In Silico Analysis of Missense Mutations as a First Step in Functional Studies: Examples from Two Sphingolipidoses


Abstract
:
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Data Identifiers
4.2. In Silico Methods
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beutler, E.; Grabowski, G.A. Gaucher disease. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3635–3668. [Google Scholar]
- Desnick, R.J.; Ioannou, Y.A.; Eng, C.M. α-Galactosidase A deficiency: Fabry disease. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3733–3774. [Google Scholar]
- Lieberman, R.L.; D’Aquino, J.A.; Ringe, D.; Petsko, G.A. Effects of pH and iminosugar pharmacological chaperones on lysosomal glycosidase structure and stability. Biochemistry 2009, 48, 4816–4827. [Google Scholar] [CrossRef] [PubMed]
- Dvir, H.; Harel, M.; McCarthy, A.A.; Toker, L.; Silman, I.; Futerman, A.H.; Sussman, J.L. X-ray structure of human acid-beta-glucosidase, the defective enzyme in Gaucher disease. EMBO Rep. 2003, 4, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Garman, S.C.; Garboczi, D.N. The molecular defect leading to Fabry disease: Structure of human alpha-galactosidase. J. Mol. Biol. 2004, 337, 319–335. [Google Scholar] [CrossRef] [PubMed]
- Amberg, A. In Silico Methods. In Drug Discovery and Evaluation: Safety and Pharmacokinetic Assays; Vogel, H.G., Maas, J., Hock, F.J., Mayer, D., Eds.; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar] [CrossRef]
- Tchernitchko, D.; Goossens, M.; Wajcman, H. In silico prediction of the deleterious effect of a mutation: Proceed with caution in clinical genetics. Clin. Chem. 2004, 50, 1974–1978. [Google Scholar] [CrossRef] [PubMed]
- Rigsby, R.E.; Parker, A.B. Using the PyMOL application to reinforce visual understanding of protein structure. Biochem. Mol. Biol. Educ. 2016, 44, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Hecht, M.; Bromberg, Y.; Rost, B. News from the protein mutability landscape. J. Mol. Biol. 2013, 425, 3937–3948. [Google Scholar] [CrossRef] [PubMed]
- Hecht, M.; Bromberg, Y.; Rost, B. Better prediction of functional effects for sequence variants. BMC Genom. 2015, 16 (Suppl. 8), S1. [Google Scholar] [CrossRef] [PubMed]
- Bromberg, Y.; Rost, B. SNAP: Predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007, 35, 3823–3835. [Google Scholar] [CrossRef] [PubMed]
