A Rapid Method for the Detection of Sarcosine Using SPIONs/Au/CS/SOX/NPs for Prostate Cancer Sensing

 ,
,
Abstract
:1. Introduction
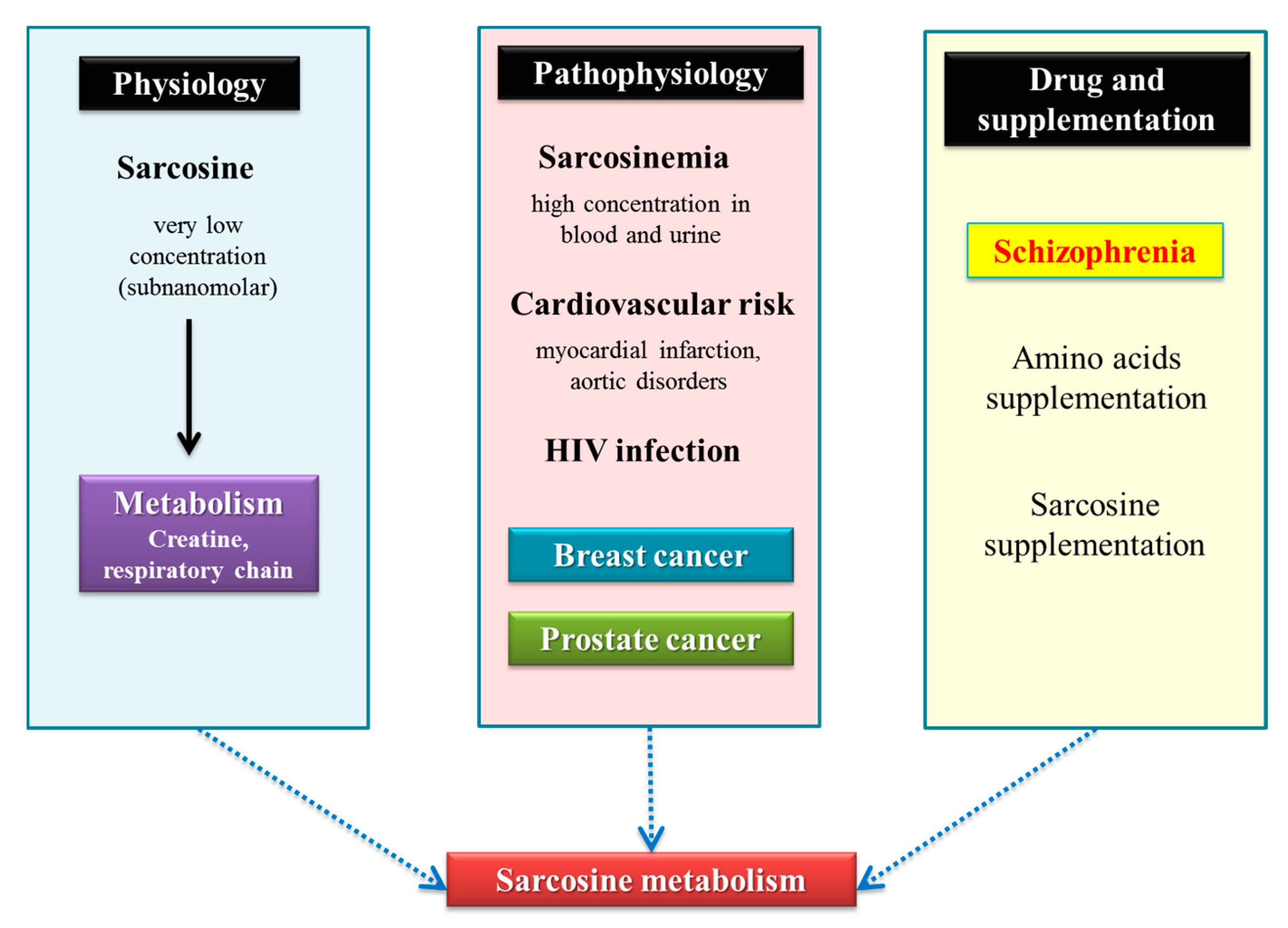
1.1. Basal Metabolism
1.2. Sarcosinemia and Sarcosinuria
1.3. Homocysteine Metabolism
1.4. Prostate Tumors
1.5. Nanoscale Materials in Cancer Diagnosis
2. Results
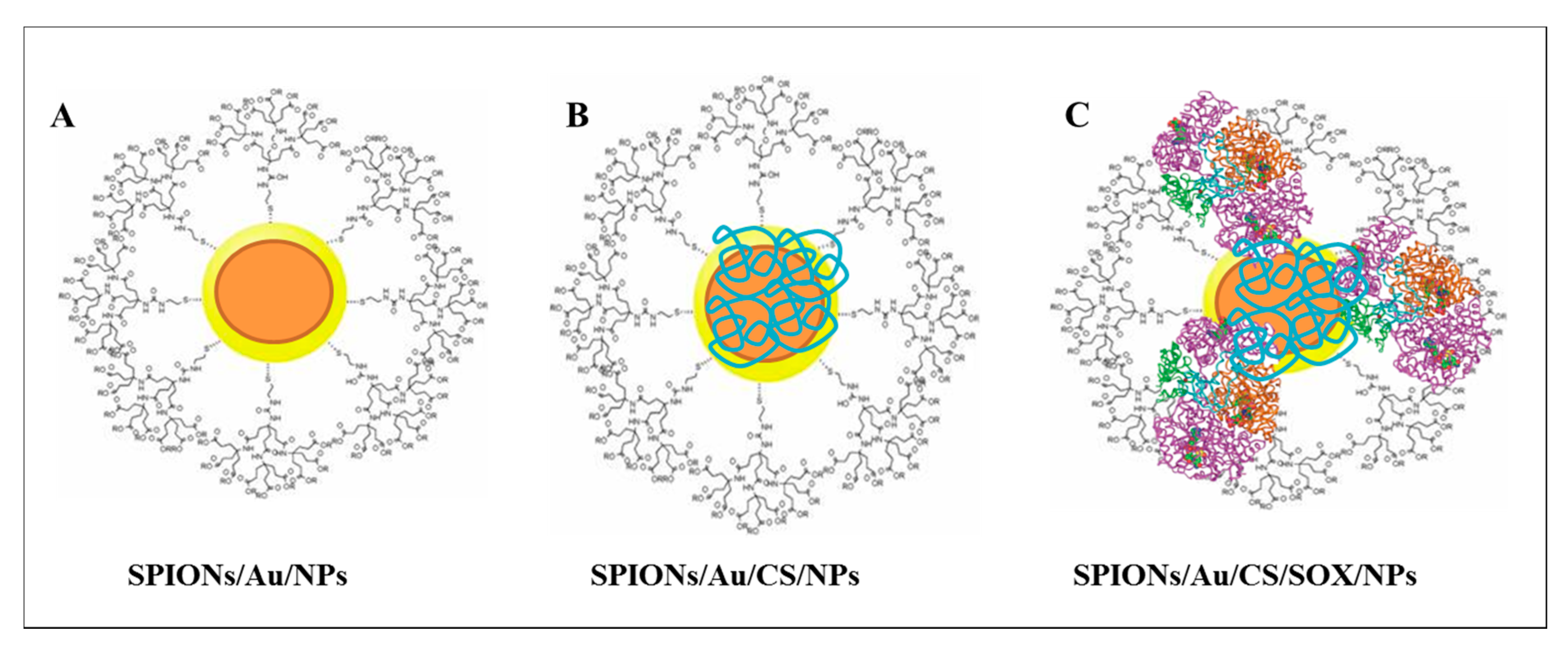
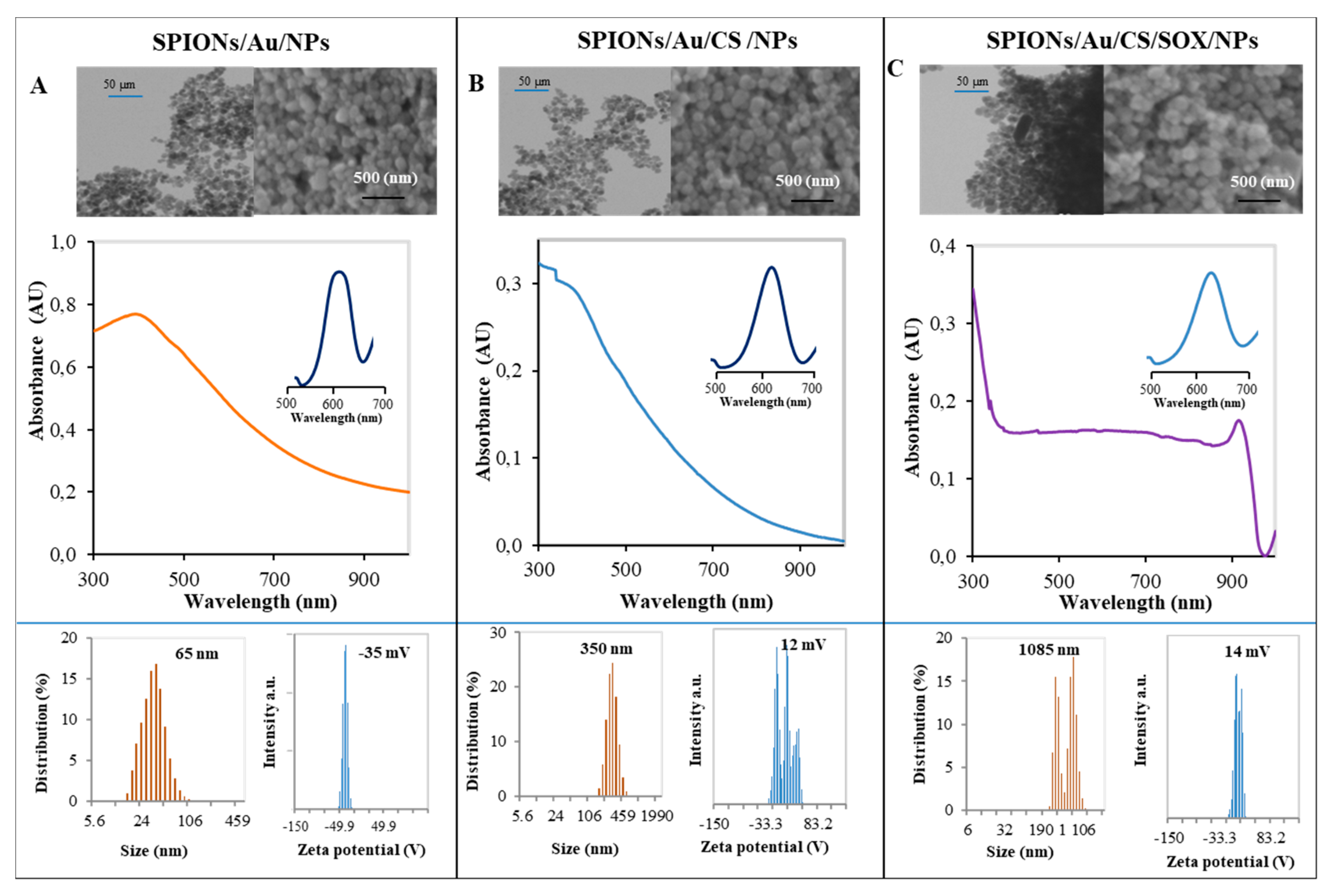
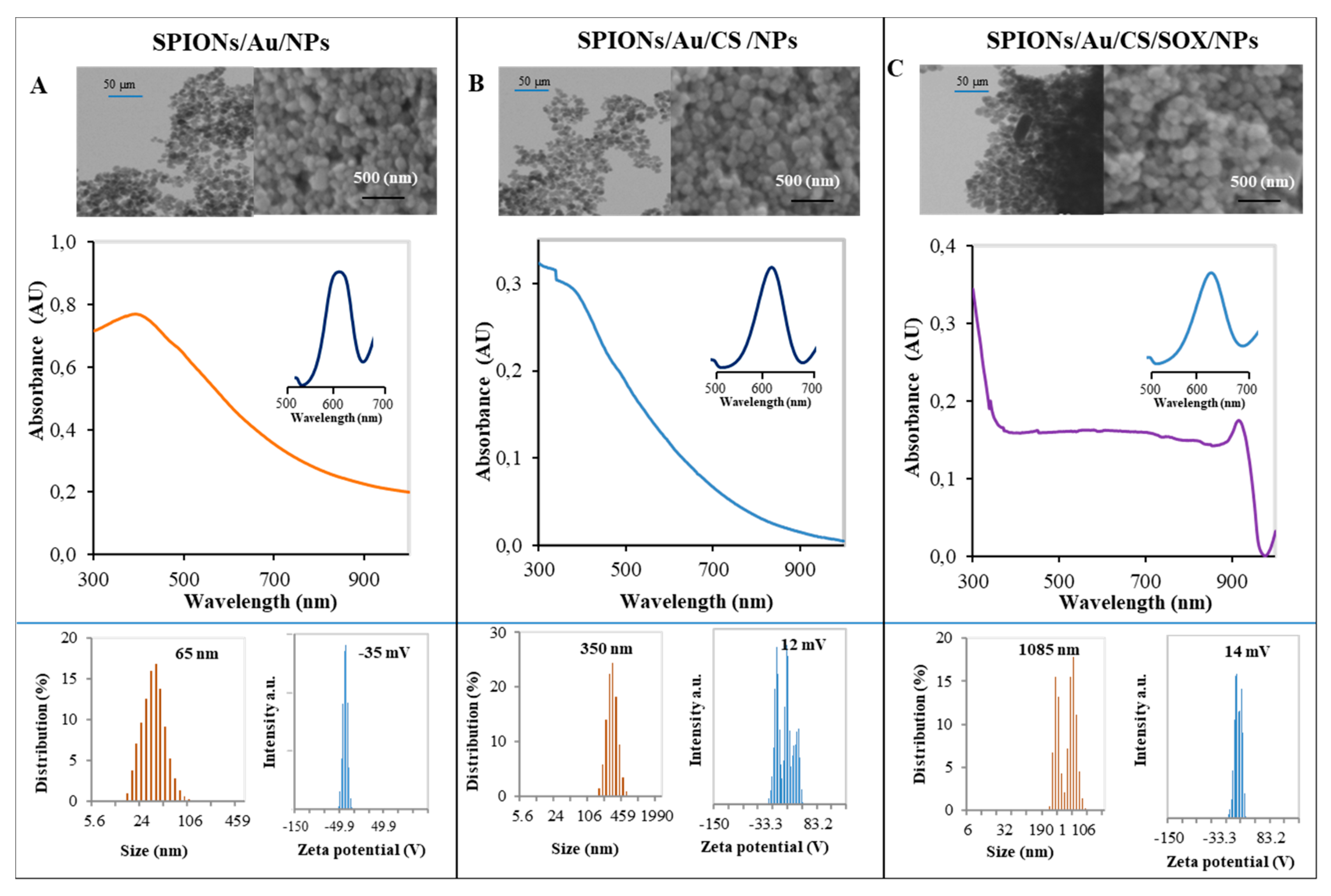
2.1. Preparation and Physicochemical Characterization of Nanoconstructs
2.2. Pseudo-Peroxidase Activity of the Gold SPIONs
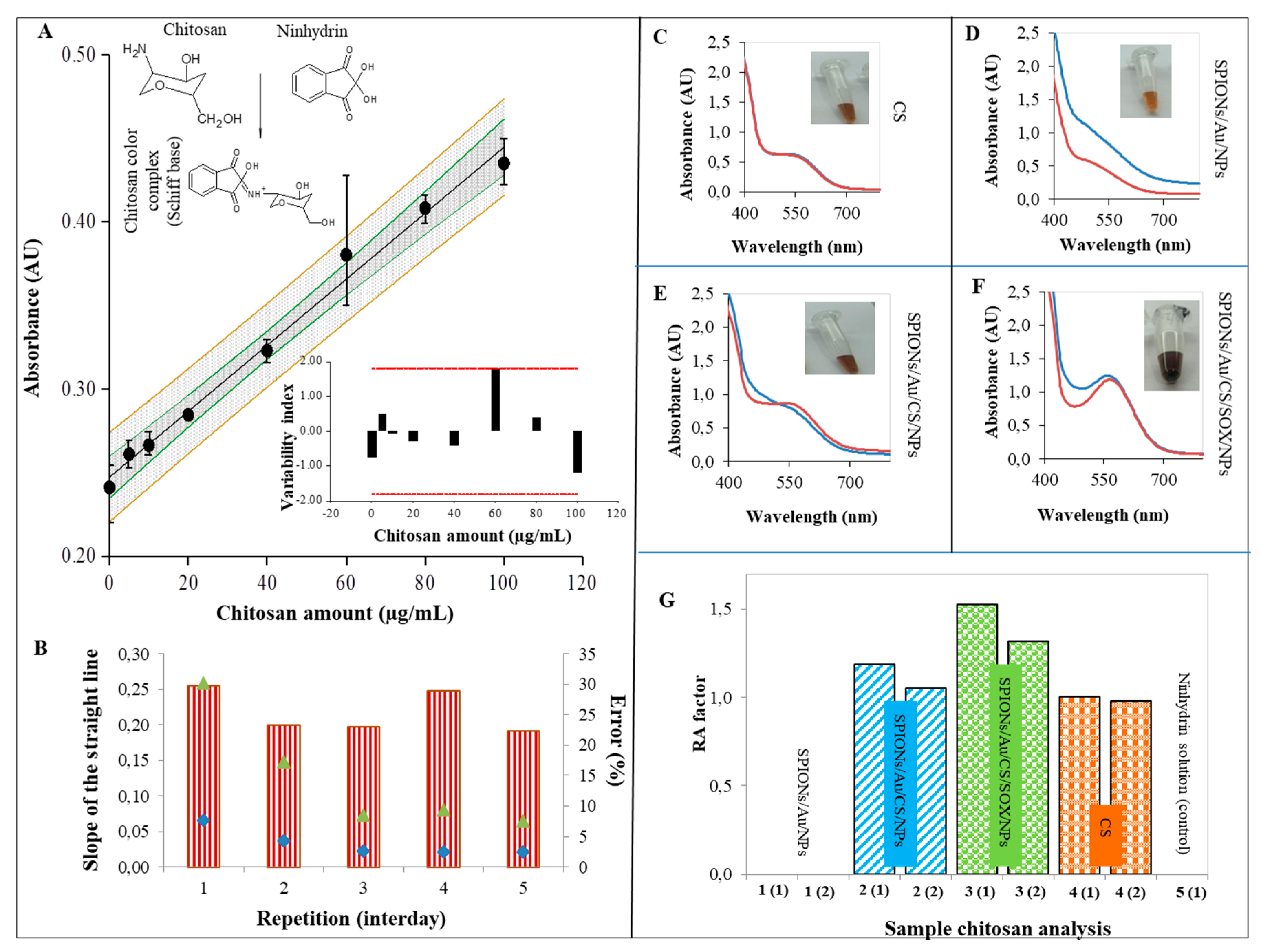
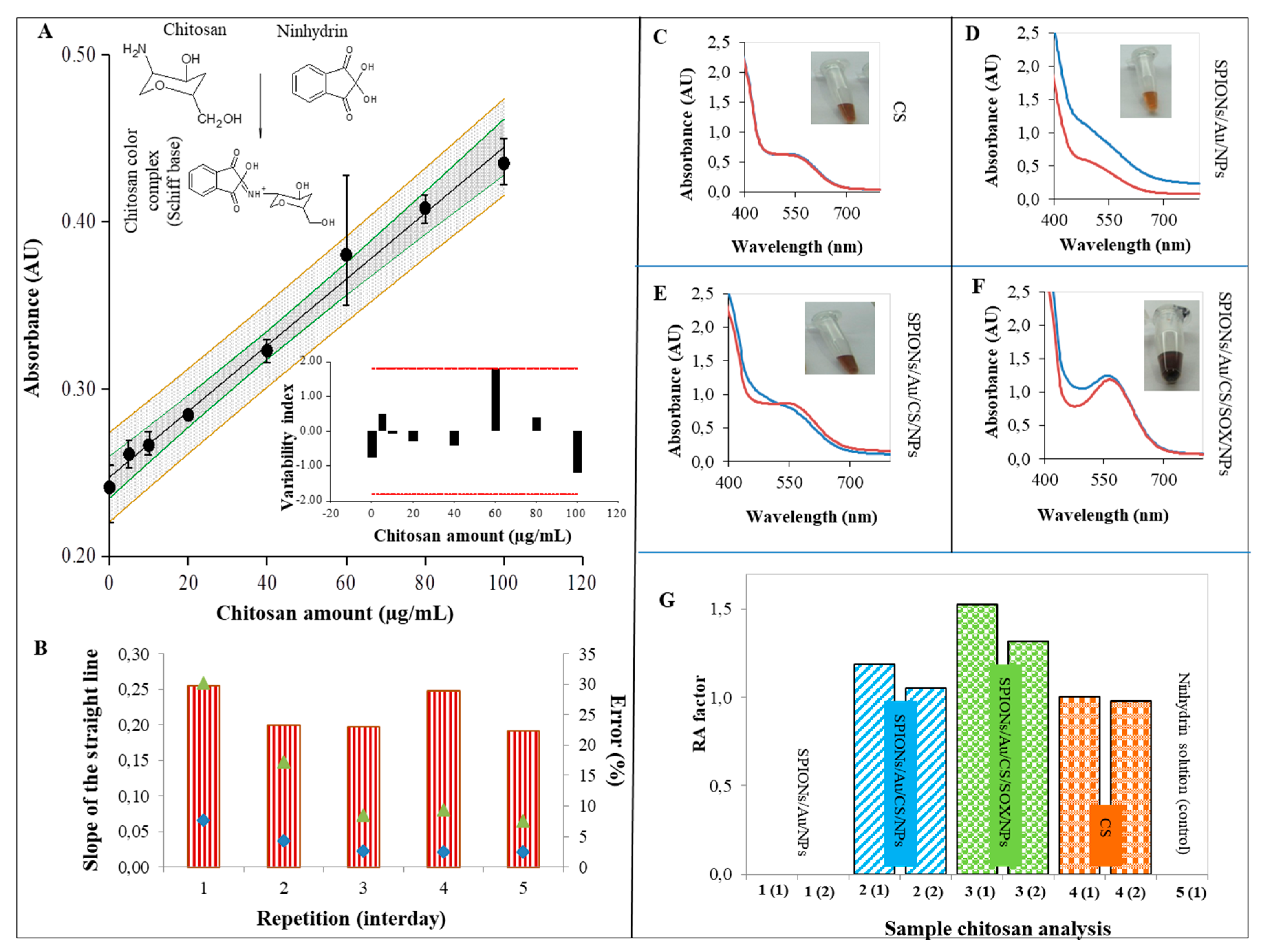
2.3. Characterization of SPIONs Using Ninhydrin Assay
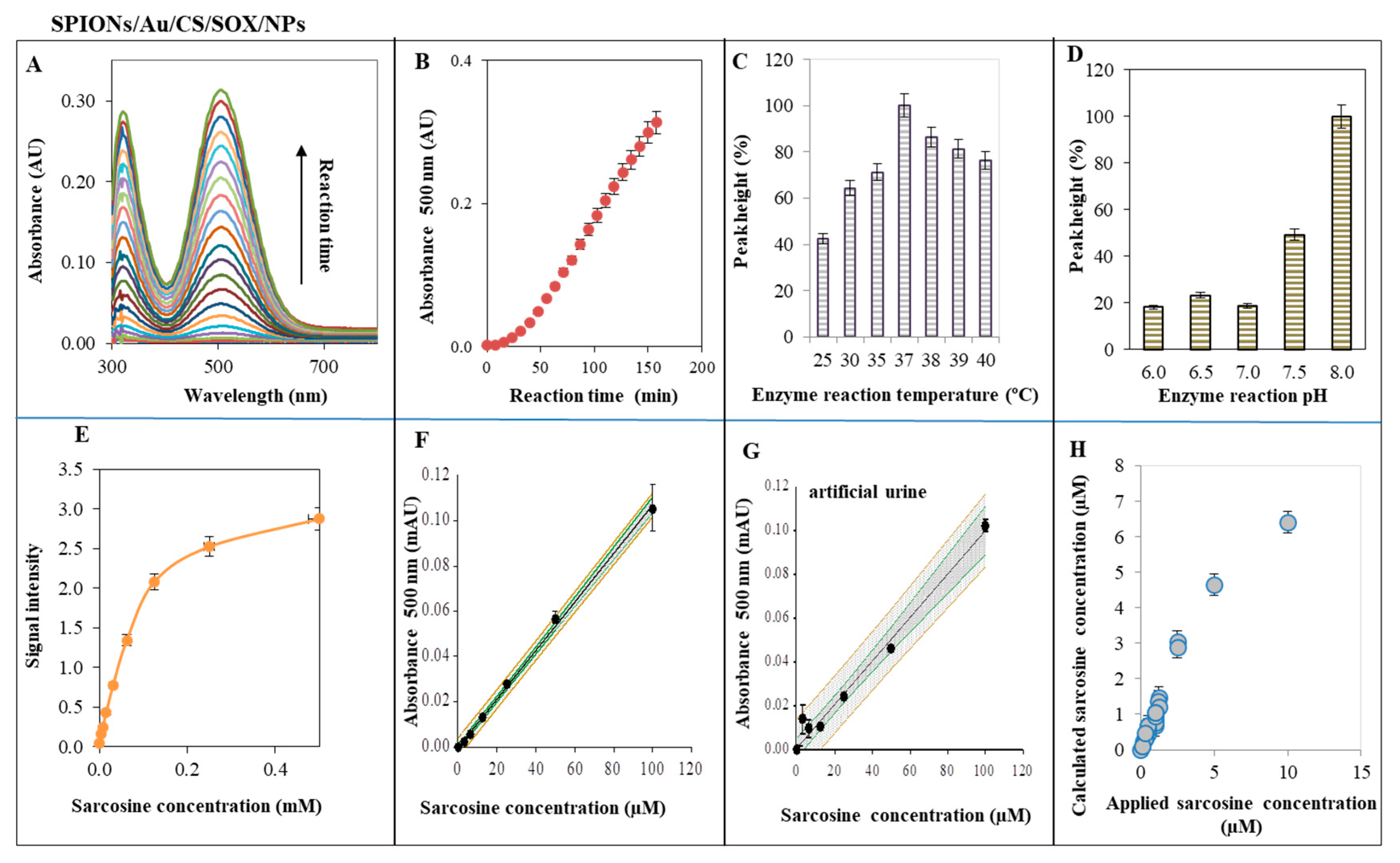
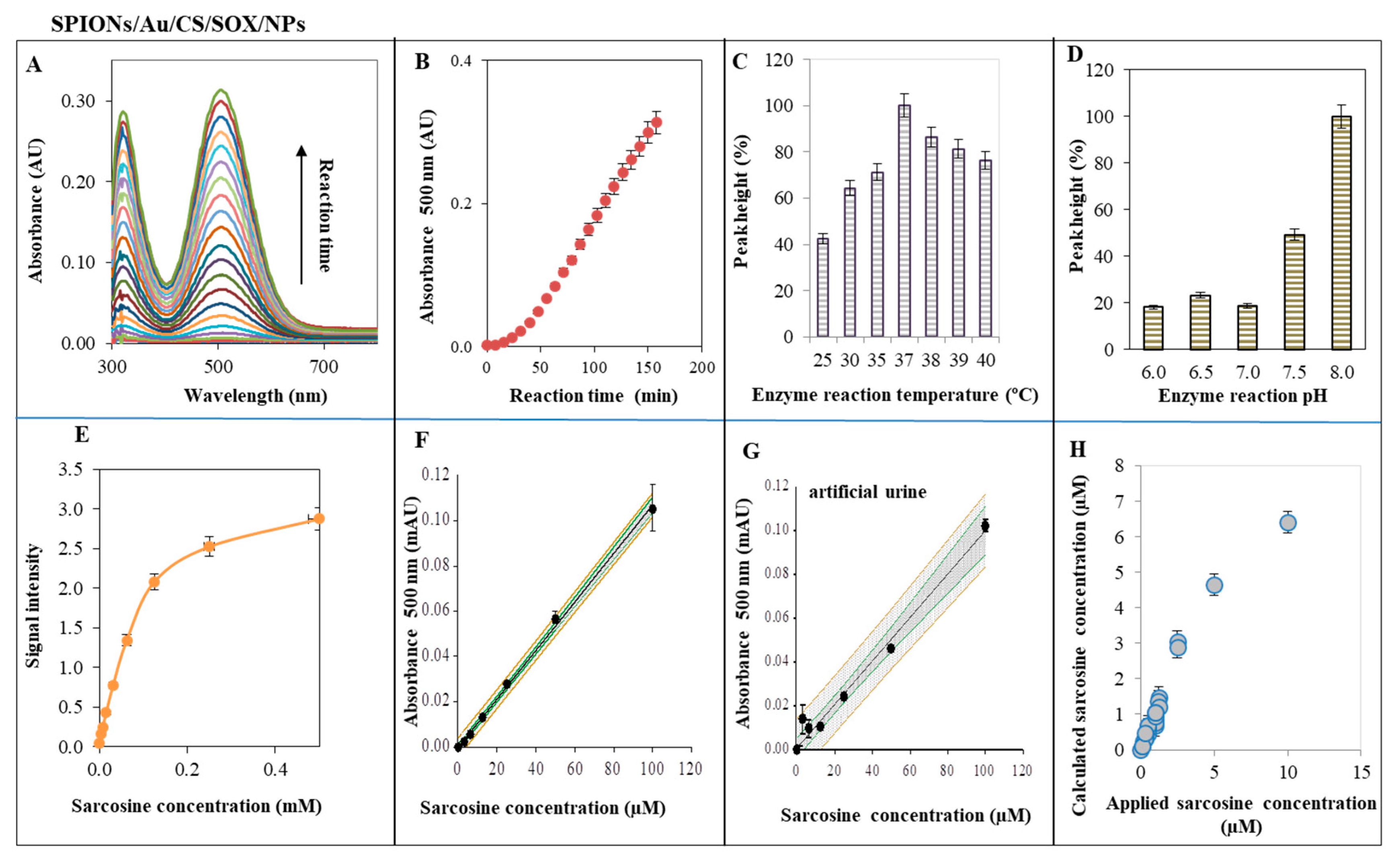
2.4. Characterization of SPIONs for Sarcosine Detection—SOX Activity of SPIONs/Au/CS/SOX/NPs
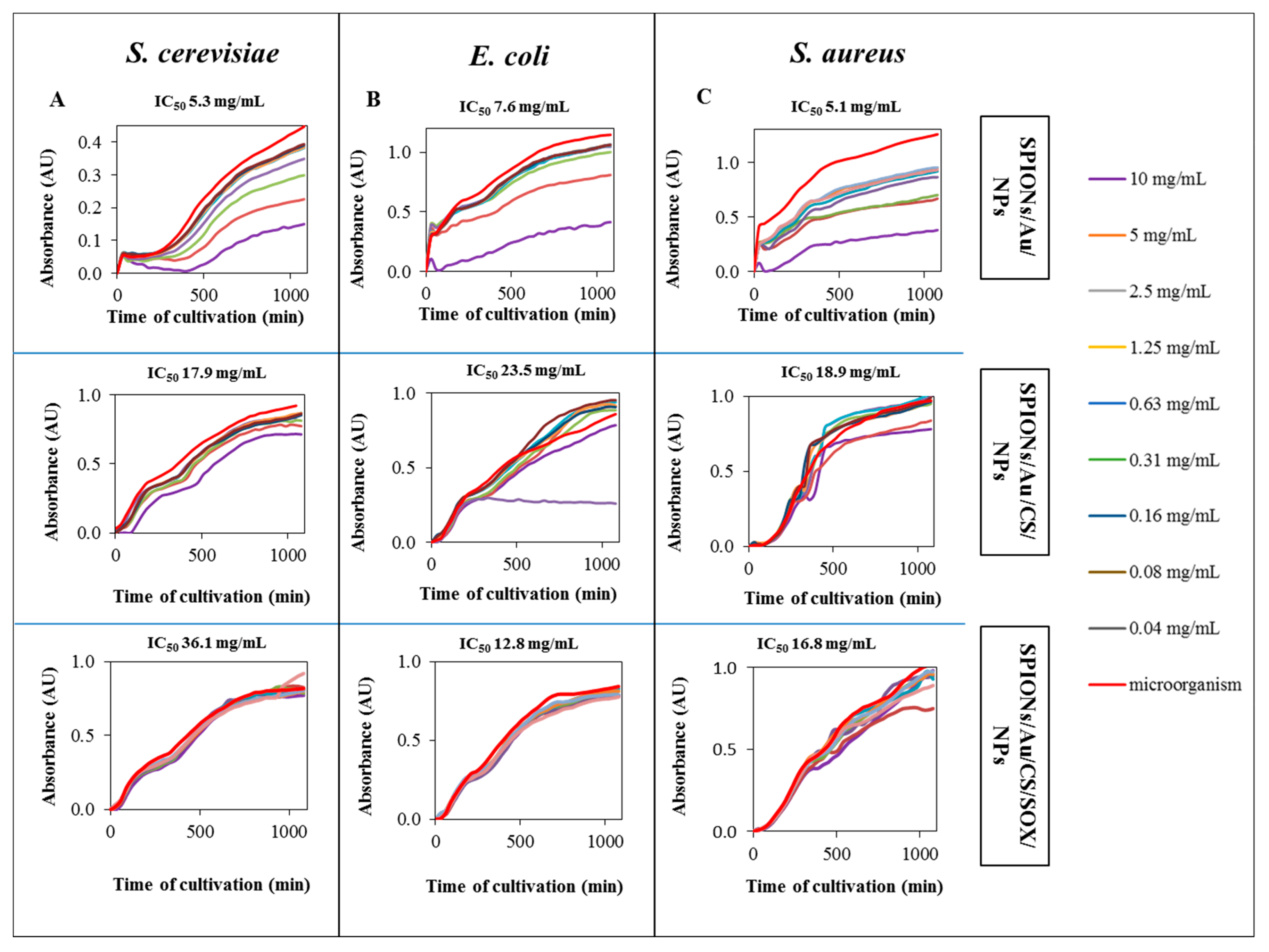
2.5. Assessing Toxicity of SPIONs
