Regulation of Protein Structural Changes by Incorporation of a Small-Molecule Linker

 , ,
, ,
Abstract
:
1. Introduction
2. Results
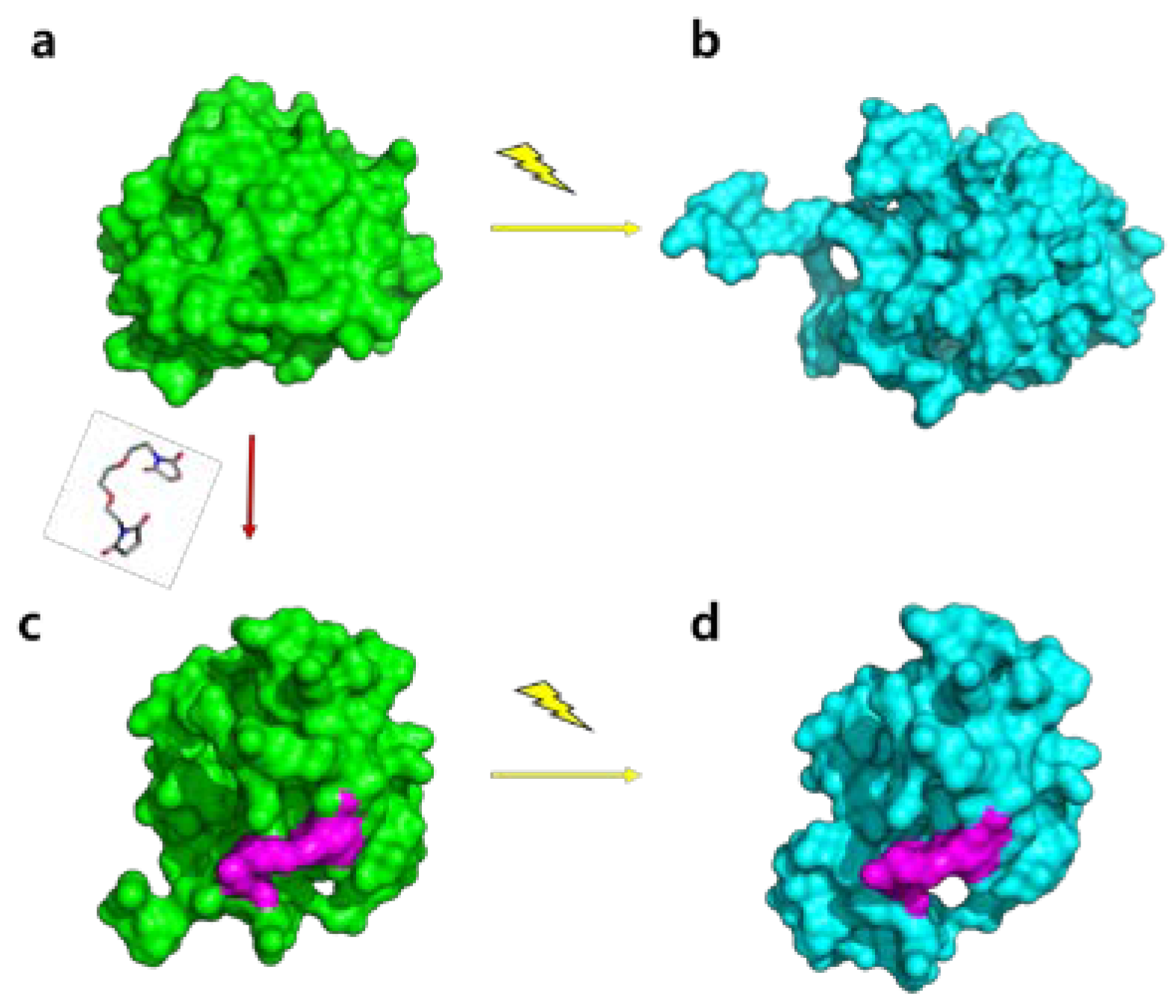
2.1. Sample Characterization
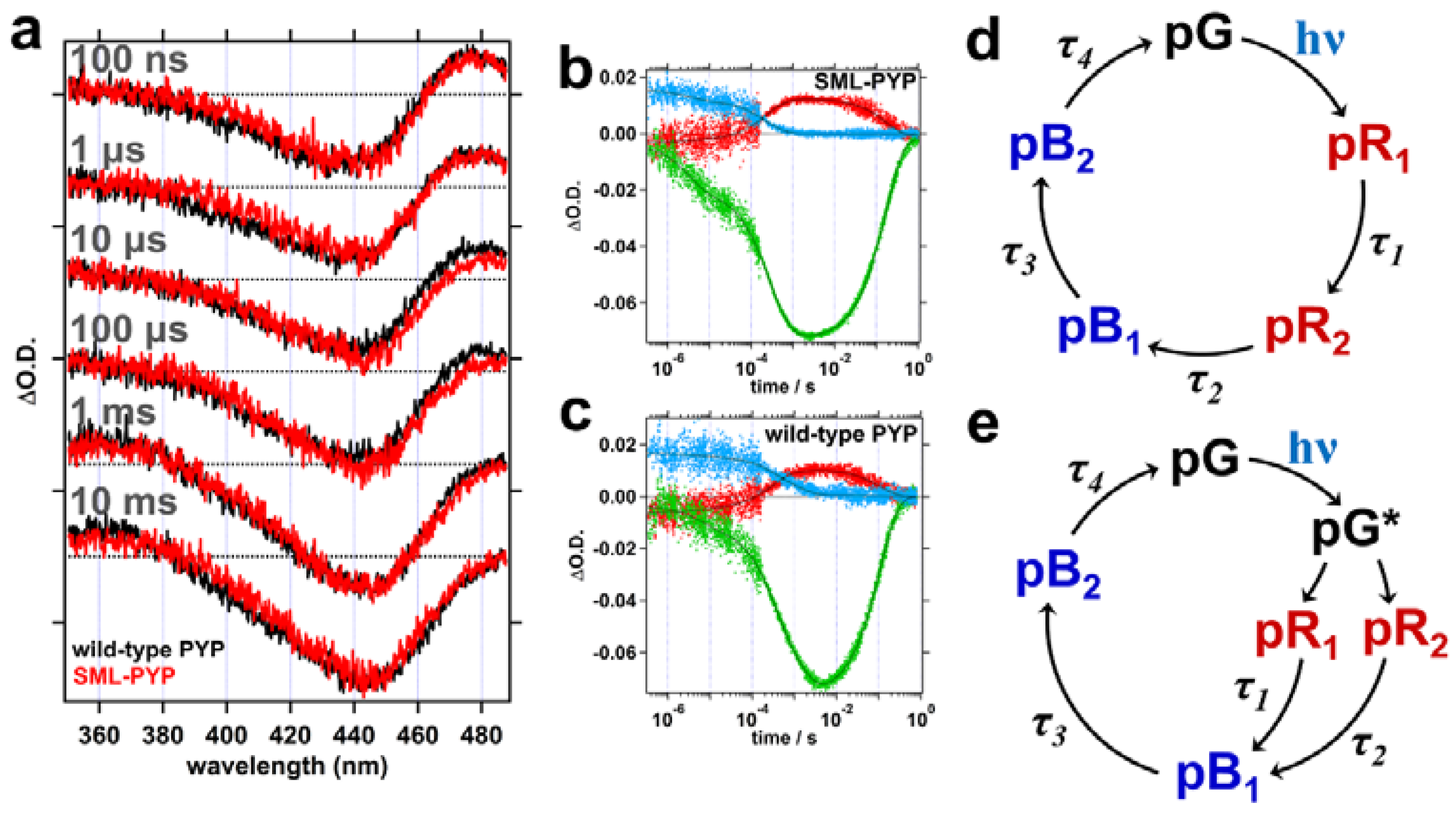
2.2. Transient Absorption (TA) Spectroscopy
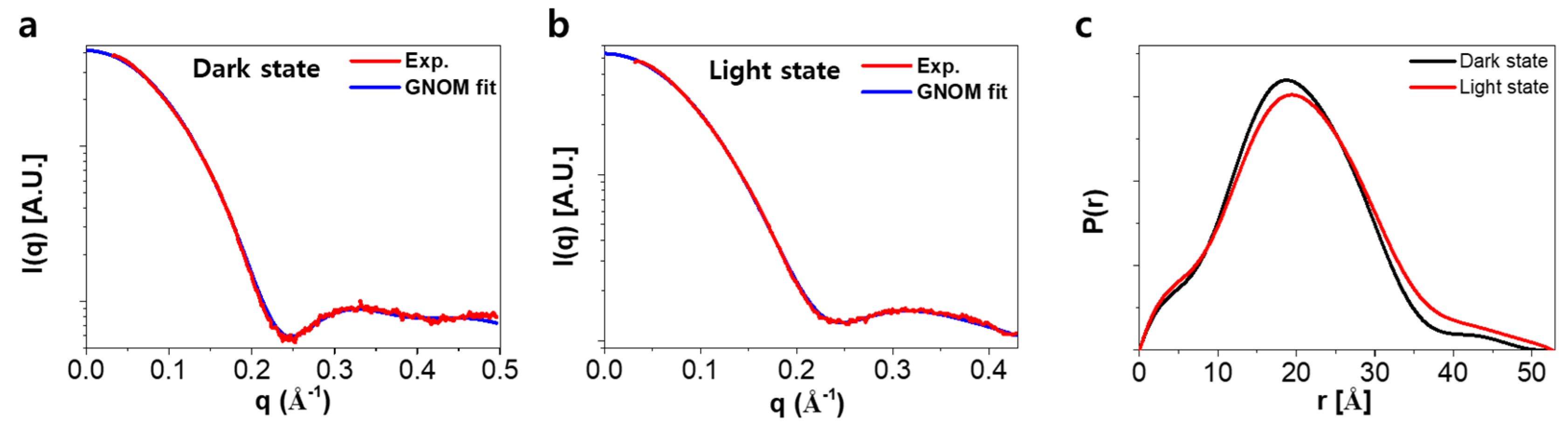
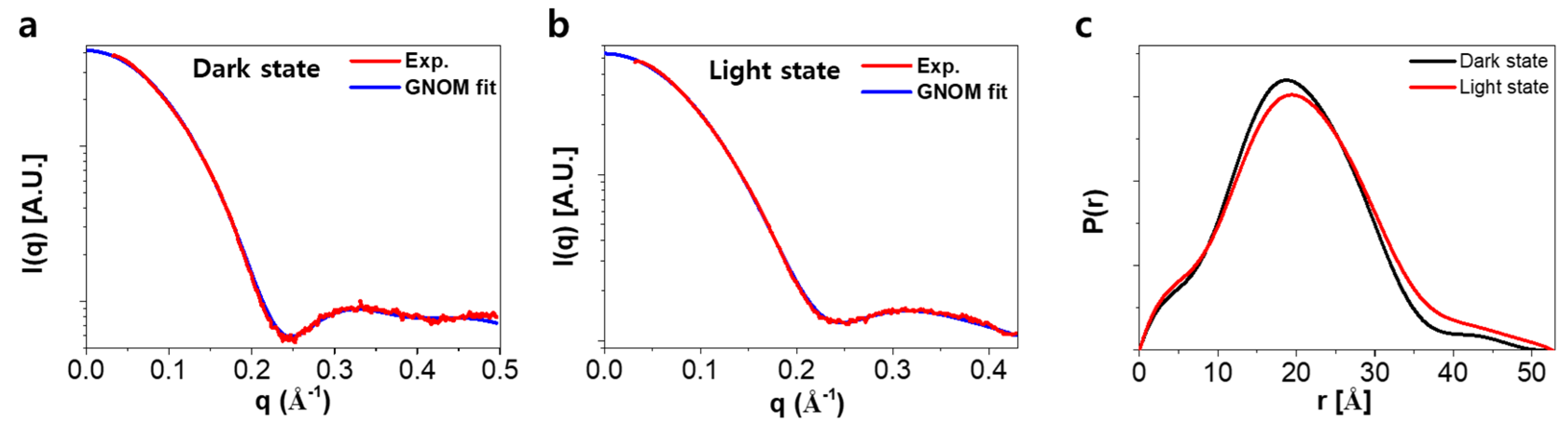
2.3. Analysis of SAXS Data for the Ground and the Light-Activated States of SML-PYP
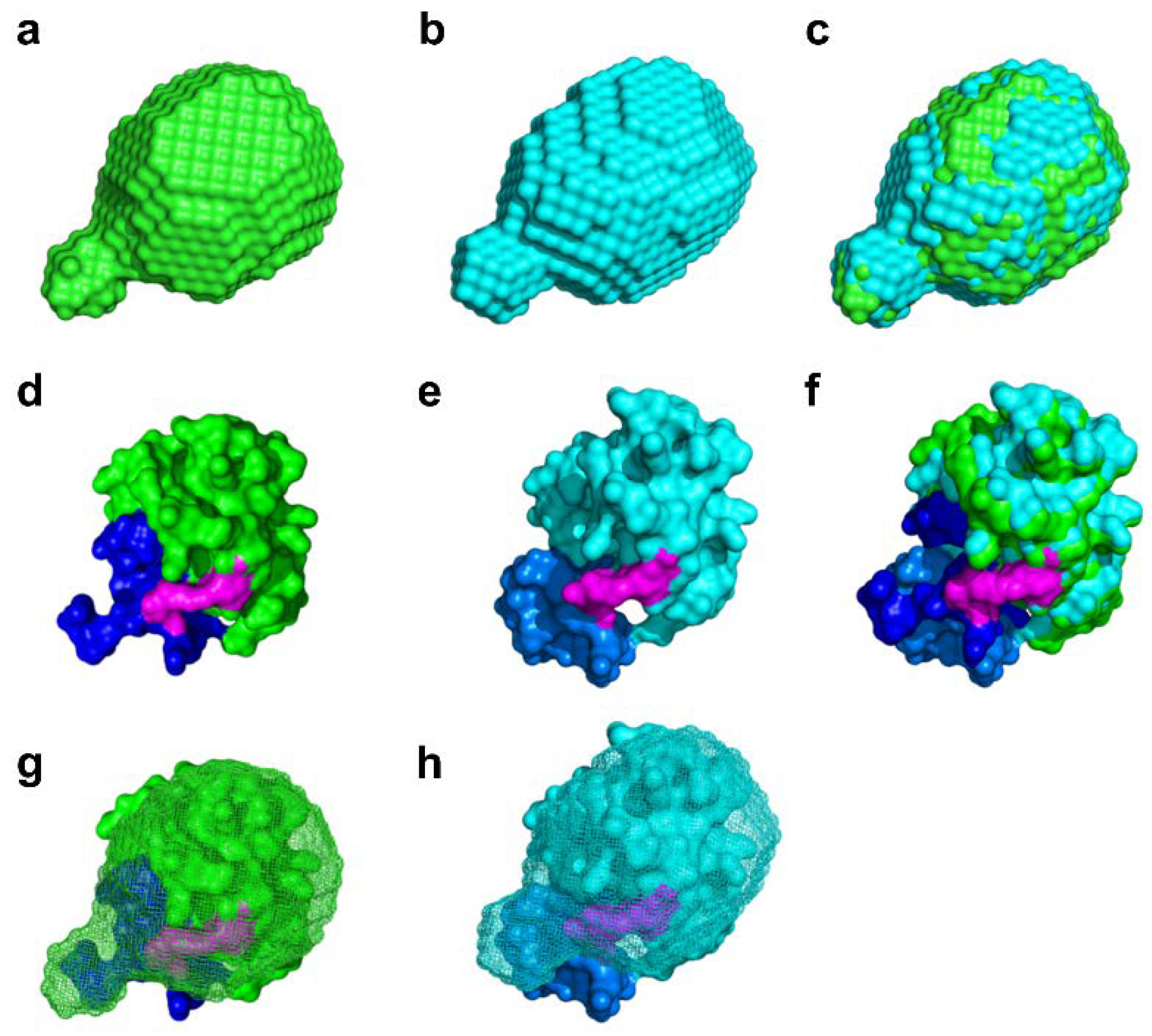
2.4. Molecular Shape Reconstruction from the SAXS Data
2.5. Experiment-Restrained Rigid-Body MD Simulation
3. Discussion
4. Materials and Methods
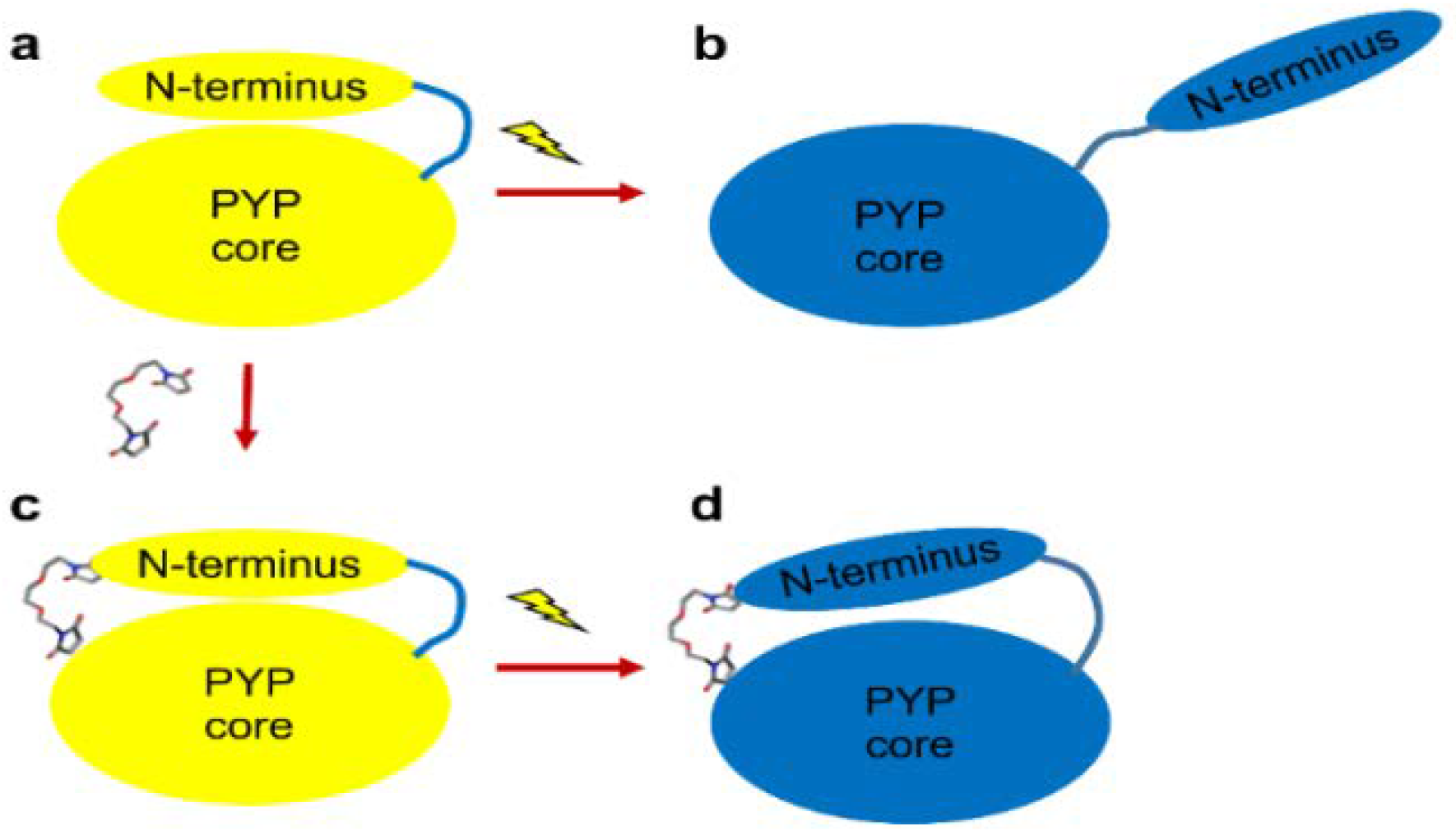
4.1. Preparation of PYP with Blocked N-terminal Movement
4.2. Transient Absorption (TA) Measurement
4.3. SAXS
4.4. SAXS Analysis and Molecular Shape Reconstruction
4.5. SAXS Curve Analysis with the Experiment-Restrained Rigid-Body MD Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PYP | Photoactive yellow protein |
| SML | Small-molecule linker |
| BM(PEG)2 | 1,8-Bismaleimidodiethyleneglycol |
| SML-PYP | Cross-linker (BM(PEG)2)-conjugated PYP |
| MD | Molecular dynamics |
| CD | Circular dichroism |
| Rg | Radius of gyration |
| SAXS | Small-angle X-ray solution scattering |
| MALDI/TOF | Matrix-assisted laser desorption/ionization time-of-flight |
| TA | Transient absorption |
References
