AMP-activated Protein Kinase Controls Immediate Early Genes Expression Following Synaptic Activation Through the PKA/CREB Pathway

 ,
,
Abstract
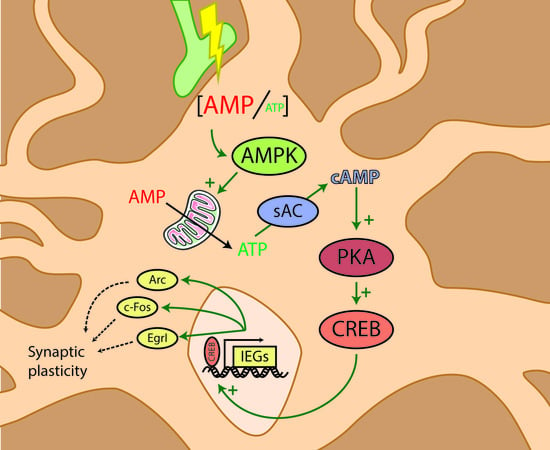
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. AMPK Activity is Required for Synaptic Activity-Induced IEGs Transcriptional Regulation
2.2. PKA Pathway Is Activated Following SA and Is Required for The Expression of IEGs
2.3. PKA Activation Following Synaptic Activation is Mediated by the Soluble AC
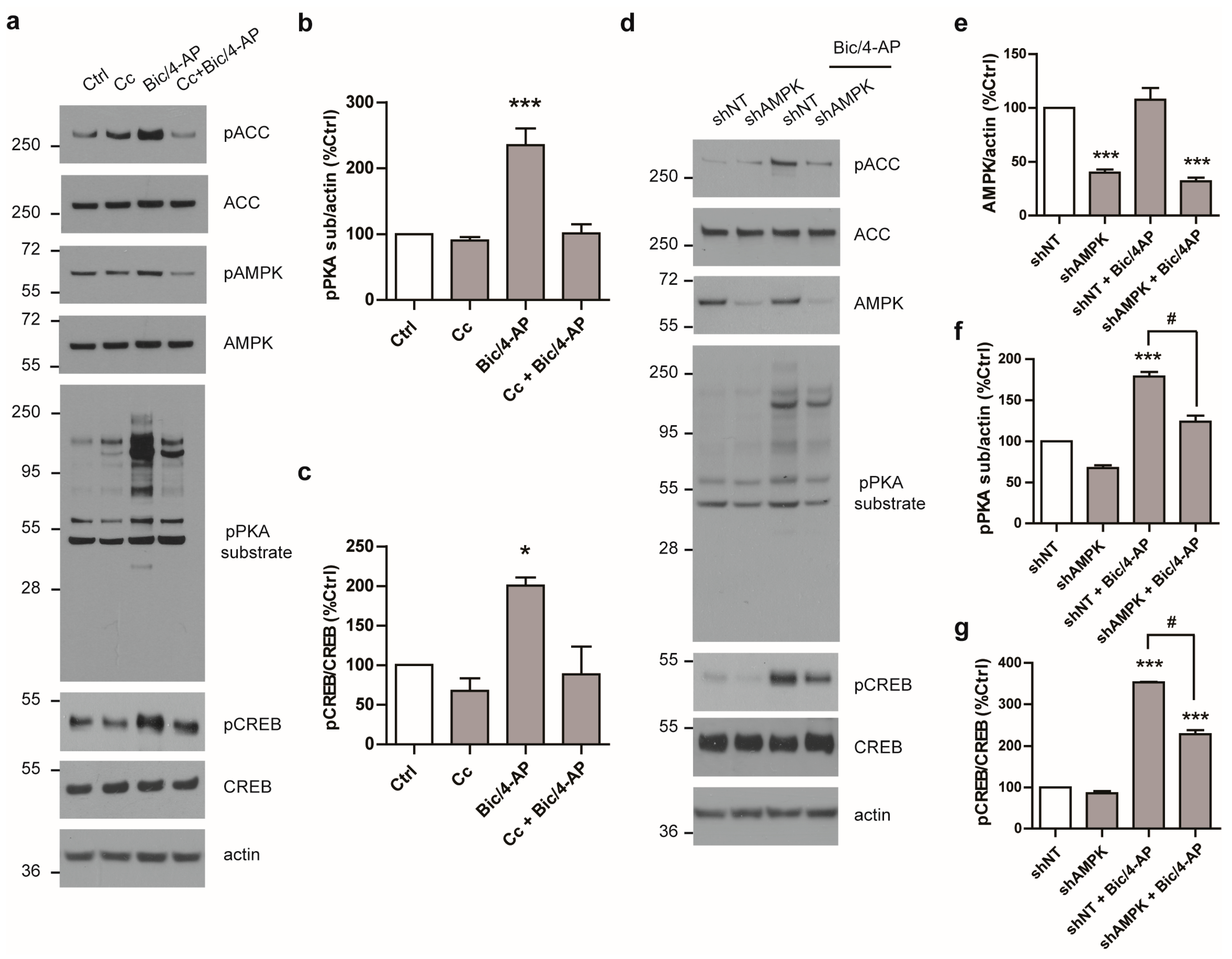
2.4. AMPK Regulates PKA Activation Following SA
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents/Antibodies
4.2. Primary Neuronal Cell Culture and Treatments
4.3. Immunoblotting
4.4. Quantitative Real-Time RT-PCR for the Expression of IEGs
4.5. Statistical Analyses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | Adenylyl cyclase |
| ACC | Acetyl-CoA carboxylase |
| AMPK | AMP-activated protein kinase |
| Arc | Activity-regulated cytoskeleton-associated protein |
| Bic/4-AP | Bicuculline/4-aminopyridine |
| cAMP | 3′,5′-cyclic AMP |
| Cc | Compound C |
| CREB | cAMP response element binding |
| DIV | Days in vitro |
| GPCRs | G protein-coupled receptors |
| IEGs | Immediate Early Genes |
| KO | Knock-out |
| PKA | cAMP-dependent protein kinase |
| SA | Synaptic activation |
| sAC | Soluble adenylyl cyclase |
| shRNA | Short hairpin RNA |
References
- Guzowski, J.F.; Timlin, J.A.; Roysam, B.; McNaughton, B.L.; Worley, P.F.; Barnes, C.A. Mapping behaviorally relevant neural circuits with immediate-early gene expression. Curr. Opin. Neurobiol. 2005, 15, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, J.D.; Rumbaugh, G.; Wu, J.; Chowdhury, S.; Plath, N.; Kuhl, D.; Huganir, R.L.; Worley, P.F. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron 2006, 52, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Peebles, C.L.; Yoo, J.; Thwin, M.T.; Palop, J.J.; Noebels, J.L.; Finkbeiner, S. Arc regulates spine morphology and maintains network stability in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 18173–18178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukazawa, Y.; Saitoh, Y.; Ozawa, F.; Ohta, Y.; Mizuno, K.; Inokuchi, K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron 2003, 38, 447–460. [Google Scholar] [CrossRef]
- Plath, N.; Ohana, O.; Dammermann, B.; Errington, M.L.; Schmitz, D.; Gross, C.; Mao, X.; Engelsberg, A.; Mahlke, C.; Welzl, H.; et al. Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron 2006, 52, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.W.; Errington, M.L.; French, P.J.; Fine, A.; Bliss, T.V.; Garel, S.; Charnay, P.; Bozon, B.; Laroche, S.; Davis, S. A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat. Neurosci. 2001, 4, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Paylor, R.; Johnson, R.S.; Papaioannou, V.; Spiegelman, B.M.; Wehner, J.M. Behavioral assessment of c-fos mutant mice. Brain Res. 1994, 651, 275–282. [Google Scholar] [CrossRef]
- Alberini, C.M. Transcription factors in long-term memory and synaptic plasticity. Physiol. Rev. 2009, 89, 121–145. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, G.A.; Montminy, M.R. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 1989, 59, 675–680. [Google Scholar] [CrossRef]
