The Bromodomain Inhibitor N-Methyl pyrrolidone Prevents Osteoporosis and BMP-Triggered Sclerostin Expression in Osteocytes



Abstract
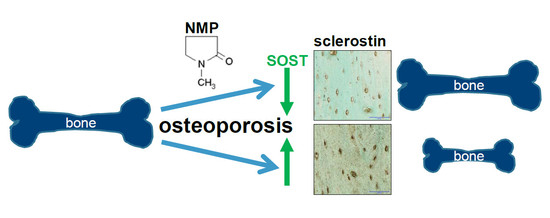
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
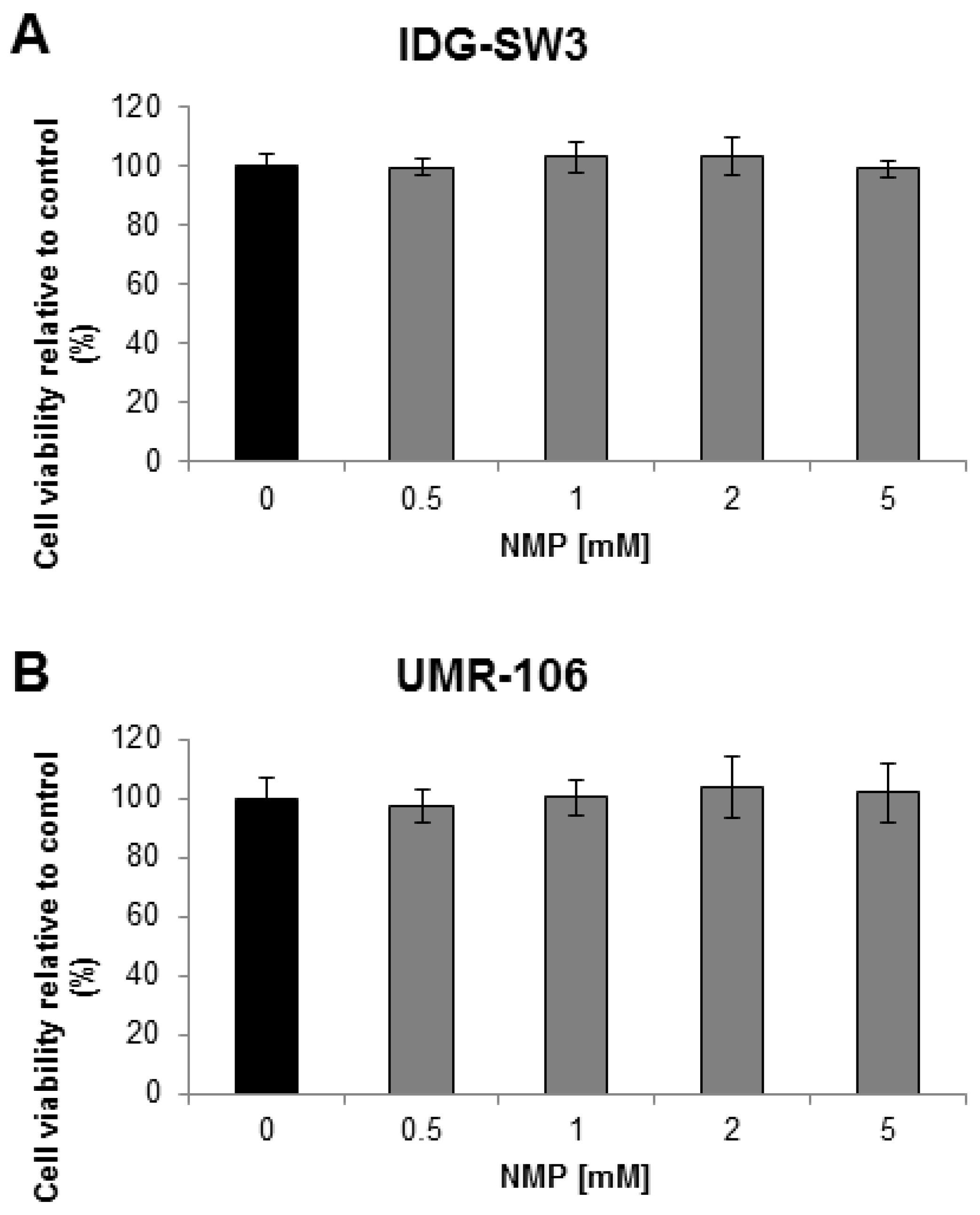
2.1. NMP Has No Toxic Effect on Osteocyte Like Cells
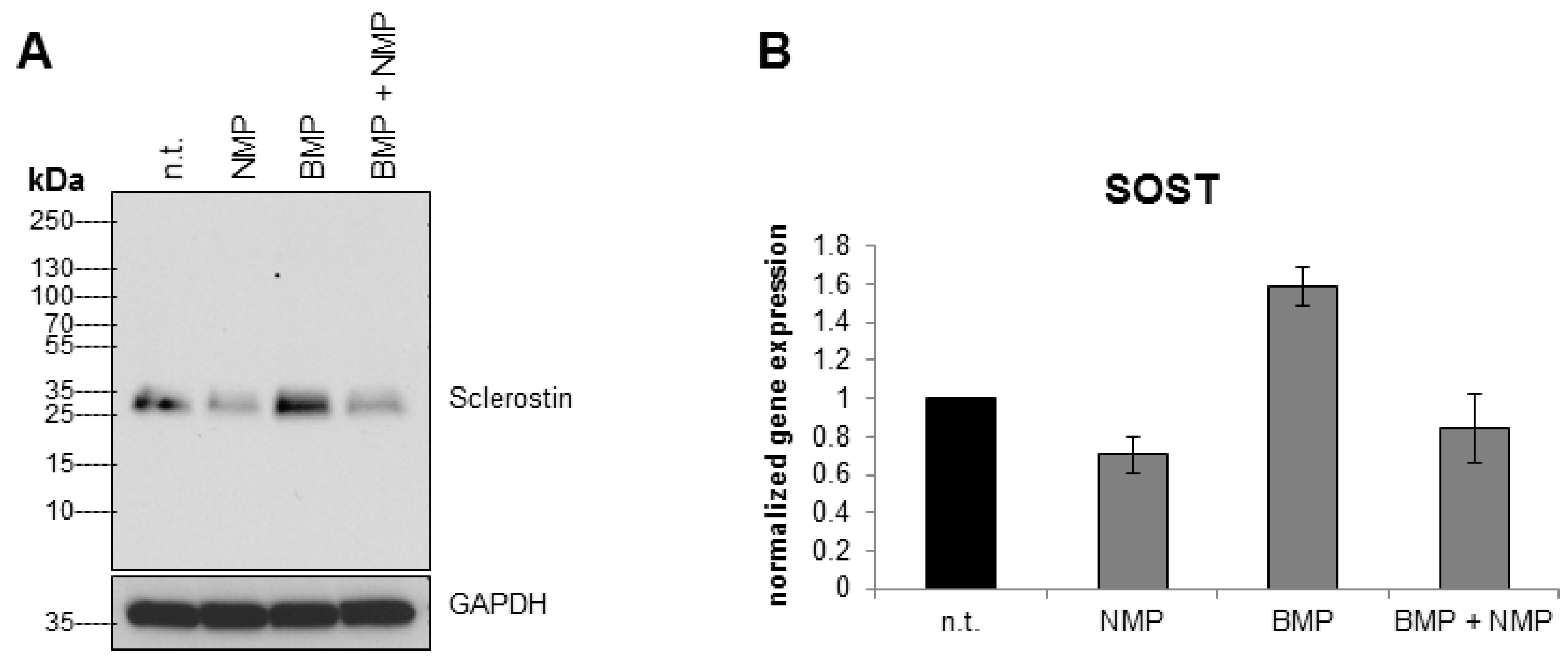
2.2. NMP Decreases Sclerostin Expression in Osteocyte Like Cells
2.3. NMP Prevents BMP-2 Induced Increase in Sclerostin Expression
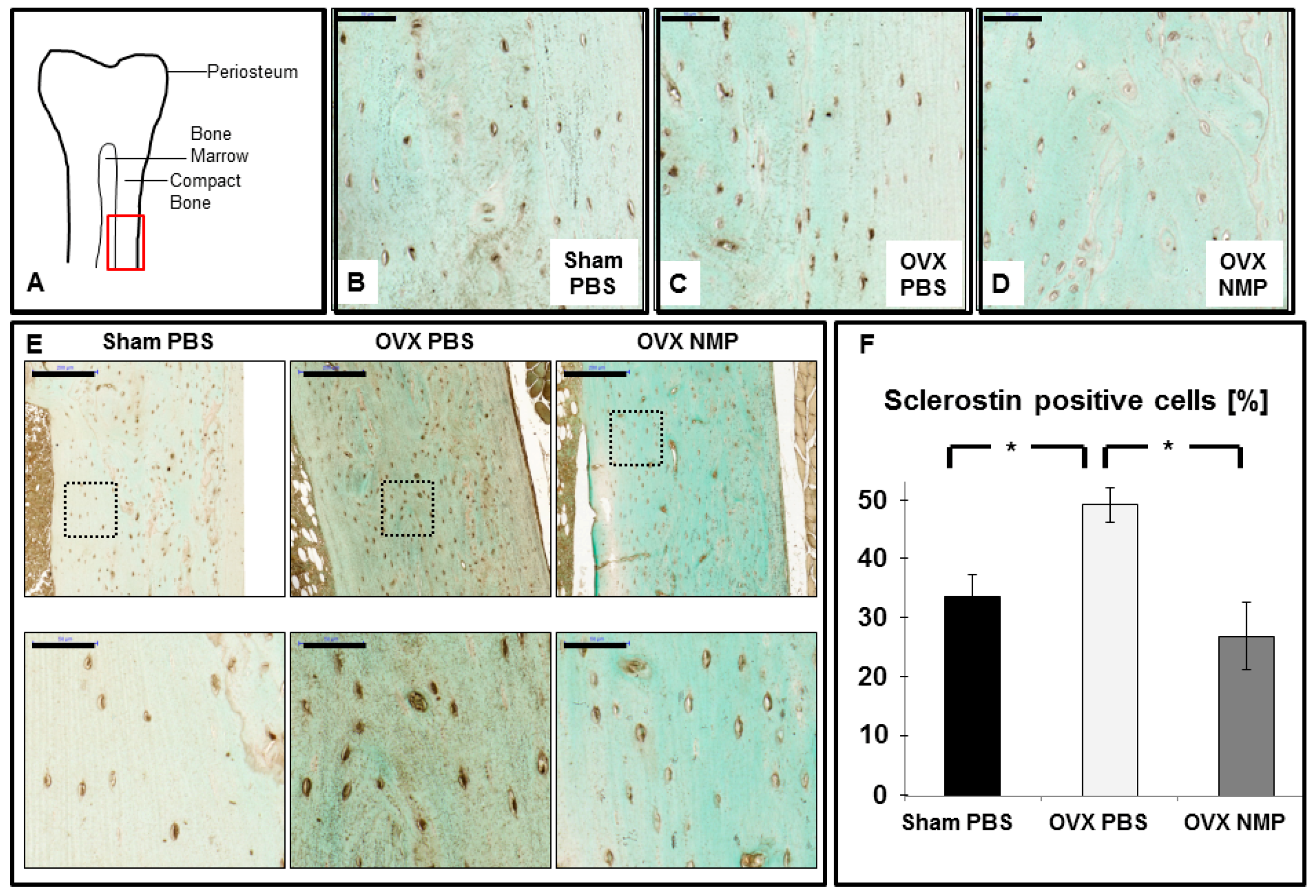
