Intravenous Delivery of piggyBac Transposons as a Useful Tool for Liver-Specific Gene-Switching

 , and
, and
Abstract
:1. Introduction
2. Results
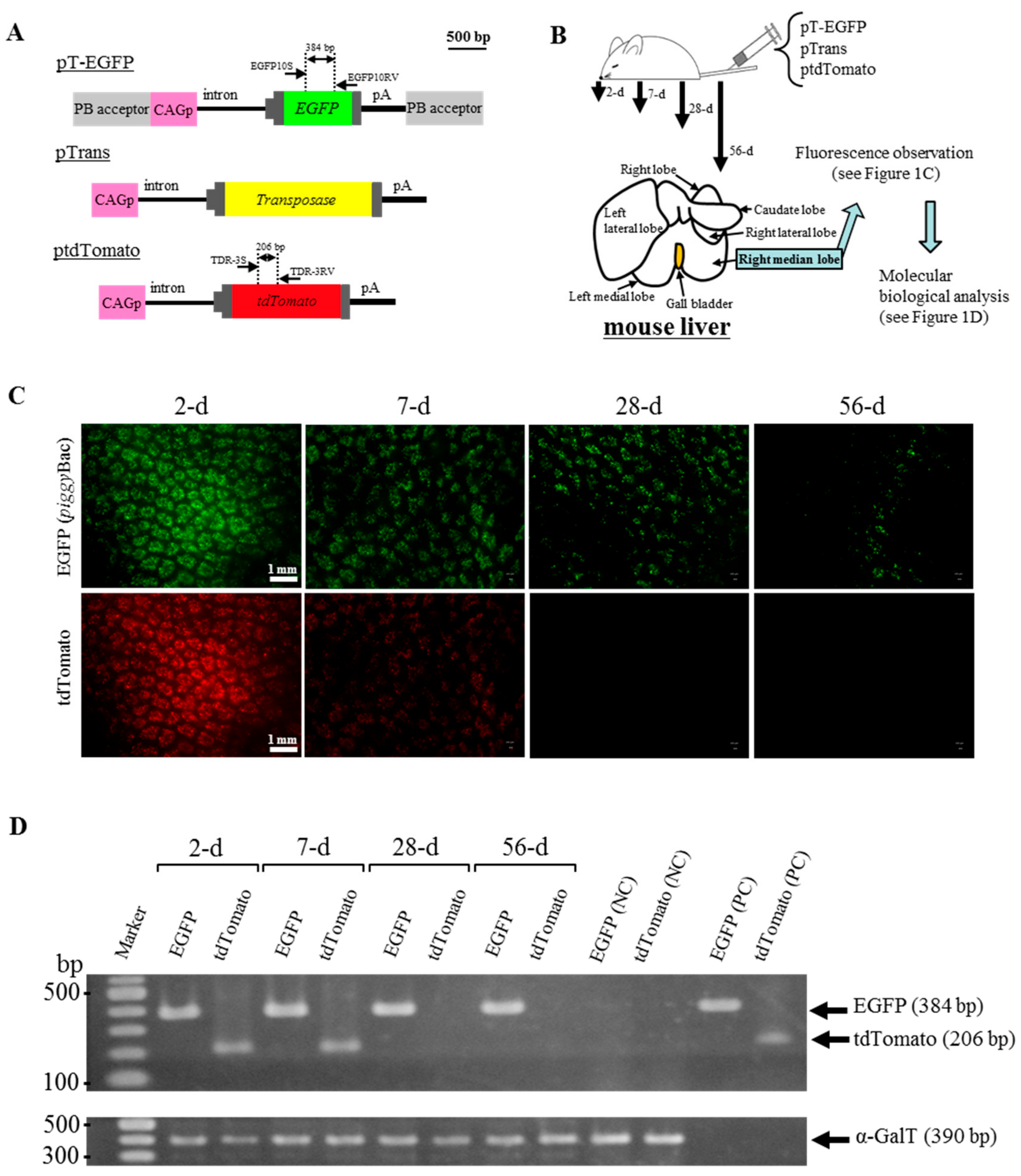
2.1. HGD with PB Transposons Confers Continuous Expression of GOI in Murine Liver
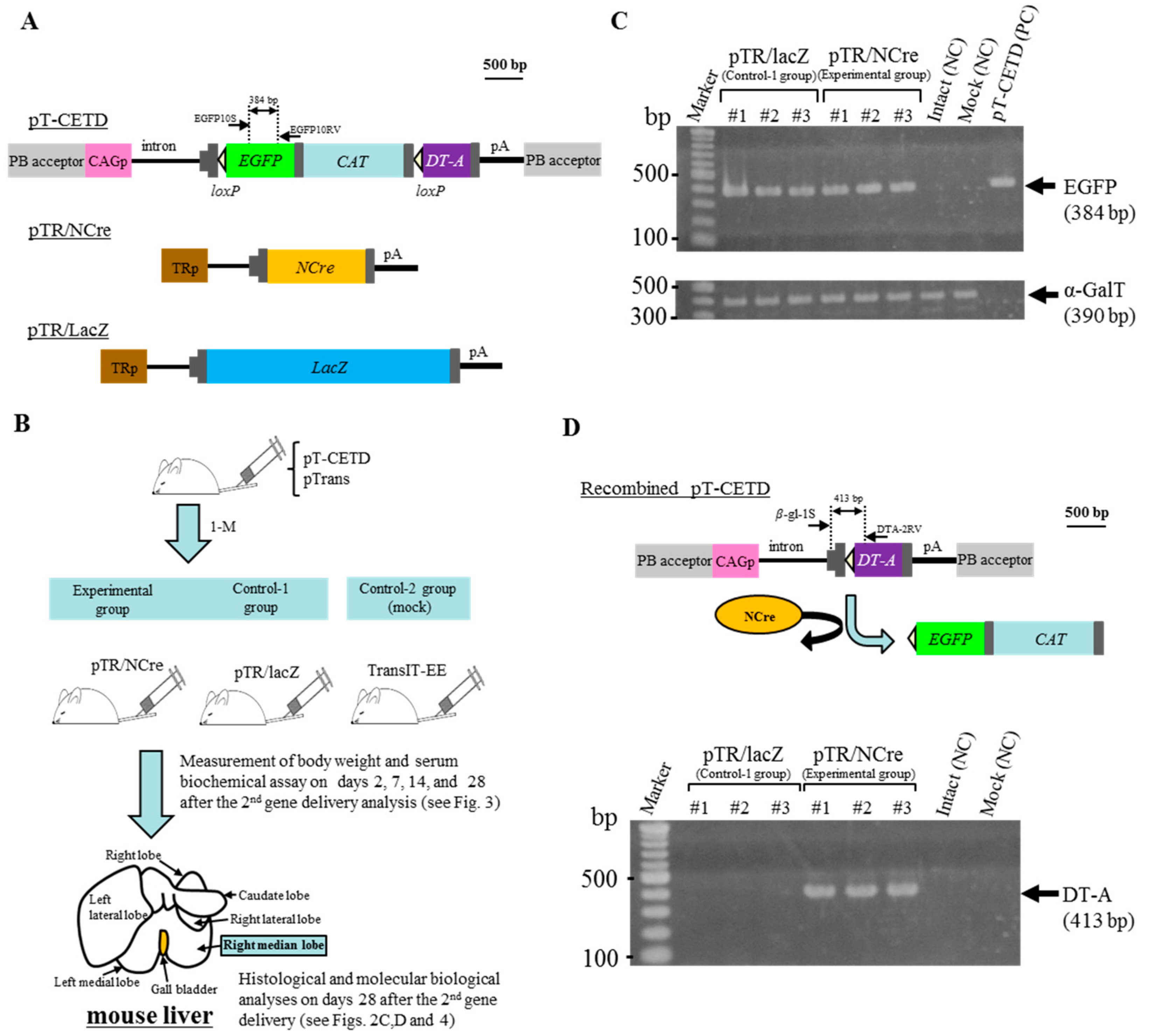
2.2. Liver-Specific Gene Switching In Vivo Using PB and Cre/loxP Systems
2.3. Serum Abnormality after DT-A Expression from Recombined pT-CETD
2.4. Pathological Abnormality in Liver after DT-A Expression from Recombined pT-CETD
3. Discussion
4. Materials and Methods
4.1. Plasmid Vectors
4.2. Mice
4.3. In Vivo Gene Delivery by the Intravenous Injection of Plasmids
4.4. Biochemical Examination
4.5. Microscopic Observation
4.6. Detection of Transgenes by PCR
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| α-GalT | α-1,3-galactosyltransferase |
| ALT | alanine transaminase |
| ANOVA | one-way factorial analysis of variance |
| AST | aspartate transaminase |
| CAG | unit comprising cytomegalovirus enhancer, chicken β-actin promoter, second intron of rabbit β-globin gene |
| CAT | chloramphenicol acetyltransferase |
| CETD | unit comprising CAG, loxP-flanked EGFP cDNA, CAT gene, DT-A gene, and poly(A) sites |
| CMV | cytomegalovirus |
| DT | diphtheria toxin |
| DT-A | cDNA for diphtheria toxin-A chain |
| EEF2 | ribosylating translation elongation factor 2 |
| EGFP | enhanced green fluorescent protein |
| EtBr | ethidium bromide |
| GOI | genes of interest |
| GOT | glutamic oxaloacetic transaminase |
| GPT | glutamic pyruvic transaminase |
| H&E | hematoxylin & eosin |
| HGD | hydrodynamics-based gene delivery |
| ICR | Institute of Cancer Research |
| IP injection | intraperitoneal injection |
| iPS cell | inducible pluripotent stem cell |
| ITR | inverted terminal repeat sequence |
| lacZ | β-galactosidase |
| NC | negative control |
| NCre | unit comprising nuclear localization signal gene, Cre gene |
| pA, | poly(A) sites |
| PB | piggyBac |
| PB acceptor | acceptor site in PB system |
| PC | positive control |
| PCR | polymerase chain reaction |
| pT | PB-based plasmid |
| SCID | severe combined immunodeficient |
| SD | standard deviation |
| tdTomato | tandem dimeric Tomato |
| TR | transthyretin promoter |
| TSTA | two-step amplification |
References
- Doyle, A.; McGarry, M.P.; Lee, N.A.; Lee, J.J. The construction of transgenic and gene knockout/knockin mouse models of human disease. Transgenic Res. 2012, 21, 327–349. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Liu, D. Hydrodynamic gene delivery: Its principles and applications. Mol. Ther. 2007, 15, 2063–2069. [Google Scholar] [CrossRef] [PubMed]
