Therapy Escapees: Biologic and Therapeutic Implications. Transl. Oncol. 2011, 4, 390–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bregy, A.; Wong, T.M.; Shah, A.H.; Goldberg, J.M.; Komotar, R.J. Active immunotherapy using dendritic cells in the treatment of glioblastoma multiforme. Cancer Treat. Rev. 2013, 39, 891–907. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.J.; Pollack, I.F.; Okada, H. Immune-checkpoint blockade and active immunotherapy for glioma. Cancers 2013, 5, 1379–1412. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, D.T.; Fong, C.; Yew, A.; Spasic, M.; Garcia, H.M.; Kruse, C.A.; Yang, I. Passive immunotherapeutic strategies for the treatment of malignant gliomas. Neurosurg. Clin. N. Am. 2012, 23, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Schuster, M.; Nechansky, A.; Kircheis, R. Cancer immunotherapy. Biotechnol. J. 2006, 1, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Coley, W.B., II. Contribution to the Knowledge of Sarcoma. Ann. Surg. 1891, 14, 199–220. [Google Scholar] [CrossRef] [PubMed]
- Debinski, W.; Obiri, N.I.; Pastan, I.; Puri, R.K. A Novel Chimeric Protein Composed of Interleukin 13 and Pseudomonas Exotoxin Is Highly Cytotoxic to Human Carcinoma Cells Expressing Receptors for Interleukin 13 and Interleukin 4. J. Biol. Chem. 1995, 270, 16775–16780. [Google Scholar] [CrossRef] [PubMed]
- Debinski, W. An immune regulatory cytokine receptor and glioblastoma multiforme: An unexpected link. Crit. Rev. Oncog. 1998, 9, 255–268. [Google Scholar] [PubMed]
- Mut, M.; Sherman, J.H.; Shaffrey, M.E.; Schiff, D. Cintredekin besudotox in treatment of malignant glioma. Expert Opin. Biol. Ther. 2008, 8, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Kunwar, S.; Prados, M.D.; Chang, S.M.; Berger, M.S.; Lang, F.F.; Piepmeier, J.M.; Sampson, J.H.; Ram, Z.; Gutin, P.H.; Gibbons, R.D.; et al. Cintredekin Besudotox Intraparenchymal Study Group Direct Intracerebral Delivery of Cintredekin Besudotox (IL13-PE38QQR) in Recurrent Malignant Glioma: A Report by the Cintredekin Besudotox Intraparenchymal Study Group. J. Clin. Oncol. 2007, 25, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Vogelbaum, M.A.; Sampson, J.H.; Kunwar, S.; Chang, S.M.; Shaffrey, M.; Asher, A.L.; Lang, F.F.; Croteau, D.; Parker, K.; Grahn, A.Y.; et al. Convection-enhanced delivery of cintredekin besudotox (interleukin-13-PE38QQR) followed by radiation therapy with and without themozolomide in newly diagnosed malignant gliomas. Neurosurgery 2007, 61, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Kunwar, S.; Chang, S.M.; Prados, M.D.; Berger, M.S.; Sampson, J.H.; Croteau, D.; Sherman, J.W.; Grahn, A.Y.; Shu, V.S.; Dul, J.L.; et al. Safety of intraparenchymal convection-enhanced delivery of cintredekin besudotox in early-phase studies. Neurosurg. Focus 2006, 20, E15. [Google Scholar] [PubMed]
- Kunwar, S.; Chang, S.; Westphal, M.; Vogelbaum, M.; Sampson, J.; Barnett, G.; Shaffrey, M.; Ram, Z.; Piepmeier, J.; Prados, M.; et al. PRECISE Study Group Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro Oncol. 2010, 12, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Archer, G.; Pedain, C.; Wembacher-Schröder, E.; Westphal, M.; Kunwar, S.; Vogelbaum, M.A.; Coan, A.; Herndon, J.E.; Raghavan, R.; et al. Investigators, P. T. Poor drug distribution as a possible explanation for the results of the PRECISE trial. J. Neurosurg. 2010, 113, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Madhankumar, A.B.; Mintz, A.; Debinski, W. Alanine-scanning Mutagenesis of α-Helix D Segment of Interleukin-13 Reveals New Functionally Important Residues of the Cytokine. J. Biol. Chem. 2002, 277, 43194–43205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debinski, W. Recombinant cytotoxins specific for cancer cells. Ann. N. Y. Acad. Sci. 1999, 886, 297–299. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.P.; Debinski, W. Mutants of interleukin 13 with altered reactivity toward interleukin 13 receptors. J. Biol. Chem. 1999, 274, 29944–29950. [Google Scholar] [CrossRef] [PubMed]
- Madhankumar, A.B.; Mintz, A.; Debinski, W. Interleukin 13 mutants of enhanced avidity toward the glioma-associated receptor, IL13Ralpha2. Neoplasia 2004, 6, 15–22. [Google Scholar] [CrossRef]
- Sonawane, P.; Choi, Y.A.; Pandya, H.; Herpai, D.M.; Fokt, I.; Priebe, W.; Debinski, W. Novel Molecular Multilevel Targeted Antitumor Agents. Cancer Transl. Med. 2017, 3, 69–79. [Google Scholar] [PubMed]
- Balyasnikova, I.V; Wainwright, D.A.; Solomaha, E.; Lee, G.; Han, Y.; Thaci, B.; Lesniak, M.S. Characterization and immunotherapeutic implications for a novel antibody targeting interleukin (IL)-13 receptor α2. J. Biol. Chem. 2012, 287, 30215–30227. [Google Scholar] [CrossRef] [PubMed]
- Madhankumar, A.B.; Slagle-Webb, B.; Mintz, A.; Sheehan, J.M.; Connor, J.R. Interleukin-13 receptor-targeted nanovesicles are a potential therapy for glioblastoma multiforme. Mol. Cancer Ther. 2006, 5, 3162–3169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madhankumar, A.B.; Slagle-Webb, B.; Wang, X.; Yang, Q.X.; Antonetti, D.A.; Miller, P.A.; Sheehan, J.M.; Connor, J.R. Efficacy of interleukin-13 receptor-targeted liposomal doxorubicin in the intracranial brain tumor model. Mol. Cancer Ther. 2009, 8, 648–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Shi, W.; Song, W.; Wang, L.; Liu, X.; Chen, J.; Huang, R. Tumor cell targeted delivery by specific peptide-modified mesoporous silica nanoparticles. J. Mater. Chem. 2012, 22, 14608. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, K.; Zhao, J.; Liu, X.; Bu, J.; Yan, X.; Huang, R. Multifunctional Mesoporous Silica-Coated Graphene Nanosheet Used for Chemo-Photothermal Synergistic Targeted Therapy of Glioma. J. Am. Chem. Soc. 2013, 135, 4799–4804. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Ulasov, I.V.; Tyler, M.A.; Han, Y.; Glasgow, J.N.; Lesniak, M.S. Novel Recombinant Adenoviral Vector That Targets The Interleukin-13 Receptor α2 Chain Permits Effective Gene Transfer to Malignant Glioma. Hum. Gene Ther. 2007, 18, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Ou, W.; Marino, M.P.; Suzuki, A.; Joshi, B.; Husain, S.R.; Maisner, A.; Galanis, E.; Puri, R.K.; Reiser, J. Specific Targeting of Human Interleukin (IL)-13 Receptor α2-Positive Cells with Lentiviral Vectors Displaying IL-13. Hum. Gene Ther. Methods 2012, 23, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, G.; Roizman, B. Construction and properties of a herpes simplex virus 1 designed to enter cells solely via the IL-13alpha2 receptor. Proc. Natl. Acad. Sci. USA. 2006, 103, 5508–5513. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, K.; Hanauer, J.R.; Prüfer, S.; Münch, R.C.; Völker, I.; Filippis, C.; Jost, C.; Hanschmann, K.-M.; Cattaneo, R.; Peng, K.-W.; et al. DARPin-targeting of measles virus: Unique bispecificity, effective oncolysis, and enhanced safety. Mol. Ther. 2013, 21, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Candolfi, M.; Xiong, W.; Yagiz, K.; Liu, C.; Muhammad, A.K.M.G.; Puntel, M.; Foulad, D.; Zadmehr, A.; Ahlzadeh, G.E.; Kroeger, K.M.; et al. Therapy-mediated delivery of targeted cytotoxins for glioma therapeutics. Proc. Natl. Acad. Sci. USA 2010, 107, 20021–20026. [Google Scholar] [CrossRef] [PubMed]