2.4. Increased Sclerostin Expression in Osteoporosis Induced Animals Can Be Prevented by NMP
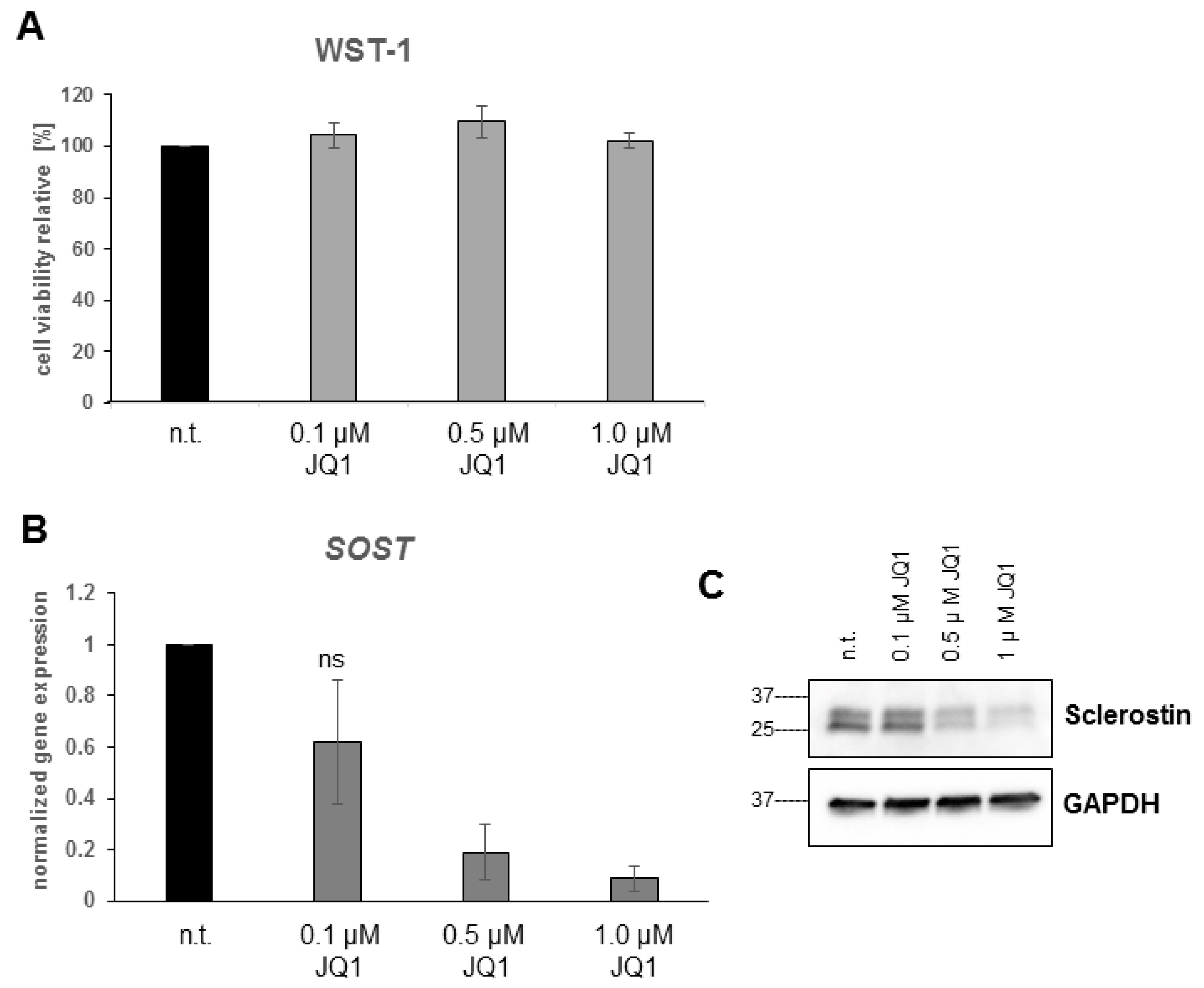
2.5. Bromodomain Inhibition Decreases Sclerostin Expression
3. Discussion
4. Materials and Methods
4.1. Cell Culture: Cell Lines
4.2. Cell Viability Assay
4.3. Quantitative Real-Time RT-PCR
4.4. Western Blot
4.5. Osteoporosis Rat Model
4.6. Immunohistochemistry (IHC)
4.7. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALP | Alkaline phosphatase |
| BCA | BiCinchoninic acid Assay |
| BMP | Bone morphogenetic protein |
| bp | Base pair |
| BRD2 | Bromodomain-containing protein 2 |
| COX | Cyclooxygenase |
| FDA | Food and Drug Administration (USA) |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| GFP | Green fluorescent protein |
| JQ1 | (6S)-4-(4-Chlorophenyl)-2,3,9-trimethyl-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepine-6-acetic acid 1,1-dimethylethyl ester |
| LEP | Leptin |
| NMP | N-methyl pyrrolidone |
| NO | Nitric oxide |
| Osx | Osterix |
| OVX | Ovariectomy |
| PGE | Prostaglandine |
| PTH | Parathyroid Hormone |
| PVDF | Polyvinylidene difluoride |
| RANKL | Receptor Activator of Nuclear factor Kappa-B Ligand |
| SOST | Sclerosteosis gene |
| WST | (4-[3-(4-Iodophenyl)-2-(4-nitro-phenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate) |
References
- NIH, Osteoporosis prevention, diagnosis, and therapy. JAMA 2001, 285, 785–795. [CrossRef]
- Foundation, I.O. What Is Osteoporosis. Available online: https://www.iofbonehealth.org/epidemiology (accessed on 26 August 2018).