- Nayerossadat, N.; Maedeh, T.; Ali, P.A. Viral and nonviral delivery systems for gene delivery. Adv. Biomed. Res. 2012, 1, 27. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Song, Y.; Liu, D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999, 6, 1258–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budker, V.; Budker, T.; Zhang, G.; Subbotin, V.; Loomis, A.; Wolff, J.A. Hypothesis: Naked plasmid DNA is taken up by cells in vivo by a receptor-mediated process. J. Gene Med. 2000, 2, 76–88. [Google Scholar] [CrossRef]
- Brinster, R.L.; Allen, J.M.; Behringer, R.R.; Gelinas, R.E.; Palmiter, R.D. Introns increase transcriptional efficiency in transgenic mice. Proc. Natl. Acad. Sci. USA 1988, 836–840. [Google Scholar] [CrossRef]
- Choi, T.; Huang, M.; Gorman, C.; Jaenisch, R. A generic intron increases gene expression in transgenic mice. Mol. Cell. Biol. 1991, 11, 3070–3074. [Google Scholar] [CrossRef] [PubMed]
- Iyer, M.; Wu, L.; Carey, M.; Wang, Y.; Smallwood, A.; Gambhir, S.S. Twostep transcriptional amplification as a method for imaging reporter gene expression using weak promoters. Proc. Natl. Acad. Sci. USA 2001, 98, 14595–14600. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarczyk, S.J.; Green, J.E. A single vector containing modified cre recombinase and LOX recombination sequences for inducible tissue-specific amplification of gene expression. Nucleic Acids Res. 2001, 29, E56. [Google Scholar] [CrossRef] [PubMed]
- Glover, C.P.; Bienemann, A.S.; Heywood, D.J.; Cosgrave, A.S.; Uney, J.B. Adenoviral-mediated, high-level, cell-specific transgene expression: A SYN1-WPRE cassette mediates increased transgene expression with no loss of neuron specificity. Mol. Ther. 2002, 5, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Hermening, S.; Kügler, S.; Bähr, M.; Isenmann, S. Increased protein expression from adenoviral shuttle plasmids and vectors by insertion of a small chimeric intron sequence. J. Virol. Methods 2004, 122, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Nettelbeck, D.M.; Jerome, V.; Müller, R. Astrategy for enhancing the transcriptional activity of weak cell type-specific promoters. Gene Ther. 1998, 5, 1656–1664. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Watanabe, S.; Ohtsuka, M.; Maehara, T.; Ishihara, M.; Yokomine, T.; Sato, M. Cre-loxP system as a versatile tool for conferring increased levels of tissue-specific gene expression from a weak promoter. Mol. Reprod. Dev. 2008, 75, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Maehara, T.; Watanabe, S.; Ishihara, M.; Sato, M. Improvement of hydrodynamics-based gene transfer of nonviral DNA targeted to murine hepatocytes. Biomed. Res. Int. 2013, 2013, 928790. [Google Scholar] [CrossRef] [PubMed]
- Cary, L.C.; Goebel, M.; Corsaro, B.G.; Wang, H.G.; Rosen, E.; Fraser, M.J. Transposon mutagenesis of baculoviruses: Analysis of Trichoplusia ni transposon IFP2 insertions within the FP-locus of nuclear polyhedrosis viruses. Virology 1989, 172, 156–169. [Google Scholar] [CrossRef]