- Phuphanich, S.; Wheeler, C.J.; Rudnick, J.D.; Mazer, M.; Wang, H.; Nuño, M.A.; Richardson, J.E.; Fan, X.; Ji, J.; Chu, R.M.; et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol. Immunother. 2013, 62, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, C.A.; Giacomini, C.P.; Vogel, H.; Jensen, K.C.; Florio, T.; Merlo, A.; Pollack, J.R.; Wong, A.J. EGFRvIII gene rearrangement is an early event in glioblastoma tumorigenesis and expression defines a hierarchy modulated by epigenetic mechanisms. Oncogene 2013, 32, 2670–2681. [Google Scholar] [CrossRef] [PubMed]
- Schuster, J.; Lai, R.K.; Recht, L.D.; Reardon, D.A.; Paleologos, N.A.; Groves, M.D.; Mrugala, M.M.; Jensen, R.; Baehring, J.M.; Sloan, A.; et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: The ACT III study. Neuro Oncol. 2015, 17, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Zussman, B.M.; Engh, J.A. Outcomes of the ACT III Study. Neurosurgery 2015, 76, N17. [Google Scholar] [CrossRef] [PubMed]
- Pollack, I.F.; Jakacki, R.I.; Butterfield, L.H.; Hamilton, R.L.; Panigrahy, A.; Normolle, D.P.; Connelly, A.K.; Dibridge, S.; Mason, G.; Whiteside, T.L.; et al. Immune responses and outcome after vaccination with glioma-associated antigen peptides and poly-ICLC in a pilot study for pediatric recurrent low-grade gliomas. Neuro Oncol. 2016, 18, 1157–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollack, I.F.; Jakacki, R.I.; Butterfield, L.H.; Hamilton, R.L.; Panigrahy, A.; Potter, D.M.; Connelly, A.K.; Dibridge, S.A.; Whiteside, T.L.; Okada, H. Antigen-Specific Immune Responses and Clinical Outcome After Vaccination With Glioma-Associated Antigen Peptides and Polyinosinic-Polycytidylic Acid Stabilized by Lysine and Carboxymethylcellulose in Children with Newly Diagnosed Malignant Brainstem and Nonbrainstem Gliomas. J. Clin. Oncol. 2014, 32, 2050–2058. [Google Scholar] [PubMed]
- Saka, M.; Amano, T.; Kajiwara, K.; Yoshikawa, K.; Ideguchi, M.; Nomura, S.; Fujisawa, H.; Kato, S.; Fujii, M.; Ueno, K.; et al. Vaccine therapy with dendritic cells transfected with Il13ra2 mRNA for glioma in mice. J. Neurosurg. 2010, 113, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Muul, L.M.; Solomon, D.; Rosenberg, S.A. Expansion of human tumor infiltrating lymphocytes for use in immunotherapy trials. J. Immunol. Methods 1987, 102, 127–141. [Google Scholar] [CrossRef]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer Regression in Patients After Transfer of Genetically Engineered Lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [PubMed]
- Eshhar, Z.; Gross, G. Chimeric T cell receptor which incorporates the anti-tumour specificity of a monoclonal antibody with the cytolytic activity of T cells: A model system for immunotherapeutical approach. Br. J. Cancer Suppl. 1990, 10, 27–29. [Google Scholar] [PubMed]
- Almåsbak, H.; Aarvak, T.; Vemuri, M.C. CAR T Cell Therapy: A Game Changer in Cancer Treatment. J. Immunol. Res. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V. CAR-T Cell Therapy: From the Bench to the Bedside. Cancers 2017, 9, 150. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves CAR-T Cell Therapy to Treat Adults with Certain Types of Large B-Cell Lymphoma. Available online: https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm581216.htm (accessed on 5 September 2018).
- Brown, C.E.; Starr, R.; Aguilar, B.; Brito, A.; Chang, B.; Sarkissian, A.; Weng, L.; Jensen, M.; Barish, M.E.; Badie, B.; et al. Clinical development of IL13Ra2-targeting CAR T cells for the treatment of glioblastoma. J. Immuno. Ther. Cancer 2015, 3, 114. [Google Scholar] [CrossRef]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.-C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13R 2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.-C.; Weng, L.; Chang, B.; Sarkissian, A.; Brito, A.; Sanchez, J.F.; et al. Optimization of IL13Rα2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Mol. Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Krebs, S.; Chow, K.K.H.; Yi, Z.; Rodriguez-Cruz, T.; Hegde, M.; Gerken, C.; Ahmed, N.; Gottschalk, S. T cells redirected to interleukin-13Rα2 with interleukin-13 mutein–chimeric antigen receptors have anti-glioma activity but also recognize interleukin-13Rα1. Cytotherapy 2014, 16, 1121–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagley, S.J.; Desai, A.S.; Linette, G.P.; June, C.H.; O’Rourke, D.M. CAR T-cell therapy for glioblastoma: Recent clinical advances and future challenges. Neuro Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kahlon, K.S.; Brown, C.; Cooper, L.J.N.; Raubitschek, A.; Forman, S.J.; Jensen, M.C. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004, 64, 9160–9166. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Aiyama, H.; Ishikawa, E. CAR-T cell in vivo tracking method using PET scan with the reporter gene and new investigational tracer [18 F] FHBG. Transl. Cancer Res. 2017, 6, S1003–S1005. [Google Scholar] [CrossRef]
- Yaghoubi, S.S.; Jensen, M.C.; Satyamurthy, N.; Budhiraja, S.; Paik, D.; Czernin, J.; Gambhir, S.S. Noninvasive detection of therapeutic cytolytic T cells with 18F-FHBG PET in a patient with glioma. Nat. Clin. Pract. Oncol. 2009, 6, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.; Jensen, M.C.; Lansdorp, P.M.; Gough, M.; Elliott, C.; Riddell, S.R. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J. Clin. Invest. 2008, 118, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Phase I Study of Cellular Immunotherapy for Recurrent/Refractory Malignant Glioma Using Intratumoral Infusions of GRm13Z40-2, An Allogeneic CD8+ Cytolitic T-Cell Line Genetically Modified to Express the IL 13-Zetakine and HyTK and to be Resistant to Glucocorticoids, in Combination with Interleukin-2. Available online: https://clinicaltrials.gov/ct2/show/NCT01082926 (accessed on 5 September 2018).