- Reginster, J.Y.; Burlet, N. Osteoporosis: A still increasing prevalence. Bone 2006, 38, S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Kress, B.C.; Mizrahi, I.A. Monitoring antiosteoporotic treatment of postmenopausal women using biochemical markers of bone turnover. Drugs Today 1999, 35, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Raggatt, L.J.; Partridge, N.C. Parathyroid hormone: A double-edged sword for bone metabolism. Trends Endocrinol. Metab. 2004, 15, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Dede, A.D.; Makras, P.; Anastasilakis, A.D. Investigational anabolic agents for the treatment of osteoporosis: An update on recent developments. Expert Opin. Investig. Drugs 2017, 26, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.M.; Nani, E.P.; Teixeira, L.N.; Peruzzo, D.C.; Joly, J.C.; Napimoga, M.H.; Martinez, E.F. Strontium ranelate increases osteoblast activity. Tissue Cell 2016, 48, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Reginster, J.Y.; Brandi, M.L.; Cannata-Andia, J.; Cooper, C.; Cortet, B.; Feron, J.M.; Genant, H.; Palacios, S.; Ringe, J.D.; Rizzoli, R. The position of strontium ranelate in today’s management of osteoporosis. Osteoporos. Int. 2015, 26, 1667–1671. [Google Scholar] [CrossRef] [PubMed]
- Ghayor, C.; Correro, R.M.; Lange, K.; Karfeld-Sulzer, L.S.; Gratz, K.W.; Weber, F.E. Inhibition of osteoclast differentiation and bone resorption by N-methyl pyrrolidone. J. Biol. Chem. 2011, 286, 24458–24466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miguel, B.S.; Ghayor, C.; Ehrbar, M.; Jung, R.E.; Zwahlen, R.A.; Hortschansky, P.; Schmoekel, H.G.; Weber, F.E. N-methyl pyrrolidone as a potent bone morphogenetic protein enhancer for bone tissue regeneration. Tissue Eng. Part A 2009, 15, 2955–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gjoksi, B.; Ghayor, C.; Siegenthaler, B.; Ruangsawasdi, N.; Zenobi-Wong, M.; Weber, F.E. The epigenetically active small chemical N-methyl pyrrolidone (NMP) prevents estrogen depletion induced osteoporosis. Bone 2015, 78, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Gong, F.; Chiu, L.Y.; Miller, K.M. Acetylation reader proteins: Linking acetylation signaling to genome maintenance and cancer. PLoS Genet. 2016, 12, e1006272. [Google Scholar] [CrossRef] [PubMed]
- Devaiah, B.N.; Gegonne, A.; Singer, D.S. Bromodomain 4: A cellular Swiss army knife. J. Leukoc. Biol. 2016, 100, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Mirguet, O.; Lamotte, Y.; Chung, C.W.; Bamborough, P.; Delannee, D.; Bouillot, A.; Gellibert, F.; Krysa, G.; Lewis, A.; Witherington, J.; et al. Naphthyridines as novel BET family bromodomain inhibitors. ChemMedChem 2014, 9, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Philpott, M.; Yang, J.; Tumber, T.; Fedorov, O.; Uttarkar, S.; Filippakopoulos, P.; Picaud, S.; Keates, T.; Felletar, I.; Ciulli, A.; et al. Bromodomain-peptide displacement assays for interactome mapping and inhibitor discovery. Mol. Biosyst. 2011, 7, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Shortt, J.; Hsu, A.K.; Martin, B.P.; Doggett, K.; Matthews, G.M.; Doyle, M.A.; Ellul, J.; Jockel, T.E.; Andrews, D.M.; Hogg, S.J.; et al. The drug vehicle and solvent N-methyl pyrrolidone is an immunomodulator and antimyeloma compound. Cell Rep. 2014, 7, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Bonewald, L.F. The amazing osteocyte. J. Bone Min. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franz-Odendaal, T.A.; Hall, B.K.; Witten, P.E. Buried alive: How osteoblasts become osteocytes. Dev. Dyn. 2006, 235, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Manolagas, S.C. Birth and death of bone cells: Basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis. Endocr. Rev. 2000, 21, 115–137. [Google Scholar] [CrossRef] [PubMed]