- Fraser, M.J., Jr.; Carey, L.; Boonvisudhi, K.; Wang, H.G.H. Assay for movement of Lepidepteran transposon IFP2 in insect cells using a Baculovirus genome as a target DNA. Virology 1995, 211, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.J.; Ciszczon, T.; Elick, T.; Bauser, C. Precise excision of TTAA-specific lepidopteran transposons piggyBac (IFP2) and tagalong (TFP3) from the baculovirus genome in cell lines from two species of Lepidoptera. Insect. Mol. Biol. 1996, 5, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Palavesam, A.; Esnault, C.; O’Brochta, D.A. Post-integration silencing of piggyBac transposable elements in Aedes aegypti. PLoS ONE 2013, 8, e68454. [Google Scholar] [CrossRef] [PubMed]
- Mossine, V.V.; Waters, J.K.; Hannink, M.; Mawhinney, T.P. PiggyBac transposon plus insulators overcome epigenetic silencing to provide for stable signaling pathway reporter cell lines. PLoS ONE 2013, 8, e85494. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Wu, X.; Li, G.; Han, M.; Zhuang, Y.; Xu, T. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell 2005, 122, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.P.; Yang, M.M.; Chen, Y.L. PiggyBac transposon-mediated gene transfer in Cashmere goat fetal fibroblast cells. Biosci. Biotechnol. Biochem. 2012, 76, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Rad, R.; Rad, L.; Wang, W.; Cadinanos, J.; Vassiliou, G.; Rice, S.; Campos, L.S.; Yusa, K.; Banerjee, R.; Li, M.A.; et al. PiggyBac transposon mutagenesis: A tool for cancer gene discovery in mice. Science 2010, 330, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
- Woltjen, K.; Michael, I.P.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. PiggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 2009, 458, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Kaji, K.; Norrby, K.; Paca, A.; Mileikovsky, M.; Mohseni, P.; Woltjen, K. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature 2009, 458, 771–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusa, K.; Rad, R.; Takeda, J.; Bradley, A. Generation of transgene-free induced pluripotent mouse stem cells by the piggyBac transposon. Nat. Methods 2009, 6, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Nagy, K.; Sung, H.K.; Zhang, P.; Laflamme, S.; Vincent, P.; Agha-Mohammadi, S.; Woltjen, K.; Monetti, C.; Michael, I.P.; Smith, L.C.; et al. Induced pluripotent stem cell lines derived from equine fibroblasts. Stem Cell Rev. Rep. 2011, 7, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H.; Higuchi, Y.; Kawakami, S.; Yamashita, F.; Hashida, M. piggyBac transposon-mediated long-term gene expression in mice. Mol. Ther. 2010, 18, 707–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.C.; Meir, Y.J.; Coates, C.J.; Handler, A.M.; Pelczar, P.; Moisyadi, S.; Kaminski, J.M. piggyBac is a flexible and highly active transposon as compared to sleeping beauty, Tol2, and Mos1 in mammalian cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15008–15013. [Google Scholar] [CrossRef] [PubMed]