- Amaral, O.; Marcao, A.; Sa Miranda, M.; Desnick, R.J.; Grace, M.E. Gaucher disease: Expression and characterization of mild and severe acid beta-glucosidase mutations in Portuguese type 1 patients. Eur. J. Hum. Genet. 2000, 8, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Montfort, M.; Chabas, A.; Vilageliu, L.; Grinberg, D. Functional analysis of 13 GBA mutant alleles identified in Gaucher disease patients: Pathogenic changes and “modifier” polymorphisms. Hum. Mutat. 2004, 23, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Ron, I.; Dagan, A.; Gatt, S.; Pasmanik-Chor, M.; Horowitz, M. Use of fluorescent substrates for characterization of Gaucher disease mutations. Blood Cells Mol. Dis. 2005, 35, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Chabas, A.; Gort, L.; Diaz-Font, A.; Montfort, M.; Santamaria, R.; Cidras, M.; Grinberg, D.; Vilageliu, L. Perinatal lethal phenotype with generalized ichthyosis in a type 2 Gaucher disease patient with the [L444P;E326K]/P182L genotype: Effect of the E326K change in neonatal and classic forms of the disease. Blood Cells Mol. Dis. 2005, 35, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Grace, M.E.; Berg, A.; He, G.S.; Goldberg, L.; Horowitz, M.; Grabowski, G.A. Gaucher disease: Heterologous expression of two alleles associated with neuronopathic phenotypes. Am. J. Hum. Genet. 1991, 49, 646–655. [Google Scholar] [PubMed]
- Liou, B.; Kazimierczuk, A.; Zhang, M.; Scott, C.R.; Hegde, R.S.; Grabowski, G.A. Analyses of variant acid beta-glucosidases: Effects of Gaucher disease mutations. J. Biol. Chem. 2006, 281, 4242–4253. [Google Scholar] [CrossRef] [PubMed]
- Lukas, J.; Giese, A.K.; Markoff, A.; Grittner, U.; Kolodny, E.; Mascher, H.; Lackner, K.J.; Meyer, W.; Wree, P.; Saviouk, V.; et al. Functional characterisation of alpha-galactosidase a mutations as a basis for a new classification system in fabry disease. PLoS Genet. 2013, 9, e1003632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.Y.; Kim, G.H.; Kim, S.S.; Ko, J.M.; Lee, J.J.; Yoo, H.W. Effects of a chemical chaperone on genetic mutations in alpha-galactosidase A in Korean patients with Fabry disease. Exp. Mol. Med. 2009, 41, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Spada, M.; Pagliardini, S.; Yasuda, M.; Tukel, T.; Thiagarajan, G.; Sakuraba, H.; Ponzone, A.; Desnick, R.J. High incidence of later-onset fabry disease revealed by newborn screening. Am. J. Hum. Genet. 2006, 79, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.; Ortiz, A.; Germain, D.P.; Viana-Baptista, M.; Caldeira-Gomes, A.; Camprecios, M.; Fenollar-Cortes, M.; Gallegos-Villalobos, A.; Garcia, D.; Garcia-Robles, J.A.; et al. The alpha-galactosidase A p.Arg118Cys variant does not cause a Fabry disease phenotype: Data from individual patients and family studies. Mol. Genet. Metab. 2015, 114, 248–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froissart, R.; Guffon, N.; Vanier, M.T.; Desnick, R.J.; Maire, I. Fabry disease: D313Y is an alpha-galactosidase A sequence variant that causes pseudodeficient activity in plasma. Mol. Genet. Metab. 2003, 80, 307–314. [Google Scholar] [CrossRef]