- Mc Garrigle, M.J.; Mullen, C.A.; Haugh, M.G.; Voisin, M.C.; McNamara, L.M. Osteocyte differentiation and the formation of an interconnected cellular network in vitro. Eur. Cells Mater. 2016, 31, 323–340. [Google Scholar] [CrossRef] [Green Version]
- Pajevic, P.D. Regulation of bone resorption and mineral homeostasis by osteocytes. IBMS BoneKEy 2009, 6, 63–70. [Google Scholar] [CrossRef]
- Palazzini, S.; Palumbo, C.; Ferretti, M.; Marotti, G. Stromal cell structure and relationships in perimedullary spaces of chick embryo shaft bones. Anat. Embryol. 1998, 197, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Knothe Tate, M.L.; Adamson, J.R.; Tami, A.E.; Bauer, T.W. The osteocyte. Int. J. Biochem. Cell Biol. 2004, 36, 1–8. [Google Scholar] [CrossRef]
- Heino, T.J.; Hentunen, T.A.; Vaananen, H.K. Osteocytes inhibit osteoclastic bone resorption through transforming growth factor-β: Enhancement by estrogen. J. Cell. Biochem. 2002, 85, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, Y.K.; Harris, S.; Ahuja, S.S.; Bonewald, L.F. MLO-Y4 osteocyte-like cells support osteoclast formation and activation. J. Bone Min. Res. 2002, 17, 2068–2079. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, N.; Standley, K.N.; Bianchi, E.N.; Stadelmann, V.; Foti, M.; Conway, S.J.; Ferrari, S.L. The matricellular protein periostin is required for sost inhibition and the anabolic response to mechanical loading and physical activity. J. Biol. Chem. 2009, 284, 35939–35950. [Google Scholar] [CrossRef] [PubMed]
- Grimston, S.K.; Watkins, M.P.; Brodt, M.D.; Silva, M.J.; Civitelli, R. Enhanced periosteal and endocortical responses to axial tibial compression loading in conditional connexin43 deficient mice. PLoS ONE 2012, 7, e44222. [Google Scholar] [CrossRef] [PubMed]
- Lara-Castillo, N.; Kim-Weroha, N.A.; Kamel, M.A.; Javaheri, B.; Ellies, D.L.; Krumlauf, R.E.; Thiagarajan, G.; Johnson, M.L. In vivo mechanical loading rapidly activates beta-catenin signaling in osteocytes through a prostaglandin mediated mechanism. Bone 2015, 76, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Zaman, G.; Saxon, L.K.; Sunters, A.; Hilton, H.; Underhill, P.; Williams, D.; Price, J.S.; Lanyon, L.E. Loading-related regulation of gene expression in bone in the contexts of estrogen deficiency, lack of estrogen receptor alpha and disuse. Bone 2010, 46, 628–642. [Google Scholar] [CrossRef] [PubMed]
- Poole, K.E.; van Bezooijen, R.L.; Loveridge, N.; Hamersma, H.; Papapoulos, S.E.; Lowik, C.W.; Reeve, J. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005, 19, 1842–1844. [Google Scholar] [CrossRef] [PubMed]
- Robling, A.G.; Niziolek, P.J.; Baldridge, L.A.; Condon, K.W.; Allen, M.R.; Alam, I.; Mantila, S.M.; Gluhak-Heinrich, J.; Bellido, T.M.; Harris, S.E.; et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of SOST/sclerostin. J. Biol. Chem. 2008, 283, 5866–5875. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Y.; Kang, H.; Liu, W.; Liu, P.; Zhang, J.; Harris, S.E.; Wu, D. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J. Biol. Chem. 2005, 280, 19883–19887. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, B.; Nichols, G., Jr. Bone matrix turnover and balance in vitro. II. The effects of aging. J. Clin. Investig. 1969, 48, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Mirza, F.S.; Padhi, I.D.; Raisz, L.G.; Lorenzo, J.A. Serum sclerostin levels negatively correlate with parathyroid hormone levels and free estrogen index in postmenopausal women. J. Clin. Endocrinol. Metab. 2010, 95, 1991–1997. [Google Scholar] [CrossRef] [PubMed]