- Ivics, Z.; Li, M.A.; Mátés, L.; Boeke, J.D.; Nagy, A.; Bradley, A.; Izsvák, Z. Transposon-mediated genome manipulation in vertebrates. Nat. Methods 2009, 6, 415–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, M.H.; Coates, C.J.; George, A.L., Jr. PiggyBac transposon-mediated gene transfer in human cells. Mol. Ther. 2007, 15, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lin, C.; Lu, D.; Ning, Z.; Cox, T.; Melvin, D.; Wang, X.; Bradley, A.; Liu, P. Chromosomal transposition of piggyBac in mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA 2008, 105, 9290–9295. [Google Scholar] [CrossRef] [PubMed]
- Bauser, C.A.; Elick, T.A.; Fraser, M.J. Proteins from nuclear extracts of two lepidopteran cell lines recognize the ends of TTAA-specific transposons piggyBac and tagalong. Insect. Mol. Biol. 1999, 8, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Saridey, S.K.; Liu, L.; Doherty, J.E.; Kaja, A.; Galvan, D.L.; Fletcher, B.S.; Wilson, M.H. piggyBac transposon-based inducible gene expression in vivo after somatic cell gene transfer. Mol. Ther. 2009, 17, 2115–2120. [Google Scholar] [CrossRef] [PubMed]
- Cooney, A.L.; Singh, B.K.; Sinn, P.L. Hybrid nonviral/viral vector systems for improved piggyBac DNA transposon in vivo delivery. Mol. Ther. 2015, 23, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Pappenheimer, A.M. Diphtheria toxin. Annu. Rev. Biochem. 1977, 46, 69–94. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Terashima, M.; Kikuchi, N.; Kimura, M.; Maehara, T.; Saito, A.; Sato, M. A new mouse model for renal lesions produced by intravenous injection of diphtheria toxin A-chain expression plasmid. BMC Nephrol. 2004, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, M.; Tanigawa, M. Production of CETD transgenic mouse line allowing ablation of any type of specific cell population. Mol. Reprod. Dev. 2005, 72, 54–67. [Google Scholar] [CrossRef] [PubMed]
- McGill, M.R. The past and present of serum aminotransferases and the future of liver injury biomarkers. EXCLI J. 2016, 15, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.R.; Zheng, Y.W.; Taniguchi, H. Generation of a Humanized Mouse Liver Using Human Hepatic Stem Cells. J. Vis. Exp. 2016, 29. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Yokoi, T. Application of chimeric mice with humanized liver for predictive ADME. Drug Metab. Rev. 2007, 39, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Maeda, K.; Koriyama, M.; Inada, E.; Saitoh, I.; Miura, H.; Ohtsuka, M.; Nakamura, S.; Sakurai, T.; Watanabe, S.; et al. The piggyBac-based gene delivery system can confer successful production of cloned porcine blastocysts with multigene constructs. Int. J. Mol. Sci. 2016, 17, 1424. [Google Scholar] [CrossRef] [PubMed]