- Yasuda, M.; Shabbeer, J.; Benson, S.D.; Maire, I.; Burnett, R.M.; Desnick, R.J. Fabry disease: Characterization of alpha-galactosidase A double mutations and the D313Y plasma enzyme pseudodeficiency allele. Hum. Mutat. 2003, 22, 486–492. [Google Scholar] [CrossRef] [PubMed]
- Heinisch, U.; Zlotogora, J.; Kafert, S.; Gieselmann, V. Multiple mutations are responsible for the high frequency of metachromatic leukodystrophy in a small geographic area. Am. J. Hum. Genet. 1995, 56, 51–57. [Google Scholar] [PubMed]
- Berná, L.; Gieselmann, V.; Poupetová, H.; Hrebícek, M.; Elleder, M.; Ledvinová, J. Novel mutations associated with metachromatic leukodystrophy: Phenotype and expression studies in nine Czech and Slovak patients. Am. J. Med. Genet. A 2004, 129A, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kafert, S.; Heinisch, U.; Wenger, D.A.; Zlotogora, J.; Gieselmann, V. Characterization of two arylsulfatase A missense mutations D335V and T274M causing late infantile metachromatic leukodystrophy. Hum. Mutat. 1996, 7, 311–317. [Google Scholar] [CrossRef]
- Marcão, A.; Amaral, O.; Pinto, E.; Pinto, R.; Sá Miranda, M.C. Metachromatic leucodystrophy in Portugal-finding of four new molecular lesions: C300F, P425T, g.1190-1191insC, and g.2408delC. Mutations in brief no. 232. Online. Hum. Mutat. 1999, 13, 337–338. [Google Scholar] [CrossRef]
- Lukatela, G.; Krauss, N.; Theis, K.; Selmer, T.; Gieselmann, V.; von Figura, K.; Saenger, W. Crystal structure of human arylsulfatase A: The aldehyde function and the metal ion at the active site suggest a novel mechanism for sulfate ester hydrolysis. Biochemistry 1998, 37, 3654–3664. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Kawame, H.; Ida, H.; Ohashi, T.; Eto, Y. Single exon mutation in arylsulfatase A gene has two effects: Loss of enzyme activity and aberrant splicing. Hum. Genet. 1994, 93, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Furuya, H.; Kukita, Y.; Nagano, S.; Sakai, Y.; Yamashita, Y.; Fukuyama, H.; Inatomi, Y.; Saito, Y.; Koike, R.; Tsuji, S.; et al. Adult onset globoid cell leukodystrophy (Krabbe disease): Analysis of galactosylceramidase cDNA from four Japanese patients. Hum. Genet. 1997, 100, 450–456. [Google Scholar] [CrossRef] [PubMed]
- De Gasperi, R.; Gama Sosa, M.A.; Sartorato, E.; Battistini, S.; Raghavan, S.; Kolodny, E.H. Molecular basis of late-life globoid cell leukodystrophy. Hum. Mutat. 1999, 14, 256–262. [Google Scholar] [CrossRef]