- Benkovic, S.J.; Raney, K.D. Mechanisms: Molecular machines. Curr. Opin. Chem. Biol. 2011, 15, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Marcos, V.; Leigh, D.A. Molecular machines with bio-inspired mechanisms. Proc. Natl. Acad. Sci. USA 2018, 115, 9397–9404. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.M.; Woolley, G.A. The effect of azobenzene cross-linker position on the degree of helical peptide photo-control. Org. Biomol. Chem. 2013, 11, 5325–5331. [Google Scholar] [CrossRef] [PubMed]
- Woolley, G.A. Protein machines: An open and shut cage. Nat. Nanotechnol. 2013, 8, 892–893. [Google Scholar] [CrossRef] [PubMed]
- Imamoto, Y.; Ito, T.; Kataoka, M.; Tokunaga, F. Reconstitution photoactive yellow protein from apoprotein and P-coumaric acid-derivatives. FEBS Lett. 1995, 374, 157–160. [Google Scholar] [CrossRef]
- Lee, K.; Kim, Y.; Jung, J.; Ihee, H.; Park, Y. Measurements of complex refractive index change of photoactive yellow protein over a wide wavelength range using hyperspectral quantitative phase imaging. Sci. Rep. 2018, 8, 3064. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Ganesan, P.; Jo, J.; Kim, S.O.; Thamilselvan, K.; Ihee, H. Chromophore-removal-induced conformational change in photoactive yellow protein determined through spectroscopic and X-ray solution scattering studies. J. Phys. Chem. B 2018, 122, 4513–4520. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Kim, T.W.; Kim, Y.; Choi, J.; Lee, S.J.; Ihee, H. Kinetics of the E46Q mutant of photoactive yellow protein investigated by transient grating spectroscopy. Chem. Phys. Lett. 2017, 683, 262–267. [Google Scholar] [CrossRef]
- Kumar, A.; Burns, D.C.; Al-Abdul-Wahid, M.S.; Woolley, G.A. A Circularly permuted photoactive yellow protein as a scaffold for photoswitch design. Biochemistry 2013, 52, 3320–3331. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.O.; Lee, J.H.; Kim, J.; Schmidt, M.; Moffat, K.; Srajer, V.; Ihee, H. Volume-conserving trans-cis isomerization pathways in photoactive yellow protein visualized by picosecond X-ray crystallography. Nat. Chem. 2013, 5, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Lee, J.H.; Choi, J.; Kim, K.H.; van Wilderen, L.J.; Guerin, L.; Kim, Y.; Jung, Y.O.; Yang, C.; Kim, J.; et al. Protein structural dynamics of photoactive yellow protein in solution revealed by pump-probe X-ray solution scattering. J. Am. Chem. Soc. 2012, 134, 3145–3153. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.L.; Lovett, J.E.; Carl, P.J.; Cammarata, M.; Lee, J.H.; Jung, Y.O.; Ihee, H.; Timmel, C.R.; van Thor, J.J. The short-lived signaling state of the photoactive yellow protein photoreceptor revealed by combined structural probes. J. Am. Chem. Soc. 2011, 133, 9395–9404. [Google Scholar] [CrossRef] [PubMed]