- Kolacsek, O.; Erdei, Z.; Apáti, A.; Sándor, S.; Izsvák, Z.; Ivics, Z.; Sarkadi, B.; Orbán, T.I. Excision efficiency is not strongly coupled to transgenic rate: Cell type-dependent transposition efficiency of sleeping beauty and piggyBac DNA transposons. Hum. Gene Ther. Methods 2014, 25, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Lin, X.; Lin, X.; Lin, T.; Chen, B.; Hao, W.; Cheng, Y.; Liu, Y.; Dian, M.; Yao, K.; et al. R/L, a double reporter mouse line that expresses luciferase gene upon Cre-mediated excision, followed by inactivation of mRFP expression. Genome 2016, 59, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.J.; Oberli, M.A.; Dorkin, J.R.; Hurtado, J.E.; Kaczmarek, J.C.; Bhadani, S.; Wyckoff, J.; Langer, R.; Jaklenec, A.; Anderson, D.G. Rapid, Single-Cell Analysis and Discovery of Vectored mRNA Transfection in vivo with a loxP-Flanked tdTomato Reporter Mouse. Mol. Ther. Nucleic Acids 2018, 10, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.J.; Drake, J.C.; Cui, D.; Zhang, M.; Perry, H.M.; Kashatus, J.A.; Kusminski, C.M.; Scherer, P.E.; Kashatus, D.F.; Okusa, M.D.; et al. Conditional MitoTimer reporter mice for assessment of mitochondrial structure, oxidative stress, and mitophagy. Mitochondrion 2017. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Sun, R.; Huang, Q.; Tian, Z. Technical Improvement and Application of Hydrodynamic Gene Delivery in Study of Liver Diseases. Front. Pharmacol. 2017, 30, 591. [Google Scholar] [CrossRef] [PubMed]
- Zang, L.; Nishikawa, M.; Ando, M.; Takahashi, Y.; Takakura, Y. Contribution of Epigenetic Modifications to the Decline in Transgene Expression from Plasmid DNA in Mouse Liver. Pharmaceutics 2015, 7, 199–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.Y.; Riu, E.; He, C.Y.; Xu, H.; Kay, M.A. Silencing of episomal transgene expression in liver by plasmid bacterial backbone DNA is independent of CpG methylation. Mol. Ther. 2008, 16, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sugden, B. Origins of bidirectional replication of Epstein-Barr virus: Models for understanding mammalian origins of DNA synthesis. J. Cell. Biochem. 2005, 94, 247–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yew, N.S.; Marshall, J.; Przybylska, M.; Wysokenski, D.M.; Ziegler, R.J.; Rafter, P.W.; Li, C.; Armentano, D.; Cheng, S.H. Increased duration of transgene expression in the lung with plasmid DNA vectors harboring adenovirus E4 open reading frame 3. Hum. Gene Ther. 1999, 10, 1833–1843. [Google Scholar] [CrossRef] [PubMed]