- Kong, S.; Sengupta, S.; Tyler, B.; Bais, A.J.; Ma, Q.; Doucette, S.; Zhou, J.; Sahin, A.; Carter, B.S.; Brem, H.; et al. Suppression of Human Glioma Xenografts with Second-Generation IL13R-Specific Chimeric Antigen Receptor-Modified T Cells. Clin. Cancer Res. 2012, 18, 5949–5960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, D.C.; Moysi, E.; Valentin, A.; Bergamaschi, C.; Devasundaram, S.; Fortis, S.P.; Bear, J.; Chertova, E.; Bess, J.; Sowder, R.; et al. Treatment with native heterodimeric IL-15 increases cytotoxic lymphocytes and reduces SHIV RNA in lymph nodes. PLoS Pathog. 2018, 14, e1006902. [Google Scholar]
- Xu, X.-J.; Song, D.-G.; Poussin, M.; Ye, Q.; Sharma, P.; Rodríguez-García, A.; Tang, Y.-M.; Powell, D.J. Multiparameter comparative analysis reveals differential impacts of various cytokines on CART cell phenotype and function ex vivo and in vivo. Oncotarget 2016, 7, 82354–82368. [Google Scholar] [CrossRef] [PubMed]
- Hurton, L.V.; Singh, H.; Najjar, A.M.; Switzer, K.C.; Mi, T.; Maiti, S.; Olivares, S.; Rabinovich, B.; Huls, H.; Forget, M.-A.; et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7788–E7797. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Wang, J.; Huang, H.; Zhao, Y. Novel immunotherapies for adult patients with B-lineage acute lymphoblastic leukemia. J. Hematol. Oncol. 2017, 10, 150. [Google Scholar] [CrossRef] [PubMed]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Barrett, D.; Teachey, D.T.; Grupp, S.A. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J. 2014, 20, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Au, R. Immunooncology: Can the Right Chimeric Antigen Receptors T-Cell Design Be Made to Cure All Types of Cancers and Will It Be Covered? J. Pharm. 2017, 2017, 7513687. [Google Scholar] [CrossRef] [PubMed]
- Mata, M.; Gerken, C.; Nguyen, P.; Krenciute, G.; Spencer, D.M.; Gottschalk, S. Inducible Activation of MyD88 and CD40 in CAR T Cells Results in Controllable and Potent Antitumor Activity in Preclinical Solid Tumor Models. Cancer Discov. 2017, 7, 1306–1319. [Google Scholar] [CrossRef] [PubMed]
- Engineering CD20-Specific Chimeric Receptor-Redirected T Cells with Inducible Co-Expression of a Caspase-9 Based Suicide Switch for Adoptive Immunotherapy of CD20 Positive Lymphoma. Mol. Ther. 2012, 20, S76. [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, H.J.; Brentjens, R.J. Overcoming Antigen Escape with CAR T-cell Therapy. Cancer Discov. 2015, 5, 1238–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, M.; Mukherjee, M.; Grada, Z.; Pignata, A.; Landi, D.; Navai, S.A.; Wakefield, A.; Fousek, K.; Bielamowicz, K.; Chow, K.K.H.; et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Invest. 2016, 126, 3036–3052. [Google Scholar] [CrossRef] [PubMed]
- Krenciute, G.; Prinzing, B.L.; Yi, Z.; Wu, M.-F.; Liu, H.; Dotti, G.; Balyasnikova, I.V; Gottschalk, S. Transgenic Expression of IL15 Improves Antiglioma Activity of IL13Rα2-CAR T Cells but Results in Antigen Loss Variants. Cancer Immunol. Res. 2017, 5, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Debinski, W. When better still might not be good enough. Transl. Cancer Res. 2017, 6, S1244–S1247. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benitez, J.A.; Ma, J.; D’Antonio, M.; Boyer, A.; Camargo, M.F.; Zanca, C.; Kelly, S.; Khodadadi-Jamayran, A.; Jameson, N.M.; Andersen, M.; et al. PTEN regulates glioblastoma oncogenesis through chromatin-associated complexes of DAXX and histone H3.3. Nat. Commun. 2017, 8, 15223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.C.; Lu, G.; Deng, Y.; Wang, C.D.; Su, X.W.; Zhou, J.Y.; Chan, T.M.; Hu, X.; Poon, W.S. Inhibition of NF-κB Pathway and Modulation of MAPK Signaling Pathways in Glioblastoma and Implications for Lovastatin and Tumor Necrosis Factor-Related Apoptosis Inducing Ligand (TRAIL) Combination Therapy. PLoS ONE 2017, 12, e0171157. [Google Scholar] [CrossRef] [PubMed]
- Gieryng, A.; Kaminska, B. Myeloid-derived suppressor cells in gliomas. Contemp. Oncol. (Poznan, Poland) 2016, 20, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Jun, Y.; Kim, S.H.; Hoang, H.-H.; Jung, Y.; Kim, S.; Kim, J.; Austin, R.H.; Lee, S.; Park, S. Rapid emergence and mechanisms of resistance by U87 glioblastoma cells to doxorubicin in an in vitro tumor microfluidic ecology. Proc. Natl. Acad. Sci. USA 2016, 113, 14283–14288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.; Pan, Y.; Gutmann, D.H. The power of the few. Genes Dev. 2017, 31, 1177–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unified nomenclature for Eph family receptors and their ligands, the ephrins. Eph Nomenclature Committee. Cell 1997, 90, 403–404. [CrossRef]
- Klein, R. Eph/ephrin signalling during development. Development 2012, 139, 4105–4109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatzizacharias, N.A.; Giaginis, C.T.; Agapitos, E.; Theocharis, S.E. The role of ephrins’ receptors and ephrins’ ligands in normal placental development and disease. Expert Opin. Ther. Targets 2014, 18, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Baumann, G.; Travieso, L.; Liebl, D.J.; Theus, M.H. Pronounced hypoxia in the subventricular zone following traumatic brain injury and the neural stem/progenitor cell response. Exp. Biol. Med. 2013, 238, 830–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frugier, T.; Conquest, A.; McLean, C.; Currie, P.; Moses, D.; Goldshmit, Y. Expression and Activation of EphA4 in the Human Brain after Traumatic Injury. J. Neuropathol. Exp. Neurol. 2012, 71, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Cavodeassi, F.; Ivanovitch, K.; Wilson, S.W. Eph/Ephrin signalling maintains eye field segregation from adjacent neural plate territories during forebrain morphogenesis. Development 2013, 140, 4193–4202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochenek, M.L.; Dickinson, S.; Astin, J.W.; Adams, R.H.; Nobes, C.D. Ephrin-B2 regulates endothelial cell morphology and motility independently of Eph-receptor binding. J. Cell Sci. 2010, 123, 1235–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, H.-Q.; Wu, X.-S.; Wei, B.; Chen, L. Eph receptors and ephrins as targets for cancer therapy. J. Cell. Mol. Med. 2012, 16, 2894–2909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Park, P.J.; Lai, W.; Maher, E.; Chakravarti, A.; Durso, L.; Jiang, X.; Yu, Y.; Brosius, A.; Thomas, M.; et al. Genome-Wide Screen Reveals Functional Gene Clusters in the Cancer Genome and Identifies EphA2 as a Mitogen in Glioblastoma. Cancer Res. 2006, 66, 10815–10823. [Google Scholar] [CrossRef] [PubMed]