- Imamoto, Y.; Kataoka, M.; Tokunaga, F.; Asahi, T.; Masuhara, H. Primary photoreaction of photoactive yellow protein studied by subpicosecond-nanosecond spectroscopy. Biochemistry 2001, 40, 6047–6052. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, K.; Imamoto, Y.; Kataoka, M.; Mihara, K.; Tokunaga, F.; Terazima, M. Structural change of site-directed mutants of PYP: New dynamics during pR state. Biophys. J. 2002, 83, 1567–1577. [Google Scholar] [CrossRef]
- Losi, A.; Gensch, T.; van der Horst, M.A.; Hellingwerf, K.J.; Braslavsky, S.E. Hydrogen-bond network probed by time-resolved optoacoustic spectroscopy: Photoactive yellow protein and the effect of e46q and e46a mutations. Phys. Chem. Chem. Phys. 2005, 7, 2229–2236. [Google Scholar] [CrossRef] [PubMed]
- Ihee, H.; Rajagopal, S.; Srajer, V.; Pahl, R.; Anderson, S.; Schmidt, M.; Schotte, F.; Anfinrud, P.A.; Wulff, M.; Moffat, K. Visualizing reaction pathways in photoactive yellow protein from nanoseconds to seconds. Proc. Natl. Acad. Sci. USA 2005, 102, 7145–7150. [Google Scholar] [CrossRef] [PubMed]
- Yeremenko, S.; van Stokkum, I.H.M.; Moffat, K.; Hellingwerf, K.J. Influence of the crystalline state on photoinduced dynamics of photoactive yellow protein studied by ultraviolet-visible transient absorption spectroscopy. Biophys. J. 2006, 90, 4224–4235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svergun, D.I. Small-angle X-ray and