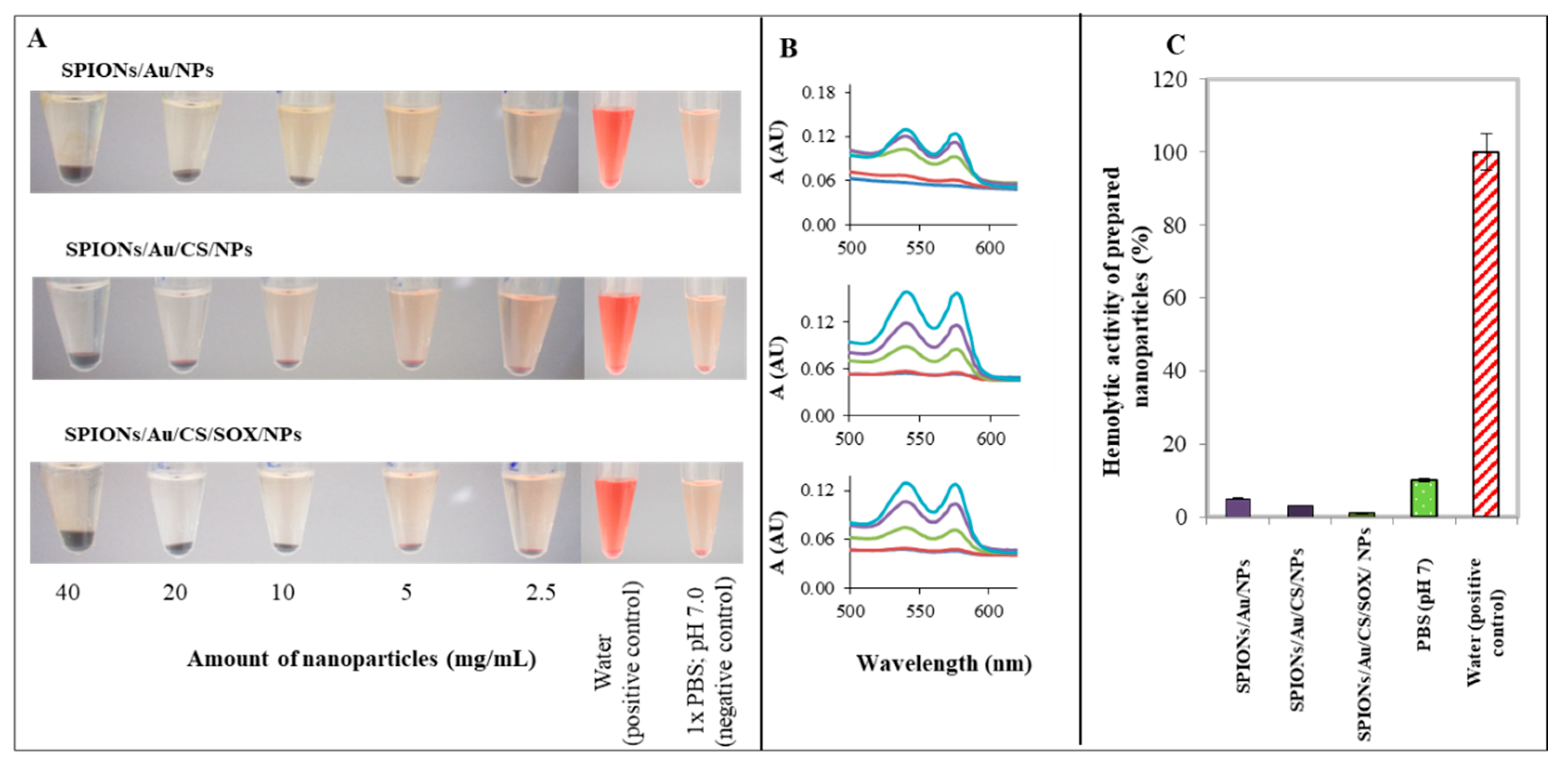
2.6. Analysis of SPIONs Hemolytic Properties In Vitro
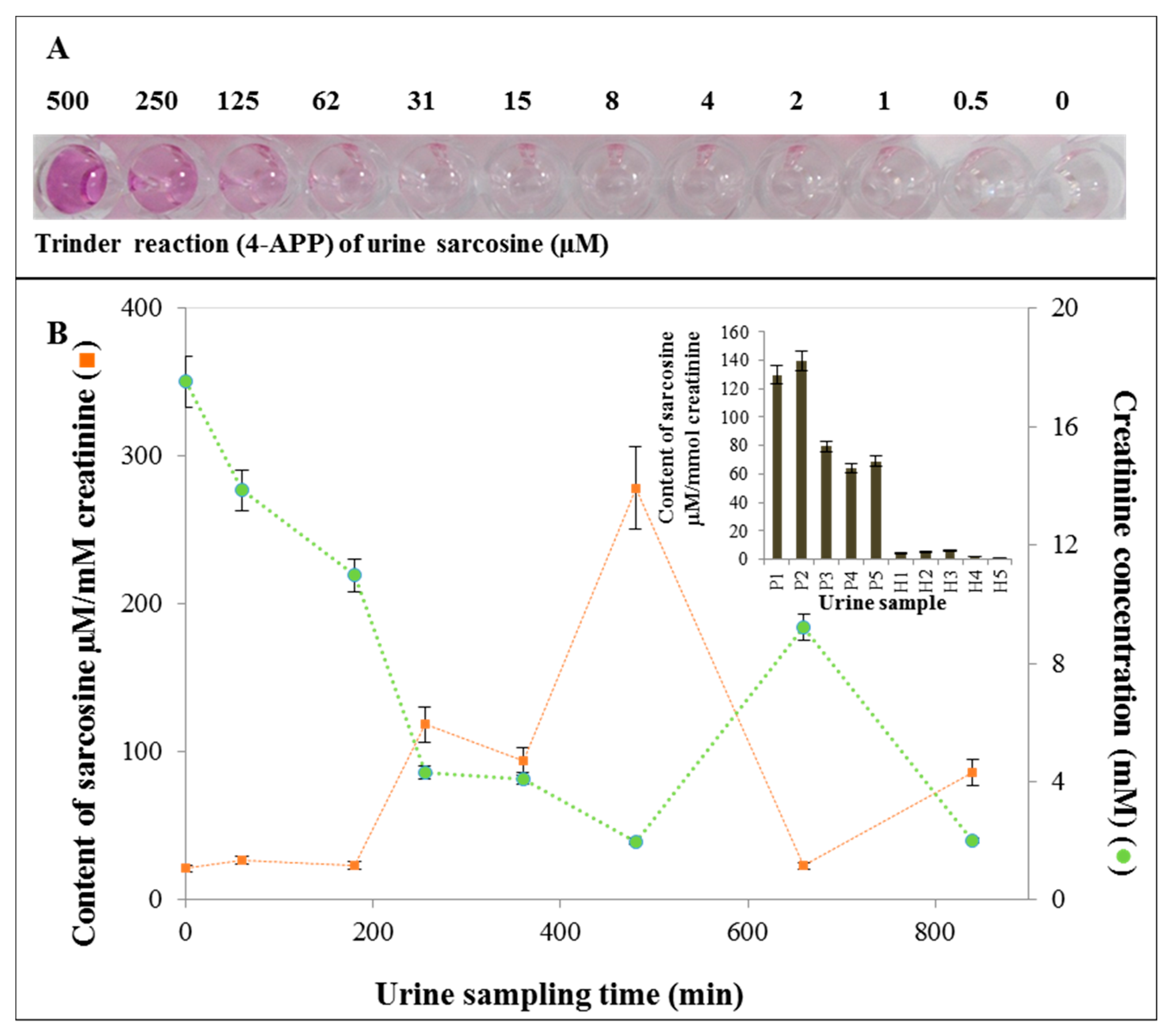
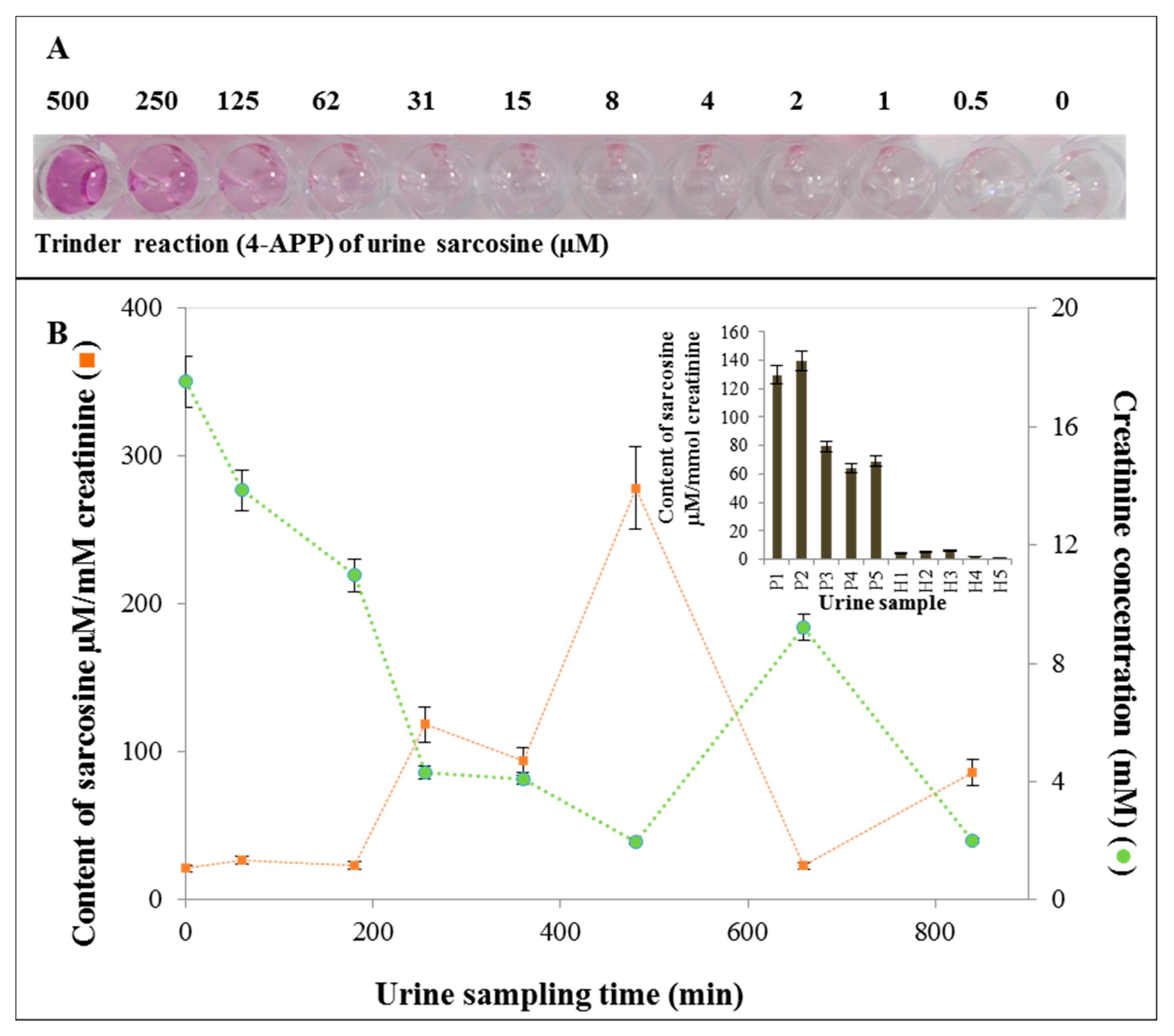
2.7. Application of SPIONs for Urine Sample Analysis
3. Discussion
3.1. Preparation and Physicochemical Characterization of Nanoconstructs
3.2. Pseudo-Peroxidase Activity of the Gold SPIONs
3.3. Characterization of SPIONs Using Ninhydrin Assay
3.4. Characterization of SPIONs for Sarcosine Detection—SOX Activity of SPIONs/Au/CS/SOX/NPs
3.5. Assessing Toxicity of SPIONs
3.6. Analysis of SPIONs Hemolytic Properties In Vitro
3.7. Application of SPIONs for Sarcosine Analysis in Urine Samples
4. Materials and Methods
4.1. Chemicals
4.2. Synthesis of SPIONs/Au/NPs
4.3. Modification of SPIONs/Au/NPs with Chitosan (CS)
4.4. Modification of SPIONs/Au/CS/NPs with Sarcosine Oxidase (SOX)
4.5. Scanning Electron Microscopy
4.6. Absorbance Measurements
4.7. Zetasizer Analysis of Nanoparticles