- Wykosky, J.; Gibo, D.M.; Stanton, C.; Debinski, W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol. Cancer Res. 2005, 3, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Wykosky, J.; Debinski, W. The EphA2 receptor and ephrinA1 ligand in solid tumors: Function and therapeutic targeting. Mol. Cancer Res. 2008, 6, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; He, S.; Fu, J.; Li, G.; Xu, R.; Lu, H.; Deng, J. Expression of EphrinB2 and EphB4 in glioma tissues correlated to the progression of glioma and the prognosis of glioblastoma patients. Clin. Transl. Oncol. 2012, 14, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-F.; Fokas, E.; Juricko, J.; You, A.; Rose, F.; Pagenstecher, A.; Engenhart-Cabillic, R.; An, H.-X. Increased expression of EphA7 correlates with adverse outcome in primary and recurrent glioblastoma multiforme patients. BMC Cancer 2008, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- Teng, L.; Nakada, M.; Furuyama, N.; Sabit, H.; Furuta, T.; Hayashi, Y.; Takino, T.; Dong, Y.; Sato, H.; Sai, Y.; et al. Ligand-dependent EphB1 signaling suppresses glioma invasion and correlates with patient survival. Neuro. Oncol. 2013, 15, 1710–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferluga, S.; Tomé, C.M.L.; Herpai, D.M.; D’Agostino, R.; Debinski, W.; Ferluga, S.; Tomé, C.M.L.; Herpai, D.M.; D’Agostino, R.; Debinski, W. Simultaneous targeting of Eph receptors in glioblastoma. Oncotarget 2016, 7, 59860–59876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiari, R.; Hames, G.; Stroobant, V.; Texier, C.; Maillère, B.; Boon, T.; Coulie, P.G.; Baer, M.; Palath, V.; Bebbington, C.; et al. Identification of a tumor-specific shared antigen derived from an Eph receptor and presented to CD4 T cells on HLA class II molecules. Cancer Res. 2000, 60, 4855–4863. [Google Scholar] [PubMed]
- Day, B.W.; Stringer, B.W.; Al-Ejeh, F.; Ting, M.J.; Wilson, J.; Ensbey, K.S.; Jamieson, P.R.; Bruce, Z.C.; Lim, Y.C.; Offenhäuser, C.; et al. EphA3 Maintains Tumorigenicity and Is a Therapeutic Target in Glioblastoma Multiforme. Cancer Cell 2013, 23, 238–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wykosky, J.; Gibo, D.M.; Debinski, W. A novel, potent, and specific ephrinA1-based cytotoxin against EphA2 receptor expressing tumor cells. Mol. Cancer Ther. 2007, 6, 3208–3218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossmeisl, J.H.; Herpai, D.; Robertson, J.L.; Dickinson, P.J.; Tatter, S.B.; Debinski, W. P08.12 Tolerability and initial efficacy of convection-enhanced delivery of combinatorial IL-13RA2 and EphA2 targeted cytotoxins to dogs with spontaneous intracranial malignant gliomas. Neuro Oncol. 2017, 19. [Google Scholar] [CrossRef]
- Yi, Z.; Prinzing, B.L.; Cao, F.; Gottschalk, S.; Krenciute, G. Optimizing EphA2-CAR T Cells for the Adoptive Immunotherapy of Glioma. Mol. Ther. Methods Clin. Dev. 2018, 9, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Debinski, W.; Priebe, W.; Tatter, S.B. Maximizing Local Access to Therapeutic Deliveries in Glioblastoma. Part I: Targeted Cytotoxic Therapy; Codon Publications: Brisbane, Australia, 2017; ISBN 9780994438126. [Google Scholar]
- Konecny, G.E.; Glas, R.; Dering, J.; Manivong, K.; Qi, J.; Finn, R.S.; Yang, G.R.; Hong, K.-L.; Ginther, C.; Winterhoff, B.; et al. Activity of the multikinase inhibitor dasatinib against ovarian cancer cells. Br. J. Cancer 2009, 101, 1699–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tryfonopoulos, D.; Walsh, S.; Collins, D.M.; Flanagan, L.; Quinn, C.; Corkery, B.; McDermott, E.W.; Evoy, D.; Pierce, A.; O’Donovan, N.; et al. Src: A potential target for the treatment of triple-negative breast cancer. Ann. Oncol. 2011, 22, 2234–2240. [Google Scholar] [CrossRef] [PubMed]
- Yeddula, N.; Xia, Y.; Ke, E.; Beumer, J.; Verma, I.M.; Berns, A.; Jacks, T.; Mccormick, F. Screening for tumor suppressors: Loss of ephrin receptor A2 cooperates with oncogenic KRas in promoting lung adenocarcinoma. Proc. Natl. Acad. Sci. USA 2015, 47, E6476–E6485. [Google Scholar] [CrossRef] [PubMed]
- Petty, A.; Myshkin, E.; Qin, H.; Guo, H.; Miao, H.; Tochtrop, G.P.; Hsieh, J.-T.; Page, P.; Liu, L.; Lindner, D.J.; et al. A Small Molecule Agonist of EphA2 Receptor Tyrosine Kinase Inhibits Tumor Cell Migration In Vitro and Prostate Cancer Metastasis In Vivo. PLoS ONE 2012, 7, e42120. [Google Scholar] [CrossRef] [PubMed]
- Rundle-Thiele, D.; Head, R.; Cosgrove, L.; Martin, J.H. Repurposing some older drugs that cross the blood-brain barrier and have potential anticancer activity to provide new treatment options for glioblastoma. Br. J. Clin. Pharmacol. 2016, 81, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Pasquale, E.B. Erratum: Eph receptor signalling casts a wide net on cell behaviour. Nat. Rev. Mol. Cell Biol. 2005, 6, 462–475. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J. Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 2015, 36, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A. Blebbing: Motility research moves in a new direction. Nat. Rev. Mol. Cell Biol. 2009, 10, 164. [Google Scholar] [CrossRef]
- Montano, N.; Cenci, T.; Martini, M.; D’Alessandris, Q.G.; Pelacchi, F.; Ricci-Vitiani, L.; Maira, G.; De Maria, R.; Larocca, L.M.; Pallini, R. Expression of EGFRvIII in glioblastoma: Prognostic significance revisited. Neoplasia 2011, 13, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Heimberger, A.B.; Suki, D.; Yang, D.; Shi, W.; Aldape, K. The natural history of EGFR and EGFRvIII in glioblastoma patients. J. Transl. Med. 2005, 3, 38. [Google Scholar] [CrossRef] [PubMed]
- Voldborg, B.R.; Damstrup, L.; Spang-Thomsen, M.; Poulsen, H.S. Epidermal growth factor receptor (EGFR) and EGFR mutations, function and possible role in clinical trials. Ann. Oncol. 1997, 8, 1197–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ennis, B.W.; Lippman, M.E.; Dickson, R.B. The EGF receptor system as a target for antitumor therapy. Cancer Invest. 1991, 9, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Libermann, T.A.; Nusbaum, H.R.; Razon, N.; Kris, R.; Lax, I.; Soreq, H.; Whittle, N.; Waterfield, M.D.; Ullrich, A.; Schlessinger, J. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature 1985, 313, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Bonavia, R.; Inda, M.M.; Vandenberg, S.; Cheng, S.-Y.; Nagane, M.; Hadwiger, P.; Tan, P.; Sah, D.W.Y.; Cavenee, W.K.; Furnari, F.B. EGFRvIII promotes glioma angiogenesis and growth through the NF-κB, interleukin-8 pathway. Oncogene 2012, 31, 4054–4066. [Google Scholar] [CrossRef] [PubMed]
- Latha, K.; Li, M.; Chumbalkar, V.; Gururaj, A.; Hwang, Y.; Dakeng, S.; Sawaya, R.; Aldape, K.; Cavenee, W.K.; Bogler, O.; et al. Nuclear EGFRvIII-STAT5b complex contributes to glioblastoma cell survival by direct activation of the Bcl-XL promoter. Int. J. Cancer 2013, 132, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Resnier, P.; David, S.; Lautram, N.; Delcroix, G.J.-R.; Clavreul, A.; Benoit, J.-P.; Passirani, C. EGFR siRNA lipid nanocapsules efficiently transfect glioma cells in vitro. Int. J. Pharm. 2013, 454, 748–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Park, J.O.; Zhang, M. Treatment of glioblastoma multiforme using a combination of small interfering RNA targeting epidermal growth factor receptor and β-catenin. J. Gene Med. 2013, 15, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.D.; Krishnan, S.; Sarkaria, J.N.; Wu, W.; Jaeckle, K.A.; Uhm, J.H.; Geoffroy, F.J.; Arusell, R.; Kitange, G.; Jenkins, R.B.; et al. North Central Cancer Treatment Group Study N0177 Phase I/II Trial of Erlotinib and Temozolomide With Radiation Therapy in the Treatment of Newly Diagnosed Glioblastoma Multiforme: North Central Cancer Treatment Group Study N0177. J. Clin. Oncol. 2008, 26, 5603–5609. [Google Scholar] [PubMed]