neutron scattering as a tool for structural systems biology. Biol. Chem. 2010, 391, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Jacques, D.A.; Trewhella, J. Small-angle scattering for structural biology-Expanding the frontier while avoiding the pitfalls. Protein Sci. 2010, 19, 642–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Putnam, C.D.; Hammel, M.; Hura, G.L.; Tainer, J.A. X-ray solution scattering (SAXS) combined with crystallography and computation: Defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 2007, 40, 191–285. [Google Scholar] [CrossRef] [PubMed]
- Svergun, D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992, 25, 495–503. [Google Scholar] [CrossRef] [Green Version]
- Franke, D.; Svergun, D.I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2009, 42, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Kim, K.H.; Kim, Y.; Kim, J.; Ihee, H. Protein tertiary structural changes visualized by time-resolved X-ray solution scattering. J. Phys. Chem. B 2009, 113, 13131–13133. [Google Scholar] [CrossRef] [PubMed]
- Amrute-Nayak, M.; Diensthuber, R.P.; Steffen, W.; Kathmann, D.; Hartmann, F.K.; Fedorov, R.; Urbanke, C.; Manstein, D.J.; Brenner, B.; Tsiavaliaris, G. Targeted optimization of a protein nanomachine for operation in biohybrid devices. Angew. Chem. Int. Ed. 2010, 49, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Ganesan, P.; Ihee, H. High-throughput