4.8. Ninhydrin Assay for Chitosan (CS) Detection
4.9. SOX Activity of SPIONs/Au/CS/SOX/NPs
4.10. Pseudo-Peroxidase Assay
4.11. Creatinine Analysis
4.12. Growth Inhibition Assay
4.13. Hemolytic Assay
4.14. Biological Samples of Urine
4.15. Data Treatment and Descriptive Statistics
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| r | correlation coefficient |
| LOD | limit of detection determined by the ISO method |
| LOQ | limit of detection determined by the second ISO method |
| RSD | relative standard deviation calculated from the individual measurements and fit into the straight line equation and compared to the input value |
| SOX | sarcosine oxidase (EC 1.5.3.1) |
| SPION | superparamagnetic iron oxide nanoparticles |
| TPP | sodium tripolyphosphate |
| TMB | 3,3′,5,5′-tetramethylbenzidine |
| CS | chitosan |
| 4-AAP | 4-aminoantipyrine |
| IC50 | the half maximal inhibitory concentration |
| RBC | red blood cell |
| AuNPs | gold nanoparticles |
References
- Bar-Joseph, I.; Pras, E.; Reznik-Wolf, H.; Marek-Yagel, D.; Abu-Horvitz, A.; Dushnitzky, M.; Goldstein, N.; Rienstein, S.; Dekel, M.; Pode-Shakked, B. Mutations in the sarcosine dehydrogenase gene in patients with sarcosinemia. Hum. Genet. 2012, 131, 1805–1810. [Google Scholar] [CrossRef] [PubMed]
- Scriver, C.R. The metabolic & molecular bases of inherited disease; McGraw-Hill: New York, NY, USA, 2001; Volume 4. [Google Scholar]
- Sreekumar, A.; Poisson, L.M.; Rajendiran, T.M.; Khan, A.P.; Cao, Q.; Yu, J.; Laxman, B.; Mehra, R.; Lonigro, R.J.; Li, Y. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 2009, 457, 910–914. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.H.; Stabler, S.P.; Lindenbaum, J. Serum betaine, N, N-dimethylglycine and N-methylglycine levels in patients with cobalamin and folate deficiency and related inborn errors of metabolism. Metabolism 1993, 42, 1448–1460. [Google Scholar] [CrossRef]