- Gallego, O.; Cuatrecasas, M.; Benavides, M.; Segura, P.P.; Berrocal, A.; Erill, N.; Colomer, A.; Quintana, M.J.; Balaña, C.; Gil, M.; et al. Efficacy of erlotinib in patients with relapsed gliobastoma multiforme who expressed EGFRVIII and PTEN determined by immunohistochemistry. J. Neurooncol. 2014, 116, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Custodio, A.; Calles, A.; Pérez-Segura, P. Response to erlotinib in recurrent glioblastoma multiforme showing coexpression of EGFRvIII and PTEN. Clin. Transl. Oncol. 2010, 12, 310–314. [Google Scholar] [PubMed]
- Uhm, J.H.; Ballman, K.V; Wu, W.; Giannini, C.; Krauss, J.C.; Buckner, J.C.; James, C.D.; Scheithauer, B.W.; Behrens, R.J.; Flynn, P.J.; et al. Phase II evaluation of gefitinib in patients with newly diagnosed Grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Fukai, J.; Nishio, K.; Itakura, T.; Koizumi, F. Antitumor activity of cetuximab against malignant glioma cells overexpressing EGFR deletion mutant variant III. Cancer Sci. 2008, 99, 2062–2069. [Google Scholar] [CrossRef] [PubMed]
- Vredenburgh, J.J.; Desjardins, A.; Herndon, J.E.; Marcello, J.; Reardon, D.A.; Quinn, J.A.; Rich, J.N.; Sathornsumetee, S.; Gururangan, S.; Sampson, J.; et al. Bevacizumab plus irinotecan in recurrent glioblastoma multiforme. J. Clin. Oncol. 2007, 25, 4722–4729. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Peng, Y.; Liao, Y.; Jiang, W.; Wei, R.; Huo, L.; Han, Z.; Duan, C.; Zhong, M. Nimotuzumab prolongs survival in patients with malignant gliomas: A phase I/II clinical study of concomitant radiochemotherapy with or without nimotuzumab. Exp. Ther. Med. 2012, 4, 151–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampson, J.H.; Akabani, G.; Archer, G.E.; Berger, M.S.; Coleman, R.E.; Friedman, A.H.; Friedman, H.S.; Greer, K.; Herndon, J.E.; Kunwar, S.; et al. Intracerebral infusion of an EGFR-targeted toxin in recurrent malignant brain tumors. Neuro. Oncol. 2008, 10, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, B.D.; O’Rourke, D.M.; Maus, M.V. Engineering Chimeric Antigen Receptor T cells to Treat Glioblastoma. J. Target. Ther. Cancer 2017, 6, 22–25. [Google Scholar] [PubMed]
- Rosenberg, S.A. Cell transfer immunotherapy for metastatic solid cancer—What clinicians need to know. Nat. Rev. Clin. Oncol. 2011, 8, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor–Modified Virus-Specific T Cells for Progressive Glioblastoma. JAMA Oncol. 2017, 3, 1094. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Wakefield, A.; Ghazi, A.; Ashoori, A.; Diouf, O.; Gerken, C.; Landi, D.; et al. Autologous HER2 CMV bispecific CAR T cells are safe and demonstrate clinical benefit for glioblastoma in a Phase I trial. J. Immunother. Cancer 2015, 3, O11. [Google Scholar] [CrossRef]
- Reardon, D.A.; Turner, S.; Peters, K.B.; Desjardins, A.; Gururangan, S.; Sampson, J.H.; McLendon, R.E.; Herndon, J.E.; Jones, L.W.; Kirkpatrick, J.P.; et al. A review of VEGF/VEGFR-targeted therapeutics for recurrent glioblastoma. J. Natl. Compr. Canc. Netw. 2011, 9, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Nazarenko, I.; Hede, S.-M.; He, X.; Hedrén, A.; Thompson, J.; Lindström, M.S.; Nistér, M. PDGF and PDGF receptors in glioma. Ups. J. Med. Sci. 2012, 117, 99–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, J.V.; Balasubramaniyan, V.; Walenkamp, A.; Kruyt, F.A.E. TGF-β as a therapeutic target in high grade gliomas—Promises and challenges. Biochem. Pharmacol. 2013, 85, 478–485. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Ranganath, K.; Hammerman, P.S.; Vaklavas, C.; Mohindra, N.; Kalyan, A.; Matsangou, M.; Costa, R.; Carneiro, B.; Villaflor, V.M.; et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: The current landscape and barriers to clinical application. Oncotarget 2017, 8, 16052–16074. [Google Scholar] [CrossRef] [PubMed]
- Awad, A.J.; Burns, T.C.; Zhang, Y.; Abounader, R. Targeting MET for glioma therapy. Neurosurg. Focus 2014, 37, E10. [Google Scholar] [CrossRef] [PubMed]
- Joshi, B.H.; Leland, P.; Silber, J.; Kreitman, R.J.; Pastan, I.; Berger, M.; Puri, R.K. IL-4 receptors on human medulloblastoma tumours serve as a sensitive target for a circular permuted IL-4-Pseudomonas exotoxin fusion protein. Br. J. Cancer 2002, 86, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Rustamzadeh, E.; Li, C.; Doumbia, S.; Hall, W.A.; Vallera, D.A. Targeting the over-expressed urokinase-type plasminogen activator receptor on glioblastoma multiforme. J. Neurooncol. 2003, 65, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Cherry, A.E.; Stella, N. G protein-coupled receptors as oncogenic signals in glioma: Emerging therapeutic avenues. Neuroscience 2014, 278, 222–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.; Wu, H.; Li, Y.; Huang, Y.; Yao, L.; Chen, X.; Han, X.; Zhou, Y.; Du, Z. Targeting transferrin receptor delivery of temozolomide for a potential glioma stem cell-mediated therapy. Oncotarget 2017, 8, 74451–74465. [Google Scholar] [CrossRef] [PubMed]
- Kumari, S.; Ahsan, S.M.; Kumar, J.M.; Kondapi, A.K.; Rao, N.M. Overcoming blood brain barrier with a dual purpose Temozolomide loaded Lactoferrin nanoparticles for combating glioma (SERP-17-12433). Sci. Rep. 2017, 7, 6602. [Google Scholar] [CrossRef] [PubMed]
- Maletínská, L.; Blakely, E.A.; Bjornstad, K.A.; Deen, D.F.; Knoff, L.J.; Forte, T.M. Human glioblastoma cell lines: Levels of low-density lipoprotein receptor and low-density lipoprotein receptor-related protein. Cancer Res. 2000, 60, 2300–2303. [Google Scholar] [PubMed]
- Guo, J.; Schlich, M.; Cryan, J.F.; O’Driscoll, C.M. Targeted Drug Delivery via Folate Receptors for the Treatment of Brain Cancer: Can the Promise Deliver? J. Pharm. Sci. 2017, 106, 3413–3420. [Google Scholar] [CrossRef] [PubMed]
- Paolillo, M.; Serra, M.; Schinelli, S. Integrins in glioblastoma: Still an attractive target? Pharmacol. Res. 2016, 113, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Shen, Y.L.; Keegan, P.; Pazdur, R. FDA Drug Approval Summary: Bevacizumab (Avastin(R)) as Treatment of Recurrent Glioblastoma Multiforme. Oncologist 2009, 14, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Soffietti, R.; Trevisan, E.; Bertero, L.; Cassoni, P.; Morra, I.; Fabrini, M.G.; Pasqualetti, F.; Lolli, I.; Castiglione, A.; Ciccone, G.; et al. Bevacizumab and fotemustine for recurrent glioblastoma: A phase II study of AINO (Italian Association of Neuro-Oncology). J. Neurooncol. 2014, 116, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Balaña, C.; Estival, A.; Pineda, E.; Sepúlveda, J.; Mesía, C.; del Barco, S.; Gil-Gil, M.; Hardy, M.; Indacoechea, A.; Cardona, A.F. Prolonged survival after bevacizumab rechallenge in glioblastoma patients with previous response to bevacizumab. Neuro Oncol. Pract. 2016, 4, 15–23. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Neyns, B.; Goldbrunner, R.; Schlegel, U.; Clement, P.M.J.; Grabenbauer, G.G.; Ochsenbein, A.F.; Simon, M.; Dietrich, P.-Y.; et al. Phase I/IIa Study of Cilengitide and Temozolomide With Concomitant Radiotherapy Followed by Cilengitide and Temozolomide Maintenance Therapy in Patients With Newly Diagnosed Glioblastoma. J. Clin. Oncol. 2010, 28, 2712–2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, D.A.; Neyns, B.; Weller, M.; Tonn, J.C.; Nabors, L.B.; Stupp, R. Cilengitide: An RGD pentapeptide ανβ3 and ανβ5 integrin inhibitor in development for glioblastoma and other malignancies. Future Oncol. 2011, 7, 339–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timotheadou, E. New agents targeting angiogenesis in glioblastoma. Chemother. Res. Pract. 2011, 2011, 878912. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, T.T.; Mulholland, P.; Neyns, B.; Nabors, L.B.; Campone, M.; Wick, A.; Mason, W.; Mikkelsen, T.; Phuphanich, S.; Ashby, L.S.; et al. Phase III Randomized Trial Comparing the Efficacy of Cediranib As Monotherapy, and in Combination With Lomustine, Versus Lomustine Alone in Patients With Recurrent Glioblastoma. J. Clin. Oncol. 2013, 31, 3212–3218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zustovich, F.; Landi, L.; Lombardi, G.; Porta, C.; Galli, L.; Fontana, A.; Amoroso, D.; Galli, C.; Andreuccetti, M.; Falcone, A.; et al. Sorafenib plus daily low-dose temozolomide for relapsed glioblastoma: A phase II study. Anticancer Res. 2013, 33, 3487–3494. [Google Scholar] [CrossRef] [PubMed]