- Modder, U.I.; Clowes, J.A.; Hoey, K.; Peterson, J.M.; McCready, L.; Oursler, M.J.; Riggs, B.L.; Khosla, S. Regulation of circulating sclerostin levels by sex steroids in women and in men. J. Bone Min. Res. 2011, 26, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Modder, U.I.; Roforth, M.M.; Hoey, K.; McCready, L.K.; Peterson, J.M.; Monroe, D.G.; Oursler, M.J.; Khosla, S. Effects of estrogen on osteoprogenitor cells and cytokines/bone-regulatory factors in postmenopausal women. Bone 2011, 49, 202–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boschert, V.; Frisch, C.; Back, J.W.; van Pee, K.; Weidauer, S.E.; Muth, E.M.; Schmieder, P.; Beerbaum, M.; Knappik, A.; Timmerman, P.; et al. The sclerostin-neutralizing antibody AbD09097 recognizes an epitope adjacent to sclerostin’s binding site for the Wnt co-receptor LRP6. Open Biol. 2016, 6, 160120. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.G.; Bilezikian, J.P.; Lewiecki, E.M. Update on romosozumab: A humanized monoclonal antibody to sclerostin. Expert Opin. Biol. Ther. 2014, 14, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Recker, R.R.; Benson, C.T.; Matsumoto, T.; Bolognese, M.A.; Robins, D.A.; Alam, J.; Chiang, A.Y.; Hu, L.; Krege, J.H.; Sowa, H.; et al. A randomized, double-blind phase 2 clinical trial of blosozumab, a sclerostin antibody, in postmenopausal women with low bone mineral density. J. Bone Min. Res. 2015, 30, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.Y.; Yang, H.J.; Song, Y.M.; Kim, I.S.; Hwang, S.J. Estrogen modulates bone morphogenetic protein-induced sclerostin expression through the Wnt signaling pathway. Tissue Eng. Part A 2015, 21, 2076–2088. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, M.K.; Geoghegan, J.C.; Yu, C.; Winkler, D.G.; Latham, J.A. Unique regulation of SOST, the sclerosteosis gene, by BMPs and steroid hormones in human osteoblasts. Bone 2004, 35, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.M.; Rosser, J.; Dusevich, V.; Kalajzic, I.; Bonewald, L.F. Cell line IDG-SW3 replicates osteoblast-to-late-osteocyte differentiation in vitro and accelerates bone formation in vivo. J. Bone Min. Res. 2011, 26, 2634–2646. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337. [Google Scholar] [CrossRef] [PubMed]
- Galea, G.L.; Lanyon, L.E.; Price, J.S. Sclerostin’s role in bone’s adaptive response to mechanical loading. Bone 2016, 96, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Bandopadhayay, P.; Bergthold, G.; Nguyen, B.; Schubert, S.; Gholamin, S.; Tang, Y.; Bolin, S.; Schumacher, S.E.; Zeid, R.; Masoud, S.; et al. BET bromodomain inhibition of MYC-amplified medulloblastoma. Clin. Cancer Res. 2014, 20, 912–925. [Google Scholar] [CrossRef] [PubMed]
- Saenz, D.T.; Fiskus, W.; Manshouri, T.; Rajapakshe, K.; Krieger, S.; Sun, B.; Mill, C.P.; DiNardo, C.; Pemmaraju, N.; Kadia, T.; et al. BET protein bromodomain inhibitor-based combinations are highly active against post-myeloproliferative neoplasm secondary AML cells. Leukemia 2016, 31, 678. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, X.; Huang, P.; Lv, Z.; Qi, Y.; Wei, X.; Yang, P.; Zhang, F. JQ1, a small molecule inhibitor of BRD4, suppresses cell growth and invasion in oral squamous cell carcinoma. Oncol. Rep. 2016, 36, 1989–1996. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Calle, J.; Sanudo, C.; Bolado, A.; Fernandez, A.F.; Arozamena, J.; Pascual-Carra, M.A.; Rodriguez-Rey, J.C.; Fraga, M.F.; Bonewald, L.; Riancho, J.A. DNA methylation contributes to the regulation of sclerostin expression in human osteocytes. J. Bone Min. Res. 2012, 27, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Hannon, R.; Blumsohn, A.; Naylor, K.; Eastell, R. Response of biochemical markers of bone turnover to hormone replacement therapy: Impact of biological variability. J. Bone Min. Res. 1998, 13, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Morse, A.; McDonald, M.M.; Kelly, N.H.; Melville, K.M.; Schindeler, A.; Kramer, I.; Kneissel, M.; van der Meulen, M.C.; Little, D.G. Mechanical load increases in bone formation via a sclerostin-independent pathway. J. Bone Min. Res. 2014, 29, 2456–2467. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, S.; Ishii, K.; Amizuka, N.; Li, M.; Kobayashi, T.; Kohno, K.; Ito, M.; Takeshita, S.; Ikeda, K. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007, 5, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Sevetson, B.; Taylor, S.; Pan, Y. Cbfa1/RUNX2 directs specific expression of the sclerosteosis gene (SOST). J. Biol. Chem. 2004, 279, 13849–13858. [Google Scholar] [CrossRef] [PubMed]
- Loots, G.G.; Kneissel, M.; Keller, H.; Baptist, M.; Chang, J.; Collette, N.M.; Ovcharenko, D.; Plajzer-Frick, I.; Rubin, E.M. Genomic deletion of a long-range bone enhancer misregulates sclerostin in Van Buchem disease. Genome Res. 2005, 15, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Weivoda, M.M.; Oursler, M.J. Developments in sclerostin biology: Regulation of gene expression, mechanisms of action, and physiological functions. Curr. Osteoporos. Rep. 2014, 12, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Bonewald, L.F. Dynamics of the transition from osteoblast to osteocyte. Ann. N. Y. Acad. Sci. 2010, 1192, 437–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baertschi, S.; Baur, N.; Lueders-Lefevre, V.; Voshol, J.; Keller, H. Class I and IIa histone deacetylases have opposite effects on sclerostin gene regulation. J. Biol. Chem. 2014, 289, 24995–25009. [Google Scholar] [CrossRef] [PubMed]
- Imhof, A.; Yang, X.J.; Ogryzko, V.V.; Nakatani, Y.; Wolffe, A.P.; Ge, H. Acetylation of general transcription factors by histone acetyltransferases. Curr. Biol. 1997, 7, 689–692. [Google Scholar] [CrossRef]
- Kramer, I.; Baertschi, S.; Halleux, C.; Keller, H.; Kneissel, M. MEF2C deletion in osteocytes results in increased bone mass. J. Bone Min. Res. 2012, 27, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Leupin, O.; Kramer, I.; Collette, N.M.; Loots, G.G.; Natt, F.; Kneissel, M.; Keller, H. Control of the SOST bone enhancer by PTH using MEF2 transcription factors. J. Bone Min. Res. 2007, 22, 1957–1967. [Google Scholar] [CrossRef] [PubMed]
- Wein, M.N.; Spatz, J.; Nishimori, S.; Doench, J.; Root, D.; Babij, P.; Nagano, K.; Baron, R.; Brooks, D.; Bouxsein, M.; et al. HDAC5 controls MEF2C-driven sclerostin expression in osteocytes. J. Bone Min. Res. 2015, 30, 400–411. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siegenthaler, B.; Ghayor, C.; Gjoksi-Cosandey, B.; Ruangsawasdi, N.; Weber, F.E. The Bromodomain Inhibitor N-Methyl pyrrolidone Prevents Osteoporosis and BMP-Triggered Sclerostin Expression in Osteocytes. Int. J. Mol. Sci. 2018, 19, 3332. https://doi.org/10.3390/ijms19113332
Siegenthaler B, Ghayor C, Gjoksi-Cosandey B, Ruangsawasdi N, Weber FE. The Bromodomain Inhibitor N-Methyl pyrrolidone Prevents Osteoporosis and BMP-Triggered Sclerostin Expression in Osteocytes. International Journal of Molecular Sciences. 2018; 19(11):3332. https://doi.org/10.3390/ijms19113332
Chicago/Turabian StyleSiegenthaler, Barbara, Chafik Ghayor, Bebeka Gjoksi-Cosandey, Nisarat Ruangsawasdi, and Franz E. Weber. 2018. "The Bromodomain Inhibitor N-Methyl pyrrolidone Prevents Osteoporosis and BMP-Triggered Sclerostin Expression in Osteocytes" International Journal of Molecular Sciences 19, no. 11: 3332. https://doi.org/10.3390/ijms19113332