- Yew, N.S.; Przybylska, M.; Ziegler, R.J.; Liu, D.; Cheng, S.H. High and sustained transgene expression in vivo from plasmid vectors containing a hybrid ubiquitin promoter. Mol. Ther. 2001, 4, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, T.; Haase, R.; Schleef, M.; Wagner, E.; Ogris, M. Sustained, high transgene expression in liver with plasmid vectors using optimized promoter-enhancer combinations. J. Gene Med. 2011, 13, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Herweijer, H.; Zhang, G.; Subbotin, V.M.; Budker, V.; Williams, P.; Wolff, J.A. Time course of gene expression after plasmid DNA gene transfer to the liver. J. Gene Med. 2001, 3, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Wooddell, C.I.; Reppen, T.; Wolff, J.A.; Herweijer, H. Sustained liver-specific transgene expression from the albumin promoter in mice following hydrodynamic plasmid DNA delivery. J. Gene Med. 2008, 10, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Wolff, L.J.; Wolff, J.A.; Sebestyén, M.G. Effect of tissue-specific promoters and microRNA recognition elements on stability of transgene expression after hydrodynamic naked plasmid DNA delivery. Hum. Gene Ther. 2009, 20, 374–388. [Google Scholar] [CrossRef] [PubMed]
- Kay, M.A.; He, C.Y.; Chen, Z.Y. A robust system for production of minicircle DNA vectors. Nat. Biotechnol. 2010, 28, 1287–1289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maniar, G.L.E.; Maniar, J.M.; Chen, Z.Y.; Lu, J.; Fire, A.Z.; Kay, M.A. Minicircle DNA vectors achieve sustained expression reflected by active chromatin and transcriptional level. Mol. Ther. 2013, 21, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Olivares, E.C.; Hollis, R.P.; Chalberg, T.W.; Meuse, L.; Kay, M.A.; Calos, M.P. Site-specific genomic integration produces therapeutic Factor IX levels in mice. Nat. Biotechnol. 2002, 20, 1124–1128. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, A.; Xu, H.; Huang, Z.; Engler, J.A.; Kay, M.A. A direct comparison of two nonviral gene therapy vectors for somatic integration: In vivo evaluation of the bacteriophage integrase phiC31 and the Sleeping Beauty transposase. Mol. Ther. 2005, 11, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Niwa, H.; Yamamura, K.; Miyazaki, J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 1991, 108, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Inada, E.; Saitoh, I.; Matsumoto, Y.; Ohtsuka, M.; Miura, H.; Nakamura, S.; Sakurai, T.; Watanabe, S. A combination of targeted toxin technology and the piggyBac-mediated gene transfer system enables efficient isolation of stable transfectants in nonhuman mammalian cells. Biotechnol. J. 2015, 10, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Yamaizumi, M.; Mekada, E.; Uchida, T.; Okada, Y. One molecule of diphtheria toxin fragment A introduced into a cell can kill the cell. Cell 1978, 15, 245–250. [Google Scholar] [CrossRef]
- Palmiter, R.D.; Behringer, R.P.; Quaife, C.J.; Maxwell, F.; Maxwell, I.H.; Brinster, R.L. Cell lineage ablation in transgenic mice by cell-specific expression of a toxin gene. Cell 1987, 50, 435–443. [Google Scholar] [CrossRef]