- Wetmore, C.; Daryani, V.M.; Billups, C.A.; Boyett, J.M.; Leary, S.; Tanos, R.; Goldsmith, K.C.; Stewart, C.F.; Blaney, S.M.; Gajjar, A. Phase II evaluation of sunitinib in the treatment of recurrent or refractory high-grade glioma or ependymoma in children: A children’s Oncology Group Study ACNS1021. Cancer Med. 2016, 5, 1416–1424. [Google Scholar] [CrossRef] [PubMed]
- Haberler, C.; Gelpi, E.; Marosi, C.; Rössler, K.; Birner, P.; Budka, H.; Hainfellner, J.A. Immunohistochemical Analysis of Platelet-derived Growth Factor Receptor-α, -β, c-kit, c-abl, and Arg Proteins in Glioblastoma: Possible Implications for Patient Selection for Imatinib Mesylate Therapy. J. Neurooncol. 2006, 76, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, E.; Stupp, R.; van den Bent, M.J.; van Herpen, C.; Laigle Donadey, F.; Gorlia, T.; Hegi, M.; Lhermitte, B.; Strauss, L.C.; Allgeier, A.; et al. EORTC 26083 phase I/II trial of dasatinib in combination with CCNU in patients with recurrent glioblastoma. Neuro Oncol. 2012, 14, 1503–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, E.; Walters, I.B.; Hanahan, D. Brivanib, a dual FGF/VEGF inhibitor, is active both first and second line against mouse pancreatic neuroendocrine tumors developing adaptive/evasive resistance to VEGF inhibition. Clin. Cancer Res. 2011, 17, 5299–5310. [Google Scholar] [CrossRef] [PubMed]
- Weaver, M.; Laske, D.W. Transferrin receptor ligand-targeted toxin conjugate (Tf-CRM107) for therapy of malignant gliomas. J. Neurooncol. 2003, 65, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Han, H.; He, X.; Li, S.; Wu, C.; Yu, C.; Wang, S. Expression of the galectin-9-Tim-3 pathway in glioma tissues is associated with the clinical manifestations of glioma. Oncol. Lett. 2016, 11, 1829–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Didenko, V.V; Ngo, H.N.; Minchew, C.; Baskin, D.S. Apoptosis of T lymphocytes invading glioblastomas multiforme: A possible tumor defense mechanism. J. Neurosurg. 2002, 96, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Fecci, P.E.; Ochiai, H.; Mitchell, D.A.; Grossi, P.M.; Sweeney, A.E.; Archer, G.E.; Cummings, T.; Allison, J.P.; Bigner, D.D.; Sampson, J.H. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin. Cancer Res. 2007, 13, 2158–2167. [Google Scholar] [CrossRef] [PubMed]
- Xue, S.; Song, G.; Yu, J. The prognostic significance of PD-L1 expression in patients with glioma: A meta-analysis. Sci. Rep. 2017, 7, 4231. [Google Scholar] [CrossRef] [PubMed]
- Nduom, E.K.; Weller, M.; Heimberger, A.B. Immunosuppressive mechanisms in glioblastoma. Neuro. Oncol. 2015, 17, vii9–vii14. [Google Scholar]
- Zagzag, D.; Salnikow, K.; Chiriboga, L.; Yee, H.; Lan, L.; Ali, M.A.; Garcia, R.; Demaria, S.; Newcomb, E.W. Downregulation of major histocompatibility complex antigens in invading glioma cells: Stealth invasion of the brain. Lab. Investig. 2005, 85, 328–341. [Google Scholar] [CrossRef] [PubMed]
- Silk, A.W.; Bassetti, M.F.; West, B.T.; Tsien, C.I.; Lao, C.D. Ipilimumab and radiation therapy for melanoma brain metastases. Cancer Med. 2013, 2, 899–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vom Berg, J.; Vrohlings, M.; Haller, S.; Haimovici, A.; Kulig, P.; Sledzinska, A.; Weller, M.; Becher, B. Intratumoral IL-12 combined with CTLA-4 blockade elicits T cell-mediated glioma rejection. J. Exp. Med. 2013, 210, 2803–2811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwalla, P.; Barnard, Z.; Fecci, P.; Dranoff, G.; Curry, W.T. Sequential Immunotherapy by Vaccination with GM-CSF–expressing Glioma Cells and CTLA-4 Blockade Effectively Treats Established Murine Intracranial Tumors. J. Immunother. 2012, 35, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hodi, F.S.; Buchbinder, E.I. Inhibition of Immune Checkpoints and Vascular Endothelial Growth Factor as Combination Therapy for Metastatic Melanoma: An Overview of Rationale, Preclinical Evidence, and Initial Clinical Data. Front. Oncol. 2015, 5, 202. [Google Scholar] [CrossRef] [PubMed]
- Prasad, V.; Kaestner, V. Nivolumab and pembrolizumab: Monoclonal antibodies against programmed cell death-1 (PD-1) that are interchangeable. Semin. Oncol. 2017, 44, 132–135. [Google Scholar] [CrossRef] [PubMed]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro. Oncol. 2018, 20, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Momtaz, P.; Postow, M.A. Immunologic checkpoints in cancer therapy: Focus on the programmed death-1 (PD-1) receptor pathway. Pharmgenomics Pers. Med. 2014, 7, 357–365. [Google Scholar] [PubMed]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Feng, S.; Xu, L.; Shi, W.; Wang, X.; Wang, H.; Yu, C.; Dong, T.; Xu, M.; Liang, G. Tim-3 on Peripheral CD4+ and CD8+ T Cells Is Involved in the Development of Glioma. DNA Cell Biol. 2014, 33, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Ngiow, S.F.; von Scheidt, B.; Akiba, H.; Yagita, H.; Teng, M.W.L.; Smyth, M.J. Anti-TIM3 Antibody Promotes T Cell IFN-Mediated Antitumor Immunity and Suppresses Established Tumors. Cancer Res. 2011, 71, 3540–3551. [Google Scholar] [CrossRef] [PubMed]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, M.; Sugamura, K.; Sano, K.; Nakai, M.; Sugita, K.; Hinuma, Y. High-affinity receptor-mediated internalization and degradation of interleukin 2 in human T cells. J. Exp. Med. 1986, 163, 550–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barzon, L.; Pacenti, M.; Franchin, E.; Colombo, F.; Palù, G. HSV-tk/IL-2 Gene Therapy for Glioblastoma Multiforme. Methods Mol. Biol. 2009, 542, 529–549. [Google Scholar] [PubMed]