- Fu, L.; Inui, K.; Nishigaki, T.; Tatsumi, N.; Tsukamoto, H.; Kokubu, C.; Muramatsu, T.; Okada, S. Molecular heterogeneity of Krabbe disease. J. Inherit. Metab. Dis. 1999, 22, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Matos, L.; Duarte, A.J.; Ribeiro, D.; Chaves, J.; Amaral, O.; Alves, S. Correction of a Splicing Mutation Affecting an Unverricht-Lundborg Disease Patient by Antisense Therapy. Genes 2018, 9, 455. [Google Scholar] [CrossRef] [PubMed]
- Lalioti, M.D.; Mirotsou, M.; Buresi, C.; Peitsch, M.C.; Rossier, C.; Ouazzani, R.; Baldy-Moulinier, M.; Bottani, A.; Malafosse, A.; Antonarakis, S.E. Identification of mutations in cystatin B, the gene responsible for the Unverricht-Lundborg type of progressive myoclonus epilepsy (EPM1). Am. J. Hum. Genet. 1997, 60, 342–351. [Google Scholar] [PubMed]
- Auerswald, E.A.; Nägler, D.K.; Assfalg-Machleidt, I.; Stubbs, M.T.; Machleidt, W.; Fritz, H. Hairpin loop mutations of chicken cystatin have different effects on the inhibition of cathepsin B, cathepsin L and papain. FEBS Lett. 1995, 361, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Joensuu, T.; Kuronen, M.; Alakurtti, K.; Tegelberg, S.; Hakala, P.; Aalto, A.; Huopaniemi, L.; Aula, N.; Michellucci, R.; Eriksson, K.; et al. Cystatin B: Mutation detection, alternative splicing and expression in progressive myclonus epilepsy of Unverricht-Lundborg type (EPM1) patients. Eur. J. Hum. Genet. 2007, 15, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Alakurtti, K.; Weber, E.; Rinne, R.; Theil, G.; de Haan, G.J.; Lindhout, D.; Salmikangas, P.; Saukko, P.; Lahtinen, U.; Lehesjoki, A.E. Loss of lysosomal association of cystatin B proteins representing progressive myoclonus epilepsy, EPM1, mutations. Eur. J. Hum. Genet. 2005, 13, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.; Pasmanik-Chor, M.; Ron, I.; Kolodny, E.H. The enigma of the E326K mutation in acid β-glucocerebrosidase. Mol. Genet. Metab. 2011, 104, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Pinto, E.; Freitas, J.; Duarte, A.J.; Ribeiro, I.; Ribeiro, D.; Lima, J.L.; Chaves, J.; Amaral, O. Unverricht-Lundborg disease: Homozygosity for a new splicing mutation in the cystatin B gene. Epilepsy Res. 2012, 99, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.J.; Ribeiro, D.; Chaves, J.; Amaral, O. Characterization of a rare Unverricht-Lundborg disease mutation. Mol. Genet. Metab. Rep. 2015, 4, 68–71. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, G.; de Souza, R.M.; Balestrino, R.; Schapira, A.H. Glucocerebrosidase Mutations in Parkinson Disease. J. Parkinsons Dis. 2017, 7, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Migdalska-Richards, A.; Schapira, A.H. The relationship between glucocerebrosidase mutations and Parkinson disease. J. Neurochem. 