- Breitman, M.L.; Clapoff, S.; Rossant, J.; Tsui, L.C.; Glode, L.M.; Maxwell, I.H.; Bernstein, A. Genetic ablation: Targeted expression of a toxin gene causes microphthalmia in transgenic mice. Science 1987, 238, 1563–1565. [Google Scholar] [CrossRef] [PubMed]
- Breitman, M.L.; Bryce, D.M.; Giddens, E.; Clapoff, S.; Goring, D.; Tsui, L.C.; Klintworth, G.K.; Bernstein, A. Analysis of lens cell fate and eye morphogenesis in transgenic mice ablated for cells of the lens lineage. Development 1989, 106, 457–463. [Google Scholar] [PubMed]
- Lowell, B.B.; S-Sisulic, V.; Hamann, A.; Lawitts, J.A.; Himms-Hagen, J.; Boyer, B.B.; Kozak, L.P.; Flier, J.S. Development of obesity in transgenic mice after genetic ablation of brown adipose tissue. Nature 1993, 366, 740–742. [Google Scholar] [CrossRef] [PubMed]
- Herrera, P.L.; Huarte, J.; Zufferey, R.; Nichols, A.; Mermillod, B.; Philippe, J.; Muniesa, P.; Sanvito, F.; Orci, L.; Vassalli, J.D. Ablation of islet endocrine cells by targeted expression of hormone-promoter-driven toxigenes. Proc. Natl. Acad. Sci. USA 1994, 91, 12999–13003. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Maehara, T.; Watanabe, S.; Ishihara, M.; Sato, M. Liver lobe and strain difference in gene expression after hydrodynamics-based gene delivery in mice. Anim. Biotechnol. 2015, 26, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Ohtsuka, M.; Nakamura, S. Intraoviductal instillation of a solution as an effective route for manipulating preimplantation mammalian embryos in vivo. In New Insights into Theriogenology; InTechOpen: London, UK, 2018. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Primer | Sequence (5′–3′) |
|---|---|
| EGFP-10S [41] | CCT GAA GTT CAT CTG CAC CAC |
| EGFP-10RV [41] | GTT GTG GCG GAT CTT GAA GTT |
| TDR-3S [41] | CCC GTA ATG CAG AAG AAG ACC |
| TDR-3RV [41] | GTG ATG TCC AGC TTG GTG TCC |
| β-gl-1S [37] | TGT GCT GTC TCA TCA TTT TGG |
| DTA-2RV [37] | GCG AGA ACC TTC GTC AGT CCT |
| mEx4-S [70] | GCA AAT GTG GAT GCT GGG AAC |
| mEx4-RV [70] | ACA GTT TTA ATG GCC ATC TGG |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakamura, S.; Ishihara, M.; Watanabe, S.; Ando, N.; Ohtsuka, M.; Sato, M. Intravenous Delivery of piggyBac Transposons as a Useful Tool for Liver-Specific Gene-Switching. Int. J. Mol. Sci. 2018, 19, 3452. https://doi.org/10.3390/ijms19113452
Nakamura S, Ishihara M, Watanabe S, Ando N, Ohtsuka M, Sato M. Intravenous Delivery of piggyBac Transposons as a Useful Tool for Liver-Specific Gene-Switching. International Journal of Molecular Sciences. 2018; 19(11):3452. https://doi.org/10.3390/ijms19113452
Chicago/Turabian StyleNakamura, Shingo, Masayuki Ishihara, Satoshi Watanabe, Naoko Ando, Masato Ohtsuka, and Masahiro Sato. 2018. "Intravenous Delivery of piggyBac Transposons as a Useful Tool for Liver-Specific Gene-Switching" International Journal of Molecular Sciences 19, no. 11: 3452. https://doi.org/10.3390/ijms19113452