instant quantification of protein expression and purity based on photoactive yellow protein turn off/on label. Protein Sci. 2013, 22, 1109–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Jung, Y.O.; Lee, J.H.; Yang, C.; Kim, B.; Ihee, H. Folding dynamics of ferrocytochrome c in a denaturant-free environment probed by transient grating spectroscopy. ChemPhysChem 2008, 9, 2708–2714. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Muniyappan, S.; Wallis, J.T.; Royer, W.E.; Ihee, H. Protein conformational dynamics of homodimeric hemoglobin revealed by combined time-resolved spectroscopic probes. ChemPhysChem 2010, 11, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Yang, C.; Kim, J.; Ihee, H. Protein folding dynamics of cytochrome c seen by transient grating and transient absorption spectroscopies. J. Phys. Chem. B 2011, 115, 3127–3135. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Choi, J.; Ihee, H. The time scale of the quaternary structural changes in hemoglobin revealed using the transient grating technique. Phys. Chem. Chem. Phys. 2015, 17, 22571–22575. [Google Scholar] [CrossRef] [PubMed]
- Volkov, V.V.; Svergun, D.I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2003, 36, 860–864. [Google Scholar] [CrossRef]
- Kozin, M.B.; Svergun, D.I. Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 2001, 34, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Svergun, D.; Barberato, C.; Koch, M.H.J. CRYSOL—A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 1995, 28, 768–773. [Google Scholar] [CrossRef]
- James, F.; Roos, M. Minuit-system for function minimization and analysis of parameter errors and correlations. Comput. Phys. Commun. 1975, 10, 343–367. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fitting Results | SML-PYP | Wild-Type PYP |
|---|---|---|
| τ1 (μs) | 3.3 ± 1.3 | 4.4 ± 1.2 |
| τ2 (μs) | 200 ± 51 | 210 ± 49 |
| τ3 (μs) | 480 ± 140 | 920 ± 260 |
| τ4 (ms) | 170 ± 3 | 120 ± 10 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.; Yang, C.; Kim, T.W.; Thamilselvan, K.; Kim, Y.; Ihee, H. Regulation of Protein Structural Changes by Incorporation of a Small-Molecule Linker. Int. J. Mol. Sci. 2018, 19, 3714. https://doi.org/10.3390/ijms19123714
Kim Y, Yang C, Kim TW, Thamilselvan K, Kim Y, Ihee H. Regulation of Protein Structural Changes by Incorporation of a Small-Molecule Linker. International Journal of Molecular Sciences. 2018; 19(12):3714. https://doi.org/10.3390/ijms19123714
Chicago/Turabian StyleKim, Youngmin, Cheolhee Yang, Tae Wu Kim, Kamatchi Thamilselvan, Yonggwan Kim, and Hyotcherl Ihee. 2018. "Regulation of Protein Structural Changes by Incorporation of a Small-Molecule Linker" International Journal of Molecular Sciences 19, no. 12: 3714. https://doi.org/10.3390/ijms19123714