2016, 139 (Suppl. 1), 77–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berge-Seidl, V.; Pihlstrøm, L.; Maple-Grødem, J.; Forsgren, L.; Linder, J.; Larsen, J.P.; Tysnes, O.B.; Toft, M. The GBA variant E326K is associated with Parkinson’s disease and explains a genome-wide association signal. Neurosci. Lett. 2017, 658, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Thirumal Kumar, D.; Eldous, H.G.; Mahgoub, Z.A.; George Priya Doss, C.; Zayed, H. Computational modelling approaches as a potential platform to understand the molecular genetics association between Parkinson’s and Gaucher diseases. Metab. Brain Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Beroud, G.; Claustres, M.; Beroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Lin, X.; Salzberg, S.L. GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Res. 2001, 29, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Hebsgaard, S.M.; Korning, P.G.; Tolstrup, N.; Engelbrecht, J.; Rouze, P.; Brunak, S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 1996, 24, 3439–3452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y. A fast computation of pairwise sequence alignment scores between a protein and a set of single-locus variants of another protein. In Proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine, Orlando, FL, USA, 7–10 October 2012; pp. 414–417. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30 (Suppl. 1), S162–S173. [Google Scholar] [CrossRef] [PubMed]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; de Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Gene Mutants | PROVEAN | PolyPhen-2 | SNAP2 | ExPASy | Protein Function and Structure |
|---|---|---|---|---|---|
| Prediction Expected Accuracy | |||||
| GBA1 Mutants | |||||
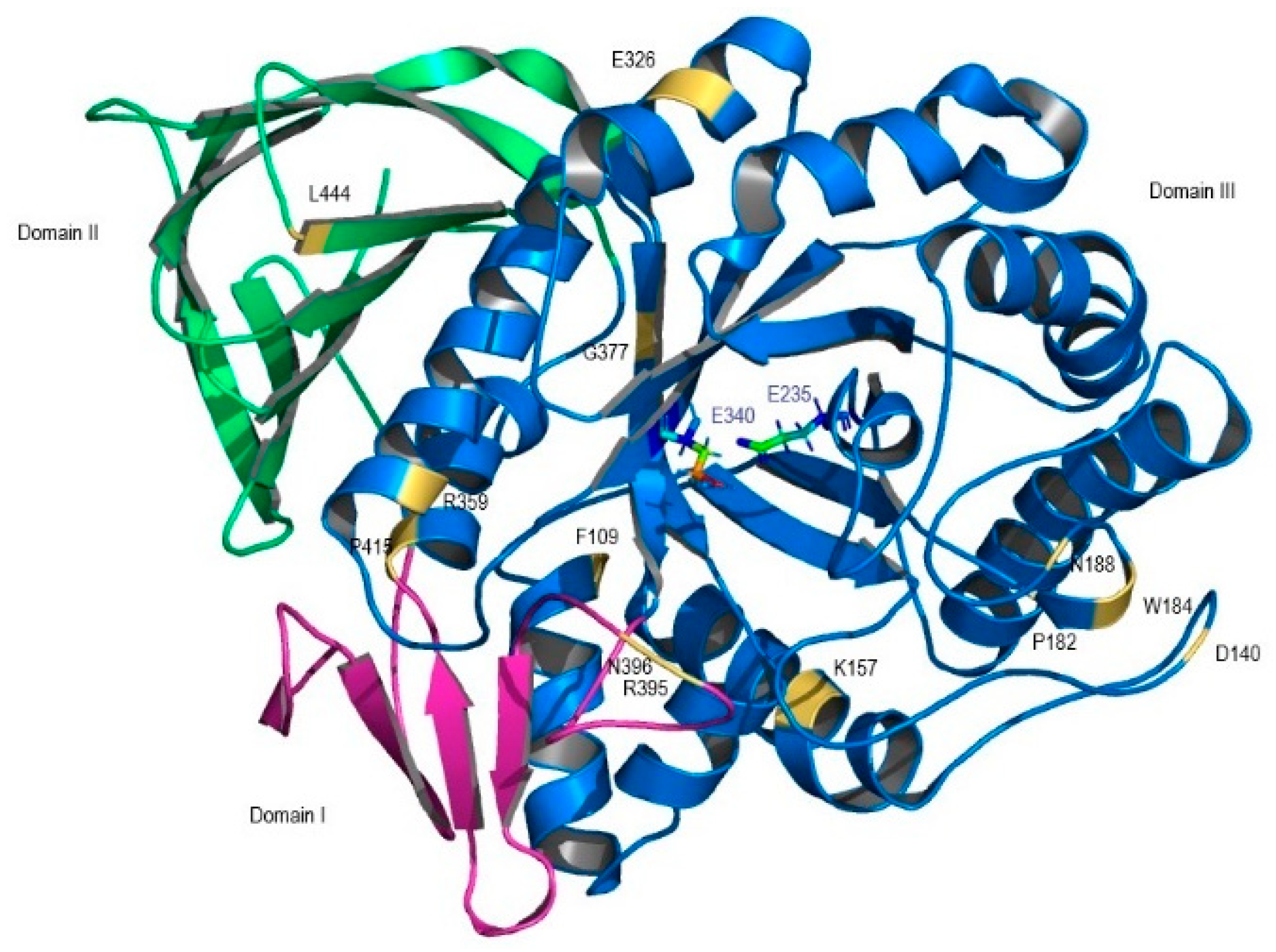
| F109V CM005404 | Deleterious | Probably damaging | Effect 75% | NA | 15% of wt activity; weakly conserved; domain III; stable protein [12] |
| P182L rs80205046 | Deleterious | Probably damaging | Effect 85% | NA | Near null activity; buried site; domain III; unstable protein [13] |
| D140H rs147138516 | Neutral | Benign | Neutral 57% | Disease | 73% of wt activity; domain III [14] |
| K157Q rs121908297 | Deleterious | Probably damaging | Neutral 57% | Disease | 9.7% of wt activity; domain III; conserved region; unstable protein [14] |
| W184R rs61748906 | Deleterious | Probably Damaging | Effect 66% | Disease | Inactive enzyme; domain III periphery; alteration of enzyme geometry; unstable protein [12] |