- Okada, H.; Giezeman-Smits, K.M.; Tahara, H.; Attanucci, J.; Fellows, W.K.; Lotze, M.T.; Chambers, W.H.; Bozik, M.E. Effective cytokine gene therapy against an intracranial glioma using a retrovirally transduced IL-4 plus HSVtk tumor vaccine. Gene Ther. 1999, 6, 219–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaji, H.; Tokuyama, T.; Yamasaki, T.; Amano, S.; Sakai, N.; Namba, H. Interferon-β and temozolomide combination therapy for temozolomide monotherapy-refractory malignant gliomas. Mol. Clin. Oncol. 2015, 3, 909–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boorjian, S.A.; Milowsky, M.I.; Kaplan, J.; Albert, M.; Cobham, M.V.; Coll, D.M.; Mongan, N.P.; Shelton, G.; Petrylak, D.; Gudas, L.J.; et al. Phase 1/2 Clinical Trial of Interferon α2b and Weekly Liposome-encapsulated All-trans Retinoic Acid in Patients with Advanced Renal Cell Carcinoma. J. Immunother. 2007, 30, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Mancone, S.; Lycan, T.; Ahmed, T.; Topaloglu, U.; Dothard, A.; Petty, W.J.; Strowd, R.E. Severe neurologic complications of immune checkpoint inhibitors: A single-center review. J. Neurol. 2018, 265, 1636–1642. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, A.; Nandhu, M.S.; Behera, P.; Chiocca, E.A.; Viapiano, M.S. Strategies in gene therapy for glioblastoma. Cancers 2013, 5, 1271–1305. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liikanen, I.; Koski, A.; Merisalo-Soikkeli, M.; Hemminki, O.; Oksanen, M.; Kairemo, K.; Joensuu, T.; Kanerva, A.; Hemminki, A. Serum HMGB1 is a predictive and prognostic biomarker for oncolytic immunotherapy. Oncoimmunology 2015, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Ge, K.; Zheng, Z. [Suicide gene therapy for glioma using HSV-tk/GCV system]. Zhonghua Zhong Liu Za Zhi 1997, 19, 353–357. [Google Scholar] [PubMed]
- Rainov, N.G. A Phase III Clinical Evaluation of Herpes Simplex Virus Type 1 Thymidine Kinase and Ganciclovir Gene Therapy as an Adjuvant to Surgical Resection and Radiation in Adults with Previously Untreated Glioblastoma Multiforme. Hum. Gene Ther. 2000, 11, 2389–2401. [Google Scholar] [CrossRef] [PubMed]
- King, G.D.; Kroeger, K.M.; Bresee, C.J.; Candolfi, M.; Liu, C.; Manalo, C.M.; Muhammad, A.G.; Pechnick, R.N.; Lowenstein, P.R.; Castro, M.G. Flt3L in Combination With HSV1-TK-mediated Gene Therapy Reverses Brain Tumor–induced Behavioral Deficits. Mol. Ther. 2008, 16, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.; Scott, K.J.; Taggart, D.; West, E.J.; Wilson, E.; Nuovo, G.J.; Thomson, S.; Corns, R.; Mathew, R.K.; Fuller, M.J.; et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Paraskevakou, G.; Liu, C.; Iankov, I.D.; Msaouel, P.; Zollman, P.; Myers, R.; Peng, K.W.; Russell, S.J.; Galanis, E. Oncolytic measles virus strains in the treatment of gliomas. Expert Opin. Biol. Ther. 2008, 8, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Aoki, H.; Kühnel, F.; Kondo, Y.; Kubicka, S.; Wirth, T.; Iwado, E.; Iwamaru, A.; Fujiwara, K.; Hess, K.R.; et al. Autophagic Cell Death of Malignant Glioma Cells Induced by a Conditionally Replicating Adenovirus. JNCI J. Natl. Cancer Inst. 2006, 98, 625–636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, J.; Wakimoto, H. Oncolytic herpes simplex virus-based strategies: Toward a breakthrough in glioblastoma therapy. Front. Microbiol. 2014, 5, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiocca, E.A.; Abbed, K.M.; Tatter, S.; Louis, D.N.; Hochberg, F.H.; Barker, F.; Kracher, J.; Grossman, S.A.; Fisher, J.D.; Carson, K.; et al. A Phase I Open-Label, Dose-Escalation, Multi-Institutional Trial of Injection with an E1B-Attenuated Adenovirus, ONYX-015, into the Peritumoral Region of Recurrent Malignant Gliomas, in the Adjuvant Setting. Mol. Ther. 2004, 10, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Panek, W.K.; Kane, J.R.; Young, J.S.; Rashidi, A.; Kim, J.W.; Kanojia, D.; Lesniak, M.S. Hitting the nail on the head: Combining oncolytic adenovirus-mediated virotherapy and immunomodulation for the treatment of glioma. Oncotarget 2017, 8, 89391–89405. [Google Scholar] [CrossRef] [PubMed]
- Cary, Z.D.; Willingham, M.C.; Lyles, D.S. Oncolytic vesicular stomatitis virus induces apoptosis in U87 glioblastoma cells by a type II death receptor mechanism and induces cell death and tumor clearance in vivo. J. Virol. 2011, 85, 5708–5717. [Google Scholar] [CrossRef] [PubMed]
- Wollmann, G.; Ozduman, K.; van den Pol, A.N. Oncolytic virus therapy for glioblastoma multiforme: Concepts and candidates. Cancer J. 2012, 18, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Msaouel, P.; Dispenzieri, A.; Galanis, E. Clinical testing of engineered oncolytic measles virus strains in the treatment of cancer: An overview. Curr. Opin. Mol. Ther. 2009, 11, 43–53. [Google Scholar] [PubMed]
- Viral Therapy in Treating Patients with Recurrent Glioblastoma Multiforme. Available online: https://clinicaltrials.gov/ct2/show/NCT00390299 (accessed on 5 September 2018).
- Gagan, J.; Van Allen, E.M. Next-generation sequencing to guide cancer therapy. Genome Med. 2015, 7, 80. [Google Scholar] [CrossRef] [PubMed]
- Kravchenko-Balasha, N.; Wang, J.; Remacle, F.; Levine, R.D.; Heath, J.R. Glioblastoma cellular architectures are predicted through the characterization of two-cell interactions. Proc. Natl. Acad. Sci. USA 2014, 111, 6521–6526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, W.; Shin, Y.S.; Xue, M.; Matsutani, T.; Masui, K.; Yang, H.; Ikegami, S.; Gu, Y.; Herrmann, K.; Johnson, D.; et al. Single-Cell Phosphoproteomics Resolves Adaptive Signaling Dynamics and Informs Targeted Combination Therapy in Glioblastoma. Cancer Cell 2016, 29, 563–573. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A schematic of various targeted therapies used to treat glioblastoma. Tumor cells express Tumor Specific Antigen/ Tumor Associated Antigen (TSA/TAA) that are lacking in healthy neighboring normal cells. These therapies specifically target the TSA/TAA to limit the cytotoxic/radio therapeutic effects to tumor cells and spare their normal counterparts.
Figure 1.
A schematic of various targeted therapies used to treat glioblastoma. Tumor cells express Tumor Specific Antigen/ Tumor Associated Antigen (TSA/TAA) that are lacking in healthy neighboring normal cells. These therapies specifically target the TSA/TAA to limit the cytotoxic/radio therapeutic effects to tumor cells and spare their normal counterparts.

Figure 2.
A schematic of the IL-13 receptor system. Normal and tumor cells express IL-4RA/IL-13RA1. IL-4RA/IL-13RA1 have IL-4RA (represented in green) and IL-13RA1 (represented in blue) subunits. IL-4 first binds to IL-4RA, then to the IL-13RA1 to form an IL-4RA/IL-13RA1 complex. IL-13 first binds to IL-13RA1, then to IL-4A to form an IL-4RA/IL-13RA1 complex in normal tissue. On the other hand, IL-13RA2 is expressed by tumor cells. IL-4 binds to IL-4RA/IL-13RA1, but not to IL-13RA2 (represented by a red encircled cross). IL-13 binds to both IL-4RA/IL-13RA1 and IL-13RA2. IL-13 binds more readily to IL-13RA2 compared to IL-4RA/IL-13RA1. By introducing mutation in the IL-13 ligand to produce IL-13M, the ligand binds mostly only to the IL-13RA2, and not to IL-4RA/IL-13RA1 complex (represented by a red encircled cross).
Figure 2.
A schematic of the IL-13 receptor system. Normal and tumor cells express IL-4RA/IL-13RA1. IL-4RA/IL-13RA1 have IL-4RA (represented in green) and IL-13RA1 (represented in blue) subunits. IL-4 first binds to IL-4RA, then to the IL-13RA1 to form an IL-4RA/IL-13RA1 complex. IL-13 first binds to IL-13RA1, then to IL-4A to form an IL-4RA/IL-13RA1 complex in normal tissue. On the other hand, IL-13RA2 is expressed by tumor cells. IL-4 binds to IL-4RA/IL-13RA1, but not to IL-13RA2 (represented by a red encircled cross). IL-13 binds to both IL-4RA/IL-13RA1 and IL-13RA2. IL-13 binds more readily to IL-13RA2 compared to IL-4RA/IL-13RA1. By introducing mutation in the IL-13 ligand to produce IL-13M, the ligand binds mostly only to the IL-13RA2, and not to IL-4RA/IL-13RA1 complex (represented by a red encircled cross).