- Naqvi, S.; Martin, K.J.; Arthur, J.S. CREB phosphorylation at Ser133 regulates transcription via distinct mechanisms downstream of cAMP and MAPK signalling. Biochem. J. 2014, 458, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell. Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Attwell, D.; Laughlin, S.B. An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow MeTable 2001, 21, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic energy use and supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [PubMed]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell. MeTable 2005, 1, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Salt, I.P.; Hawley, S.A.; Davies, S.P. AMP-activated protein kinase: An ultrasensitive system for monitoring cellular energy charge. Biochem. J. 1999, 338, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.W.; Hawley, S.A.; Green, K.A.; Anis, M.; Stewart, G.; Scullion, G.A.; Norman, D.G.; Hardie, D.G. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Invest. 2004, 113, 274–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stapleton, D.; Mitchelhill, K.I.; Gao, G.; Widmer, J.; Michell, B.J.; Teh, T.; House, C.M.; Fernandez, C.S.; Cox, T.; Witters, L.A.; Kemp, B.E. Mammalian AMP-activated protein kinase subfamily. J. Biol. Chem. 1996, 271, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Davison, M.; Woods, A.; Davies, S.P.; Beri, R.K.; Carling, D.; Hardie, D.G. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J. Biol. Chem. 1996, 271, 27879–27887. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.C.; Woods, A.; Jones, N.A.; Davison, M.D.; Carling, D. The regulation of AMP-activated protein kinase by phosphorylation. Biochem. J. 2000, 345, 437–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marinangeli, C.; Didier, S.; Ahmed, T.; Caillerez, R.; Domise, M.; Laloux, C.; Bégard, S.; Carrier, S.; Colin, M.; Marchetti, P.; et al. AMP-Activated Protein Kinase Is Essential for the Maintenance of Energy Levels during Synaptic Activation. iScience 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Hoey, S.E.; Williams, R.J.; Perkinton, M.S. Synaptic NMDA receptor activation stimulates alpha-secretase amyloid precursor protein processing and inhibits amyloid-beta production. J. Neurosci. 2009, 29, 4442–4460. [Google Scholar] [CrossRef] [PubMed]
- Steward, O.; Farris, S.; Pirbhoy, P.S.; Darnell, J.; Driesche, S.J. Localization and local translation of Arc/Arg3.1 mRNA at synapses: Some observations and paradoxes. Front. Mol. Neurosci. 2014, 7. [Google Scholar] [CrossRef] [PubMed]
- Bitterman, J.L.; Ramos-Espiritu, L.; Diaz, A.; Levin, L.R.; Buck, J. Pharmacological distinction between soluble and transmembrane adenylyl cyclases. J. Pharmacol. Exp. Ther. 2013, 347, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Tresguerres, M.; Buck, J.; Levin, L.R. Physiological carbon dioxide, bicarbonate, and pH sensing. Pflugers. Arch. 2010, 460, 953–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acin-Perez, R.; Salazar, E.; Brosel, S.; Yang, H.; Schon, E.A.; Manfredi, G. Modulation of mitochondrial protein phosphorylation by soluble adenylyl cyclase ameliorates cytochrome oxidase defects. EMBO Mol. Med. 2009, 1, 392–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acin-Perez, R.; Salazar, E.; Kamenetsky, M.; Buck, J.; Levin, L.R.; Manfredi, G. Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell. MeTable 2009, 9, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Hebert-Chatelain, E.; Desprez, T.; Serrat, R.; Bellocchio, L.; Soria-Gomez, E.; Busquets-Garcia, A.; Pagano Zottola, A.C.; Delamarre, A.; Cannich, A.; Vincent, P.; et al. A cannabinoid link between mitochondria and memory. Nature 2016, 539, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Hurley, R.L.; Barré, L.K.; Wood, S.D.; Anderson, K.A.; Kemp, B.E.; Means, A.R.; Witters, L.A. Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J. Biol. Chem. 2006, 281, 36662–36672. [Google Scholar] [CrossRef] [PubMed]
- Djouder, N.; Tuerk, R.D.; Suter, M.; Salvioni, P.; Thali, R.F.; Scholz, R.; Vaahtomeri, K.; Auchli, Y.; Rechsteiner, H.; Brunisholz, R.A.; et al. PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis. EMBO J. 2010, 29, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Bischof, G.N.; Park, D.C. Obesity and Aging: Consequences for Cognition, Brain Structure, and Brain Function. Psychosom. Med. 2015, 77, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Ngandu, T.; Fratiglioni, L.; Viitanen, M.; Kåreholt, I.; Winblad, B.; Helkala, E.L.; Tuomilehto, J.; Soininen, H.; Nissinen, A. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch. Neurol. 2005, 62, 1556–1560. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.J.; Jiang, L.; Hamza, M.; Sanchez Rangel, E.; Dai, F.; Belfort-DeAguiar, R.; Parikh, L.; Koo, B.B.; Rothman, D.L.; Mason, G.; Sherwin, R.S. Blunted rise in brain glucose levels during hyperglycemia in adults with obesity and T2DM. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vingtdeux, V.; Davies, P.; Dickson, D.W.; Marambaud, P. AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol. 2011, 121, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Domise, M.; Didier, S.; Marinangeli, C.; Zhao, H.; Chandakkar, P.; Buée, L.; Viollet, B.; Davies, P.; Marambaud, P.; Vingtdeux, V. AMP-activated protein kinase modulates tau phosphorylation and tau pathology in vivo. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Didier, S.; Sauvé, F.; Domise, M.; Buée, L.; Marinangeli, C.; Vingtdeux, V. AMP-activated Protein Kinase Controls Immediate Early Genes Expression Following Synaptic Activation Through the PKA/CREB Pathway. Int. J. Mol. Sci. 2018, 19, 3716. https://doi.org/10.3390/ijms19123716
Didier S, Sauvé F, Domise M, Buée L, Marinangeli C, Vingtdeux V. AMP-activated Protein Kinase Controls Immediate Early Genes Expression Following Synaptic Activation Through the PKA/CREB Pathway. International Journal of Molecular Sciences. 2018; 19(12):3716. https://doi.org/10.3390/ijms19123716
Chicago/Turabian StyleDidier, Sébastien, Florent Sauvé, Manon Domise, Luc Buée, Claudia Marinangeli, and Valérie Vingtdeux. 2018. "AMP-activated Protein Kinase Controls Immediate Early Genes Expression Following Synaptic Activation Through the PKA/CREB Pathway" International Journal of Molecular Sciences 19, no. 12: 3716. https://doi.org/10.3390/ijms19123716