| N188S rs364897 | Deleterious | Benign | Effect 66% | Disease | 66.6% of wt activity; domain III periphery; stable protein [13] |
| E326K rs2230288 | Neutral | Benign | Neutral 56% | Disease | 42.7–25% of wt activity; domain III; stable protein [15] |
| R359Q rs74979486 | Deleterious | Probably Damaging | Effect 75% | Disease | 4.5% of wt activity; domain III stable protein; highly conserved region [12] |
| G377S rs121908311 | Deleterious | Probably Damaging | Effect 91% | Disease | 17% of wt activity; domain III; stable protein [12] |
| R395P | Deleterious | Benign | Effect 75% | NA | 4.5% of wt activity; domain I, loop 2; stable protein [12] |
| N396T rs75385858 | Deleterious | Probably Damaging | Effect 85% | NA | 14% of wt activity; domain I; stable protein [12] |
| P415R rs121908295 | Deleterious | Probably damaging | Effect 59% | Disease | Near null activity; conserved region; unstable protein [16] |
| L444P rs421016 | Deleterious | Possibly damaging | Effect 91% | NA | 5.7–9% of wt activity; unstable protein [12,17]. |
| GLA Mutants | |||||
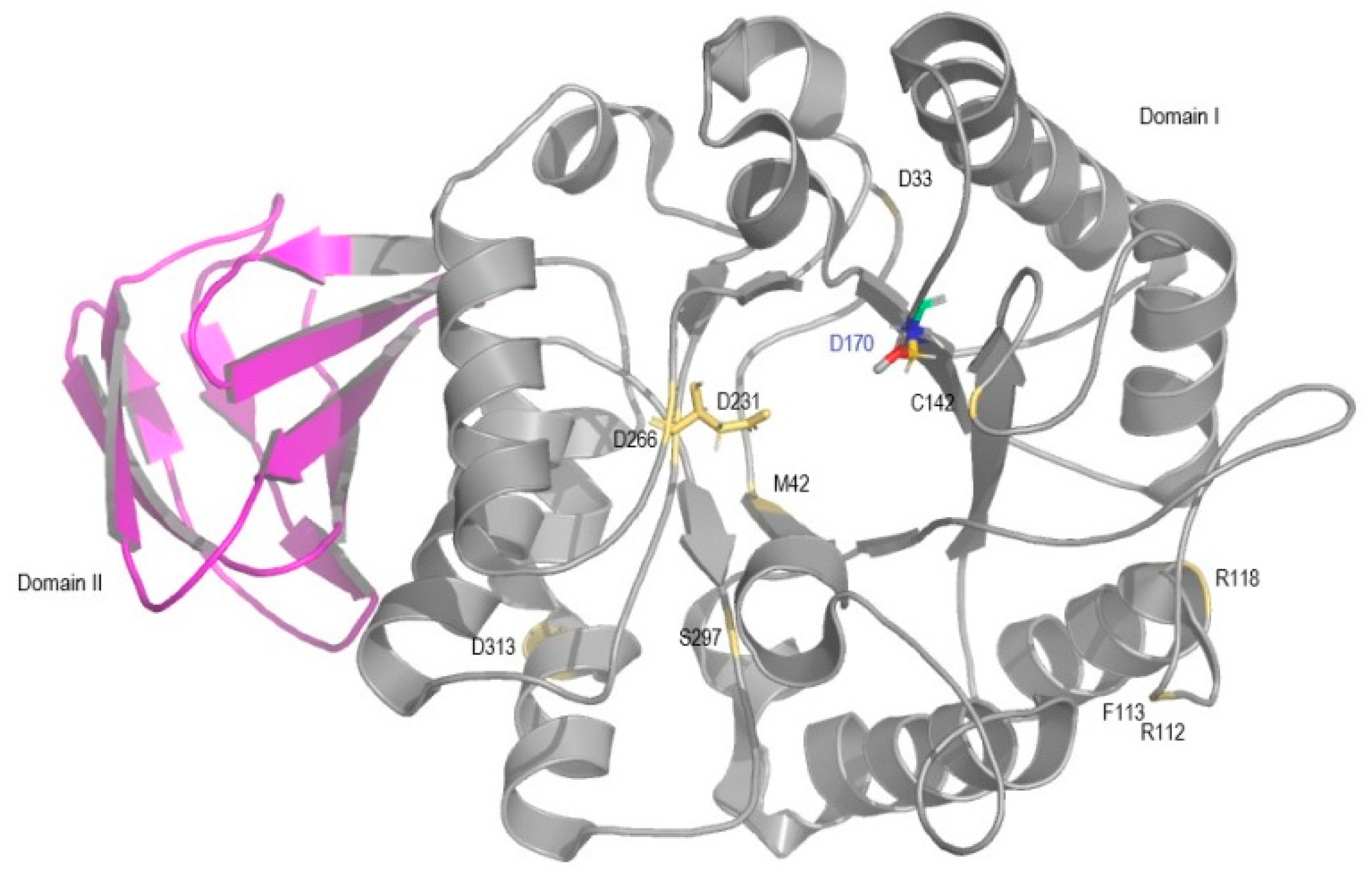
| D33G rs869312136 | Deleterious | Possibly damaging | Effect 75% | Unclassified | 37% of wt activity; periphery of domain I [18] |
| M42V | Deleterious | Probably damaging | Effect 85% | Disease | 7% of wt activity; domain I; unstable protein [19] |
| R112C rs104894834 | Deleterious | Probably damaging | Effect 91% | Disease | 5% of wt activity; periphery of domain I; unstable protein [19] |
| F113L rs869312142 | Deleterious | Probably damaging | Effect 91% | Disease | 20% of wt activity; periphery of domain I; altered alpha-GAL surface; unstable protein [19] |
| R118C rs148158093 | Deleterious | Probably Damaging | Effect 53% | NA | 29–32% of wt activity; periphery of domain I; unstable protein [20,21]. |
| C142W | Deleterious | Probably damaging | Effect 95% | NA | 5% of wt activity; domain I; near active site pocket; unstable protein [19]. |
| D231G | Deleterious | Probably damaging | Effect 95% | NA | 4% of wt activity; domain I, active site pocket; stable protein [19] |
| D266N rs869312407 | Deleterious | Probably damaging | Effect 95% | Disease | 5% of wt activity; domain I, near the active site pocket; buried; unstable protein [19] |
| S297F rs28935489 | Deleterious | Probably damaging | Effect 95% | Disease | 5% of wt activity; unstable protein [19] |
| D313Y rs28935490 | Deleterious | Probably damaging | Effect 95% | Disease | 76% of wt activity; in domain I periphery; stable protein [22,23] |
| Gene Mutants | PROVEAN | PolyPhen-2 | SNAP2 | ExPASy | Protein Function and Structure |