{kind=link}
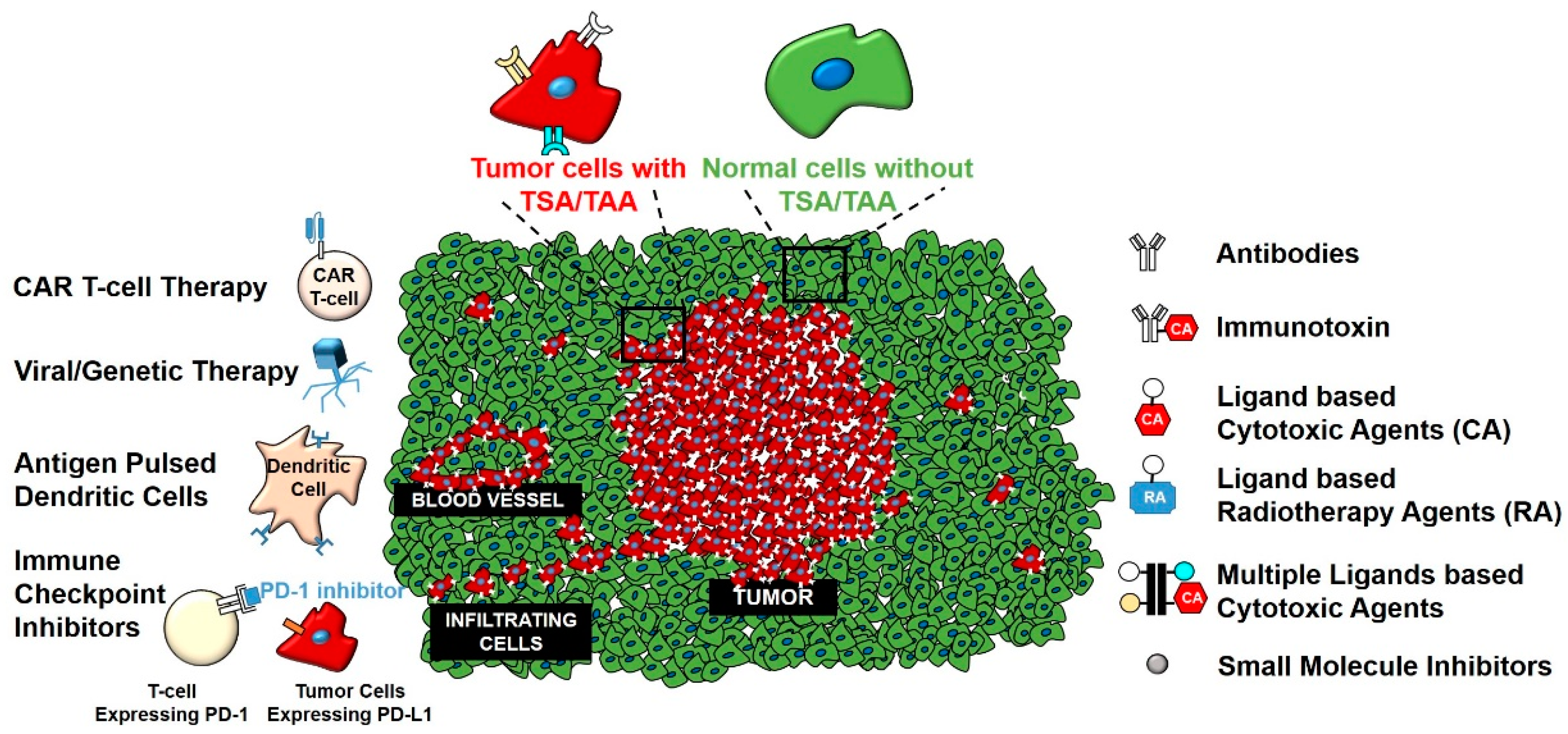{kind=link}
{kind=link}
Table 1.
An overview of the targeted therapies used to treat glioblastoma.
| Type of Therapy | Therapy | TSA/TAA Target (s) | Reference (s) |
|---|---|---|---|
| Peptide vaccines | Poly ICLC | IL-13RA2, Wilms Tumor 1, Survivin | [50] |
| CDX-110 | EGFRvIII | [116] | |
| Poly ICLC | IL-13RA2, EphA2, Survivin | [119] | |
| Cilengitide | Integrin | [226] | |
| Liposomes/Nanoparticles/Nanosheets | IL-13 based Liposome | IL-13RA2 | [104,105] |
| IL-13 based Nanoparticles | IL-13RA2 | [106,107] | |
| Transferrin based Nanoparticles | Transferrin receptor | [218] | |
| Lactoferrin based Nanoparticles | Lactoferrin receptor | [219] | |
| Radiotherapy | EphA3 mAb(IIIA4) Radiolabelled with (177 Lu) | EphA3 | [176] |
| [64Cu]Pep-1L | IL-13RA2 | [63,74] | |
| Antigen pulsed dendritic cells | Multiple Epitope Pulsed Dendritic Cells (ICT-107) | IL-13RA2, MAGE-1, TRP-2, gp-100, EGFR, AIM-2 | [114] |
| Antibodies | Cetuximab | EGFRvIII | [202] |
| Bevacizumab and Irinotecan | VEGF | [203] | |
| Nimotuzumab | EGFR | [204] | |
| Bevacizumab | VEGF | [223] | |
| Immunotoxins | Transferrin-CRM 107 | Transferrin | [235] |
| ephrin A1 based Cytotoxin | EphA2 | [177] | |
| TGF-α based Cytotoxin | EGFR | [205] | |
| IL-4 based Cytotoxin | IL-4RA | [215] | |
| Amino-terminal Fragment of uPA based Cytotoxin | uPAR | [216] | |
| IL-13 and ephrin A1 based Cytotoxins | IL-13RA2, EphA2 | [178] | |
| ephrin A5 based Cytotoxins | EphA2, EphA3, EphB2 | [174] | |
| IL-13 based Cytotoxin | IL-13RA2 | [65] | |
| CAR T-cell therapy | CAR T-cells targeting EGFRvIII | EGFRvIII | [206] |
| CAR T-cells targeting HER2 | HER2 | [208,209] | |
| CAR T-cells targeting EphA2 | EphA2 | [179] | |
| IL-13.E13K.R109K CAR T cells | IL-13RA2 | [138] | |
| TanCARs | HER2, IL-13RA2 | [150] | |
| GRm13Z40-2 | IL-13RA2 | [56,129,137] | |
| IL-13(E13Y)-zetakine CAR T cells | IL-13RA2 | [125] | |
| IL-13BBζ-CAR T cells | IL-13RA2 | [130] | |
| IL-13 (E13K, E13Y, E13K.K105R, E13Y.K105R) based CAR T-cells | IL-13RA2 | [131] | |
| IL-13RA2 with Herpes Simplex Virus and PET reporter gene | IL-13RA2 | [134,135] | |
| Viral/Genetic Therapy | Delta-24-RGD (DNX-2401) Oncolytic Adenovirus | αvβ3 and αvβ5 Integrins | [108] |
| Ad.mhIL-4.TRE.mnhIL-13-PE) Adenovirus | IL-13RA2 | [113] | |
| R5141, Recombinant Herpes Simplex Virus-1 | IL-13RA3 | [111] | |
| DARPin-targeted Measles Virus | HER2 | [112] | |
| LU-13, Adenovirus | IL-13RA2 | [109] | |
| IL-13 based Lentiviral Vectors | IL-13RA2 | [110] | |
| Oncolytic Measles Virus | CD46 | [267,274,275] | |
| Reovirus | N/A | [266] | |
| Conditional Replicating Adenovirus (CAd) | Coxsackie and adenovirus receptor | [268] | |
| Oncolytic Herpes Simplex Virus | CD133 and CD111 | [269] | |
| ONYX-015, Recombinant Adenovirus | Coxsackie and adenovirus receptor | [270] | |
| Oncolytic Vesicular Stomatitis Virus | Death receptor | [272] | |
| IL-4 and Herpes Simplex Virus Thymidine Kinase | CD133 and CD111 | [256] | |
| Herpes Simplex Virus-Thymidine Kinase Ganciclovir (HSV-TK/GCV) | CD133 and CD111 | [263] | |
| Immune checkpoint inhibitors | Ipilimumab | CTLA-4 on T-cells | [242] |
| Nivolumab and Ipilumumab | PD-1 and CTLA-4 on T-cells respectively | [247] | |
| Anti TIM-3 | TIM-3 on T-cells | [252] | |
| Anti LAG-3 and Nivolumab | LAG-3 and PD-1 on T-cells respectively | [55] | |
| anti-CTLA-4 mAb (9D9, IgG2b) | CTLA-4 on T-cells | [243] | |
| ani-CTLA-4mAb and Granulocyte-Macrophage Colony Stimulating Factor (GMCSF) | CTLA-4 on T-cells | [244] | |
| Tyrosine kinase inhibitors | Cediranib | VEGFR | [229] |
| Sorafenib | VEGFR | [230] | |
| Sunitinib | VEGFR | [231] | |
| Vandetanib | VEGFR | [228] | |
| Imatinib | PDGFR | [232] | |
| Desatinib | PDGFR | [233] | |
| Brivanib | FGFR | [234] | |
| siRNA based Therapy | Lipid Nanocapsules | EGFR | [196] |
| Transfection of siRNA | EGFR and β-catenin | [197] | |
| Small molecule inhibitors | Erlotinib | EGFR | [198] |
| Geftinib | EGFR | [201] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sharma, P.; Debinski, W. Receptor-Targeted Glial Brain Tumor Therapies. Int. J. Mol. Sci. 2018, 19, 3326. https://doi.org/10.3390/ijms19113326
AMA Style
Sharma P, Debinski W. Receptor-Targeted Glial Brain Tumor Therapies. International Journal of Molecular Sciences. 2018; 19(11):3326. https://doi.org/10.3390/ijms19113326
Chicago/Turabian StyleSharma, Puja, and Waldemar Debinski. 2018. "Receptor-Targeted Glial Brain Tumor Therapies" International Journal of Molecular Sciences 19, no. 11: 3326. https://doi.org/10.3390/ijms19113326
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.