|---|---|---|---|---|---|
| Prediction Expected Accuracy | |||||
| ARSA mutants | |||||
| G86D rs74315460 | Deleterious | Probably damaging | Effect 95% | Disease | Null activity; unstable protein [24] |
| C156R rs199476348 | Deleterious | Probably Damaging | Effect 59% | Disease | 50% of wt activity [25] |
| T274M rs74315472 | Deleterious | Probably Damaging | Effect 95% | Disease | 35% of wt activity [26] |
| C300F rs74315484 | Deleterious | Probably Damaging | Effect 95% | Disease | Null activity; disruption of disulfide bond linking major and minor β-sheets [27,28] |
| T409I rs74315481 | Neutral | Possibly damaging | Effect 75% | Disease | 60% of wt activity [29] |
| GALC Mutants | |||||
| I82M without reference SNP (rs) | Deleterious | Probably Damaging | Neutral 57% | Disease | Normal activity [30] |
| G286D rs199847983 | Deleterious | Probably Damaging | Effect 71% | Disease | 17.5% of wt activity [31] |
| Y335C rs757407613 | Deleterious | Probably Damaging | Effect 75% | Disease | 10% of wt activity [32] |
| G553R rs748573754 | Deleterious | Probably Damaging | Effect 91% | Disease | 1.8% of wt activity [31] |
| L634S rs138577661 | Deleterious | Probably Damaging | Effect 95% | Disease | 12% of wt activity [30] |
| CSTB mutants | |||||
| Q22Q rs386833443 | Neutral | NA | Neutral 82% | NA | Expected abnormal peptide with premature truncation [33] |
| G4R rs74315443 | Deleterious | Probably Damaging | Effect 85% | Disease | Binding pocket modification; interaction properties compromised [34] |
| G50E rs312262708 | Deleterious | Possibly Damaging | Effect 95% | NA | Altered stability and interaction with target proteins [35,36] |
| Q71P rs796052392 | Deleterious | Possibly Damaging | Effect 75% | NA | Changes in second binding loop; altered binding affinities [37] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duarte, A.J.; Ribeiro, D.; Moreira, L.; Amaral, O. In Silico Analysis of Missense Mutations as a First Step in Functional Studies: Examples from Two Sphingolipidoses. Int. J. Mol. Sci. 2018, 19, 3409. https://doi.org/10.3390/ijms19113409
Duarte AJ, Ribeiro D, Moreira L, Amaral O. In Silico Analysis of Missense Mutations as a First Step in Functional Studies: Examples from Two Sphingolipidoses. International Journal of Molecular Sciences. 2018; 19(11):3409. https://doi.org/10.3390/ijms19113409
Chicago/Turabian StyleDuarte, Ana Joana, Diogo Ribeiro, Luciana Moreira, and Olga Amaral. 2018. "In Silico Analysis of Missense Mutations as a First Step in Functional Studies: Examples from Two Sphingolipidoses" International Journal of Molecular Sciences 19, no. 11: 3409. https://doi.org/10.3390/ijms19113409