- Steenkamp, D.J.; Husain, M. The effect of tetrahydrofolate on the reduction of electron transfer flavoprotein by sarcosine and dimethylglycine dehydrogenases. Biochem. J. 1982, 203, 707. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, S.; Yang, W.; Yu, H.; Zhang, L.; Ma, P.; Wu, P.; Li, X.; Cho, K.; Xue, S. Plasma amino acid profile in patients with aortic dissection. Sci. Rep. 2017, 7, 40146. [Google Scholar] [CrossRef] [PubMed]
- Munshi, S.U.; Rewari, B.B.; Bhavesh, N.S.; Jameel, S. Nuclear magnetic resonance based profiling of biofluids reveals metabolic dysregulation in HIV-infected persons and those on anti-retroviral therapy. PLoS ONE 2013, 8, e64298. [Google Scholar] [CrossRef] [PubMed]
- Lane, H.-Y.; Lin, C.-H.; Huang, Y.-J.; Liao, C.-H.; Chang, Y.-C.; Tsai, G.E. A randomized, double-blind, placebo-controlled comparison study of sarcosine (N-methylglycine) and D-serine add-on treatment for schizophrenia. Int. J. Neuropsychopharmacol. 2010, 13, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Tsai, G.; Lane, H.-Y.; Yang, P.; Chong, M.-Y.; Lange, N. Glycine transporter I inhibitor, N-methylglycine (sarcosine), added to antipsychotics for the treatment of schizophrenia. Biol. Psychiatry 2004, 55, 452–456. [Google Scholar] [CrossRef] [PubMed]
- McKeever, M.; Weir, D.; Molloy, A.; Scott, J. Betaine-homocysteine methyltransferase: Organ distribution in man, pig and rat and subcellular distribution in the rat. Clin. Sci. 1991, 81, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Schwahn, B.C.; Hafner, D.; Hohlfeld, T.; Balkenhol, N.; Laryea, M.D.; Wendel, U. Pharmacokinetics of oral betaine in healthy subjects and patients with homocystinuria. Br. J. Clin. Pharmcol. 2003, 55, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Benevenga, N.J. Consideration of betaine and one-carbon sources of N5-methyltetrahydrofolate for use in homocystinuria and neural tube defects. Am. J. Clin. Nutr. 2007, 85, 946–949. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.H.; Kruger, W.D.; Wang, L.; Pasquali, M.; Elsas II, L.J. Cystathionine β-synthase deficiency: Effects of betaine supplementation after methionine restriction in B6-nonresponsive homocystinuria. Genet. Med. 2004, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.R.; Moroz, K.; Zhang, H.; Mulholland, D.; Abdel-Mageed, A.B.; Mondal, D. PRL-3 increases the aggressive phenotype of prostate cancer cells in vitro and its expression correlates with high-grade prostate tumors in patients. Int. J. Oncol. 2018, 52, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Pihikova, D.; Pakanova, Z.; Nemcovic, M.; Barath, P.; Belicky, S.; Bertok, T.; Kasak, P.; Mucha, J.; Tkac, J. Sweet characterisation of prostate specific antigen using electrochemical lectin-based immunosensor assay and MALDI TOF/TOF analysis: Focus on sialic acid. Proteomics 2016, 16, 3085–3095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.H.; Barry, M.J. Screening for prostate cancer with the prostate-specific antigen test: A review of current evidence. JAMA 2014, 311, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- De Kok, J.B.; Verhaegh, G.W.; Roelofs, R.W.; Hessels, D.; Kiemeney, L.A.; Aalders, T.W.; Swinkels, D.W.; Schalken, J.A. DD3PCA3, a very sensitive and specific marker to detect prostate tumors. Cancer Res. 2002, 62, 2695–2698. [Google Scholar] [PubMed]
- Font-Tello, A.; Juanpere, N.; de Muga, S.; Lorenzo, M.; Lorente, J.A.; Fumado, L.; Serrano, L.; Serrano, S.; Lloreta, J.; Hernández, S. Association of ERG and TMPRSS2-ERG with grade, stage, and prognosis of prostate cancer is dependent on their expression levels. Prostate 2015, 75, 1216–1226. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, K.; Tobisawa, Y.; Kaya, T.; Kaneko, T.; Hatakeyama, S.; Mori, K.; Hashimoto, Y.; Koie, T.; Suda, Y.; Ohyama, C. Wisteria Floribunda Agglutinin and its Reactive-Glycan-Carrying Prostate-Specific Antigen as a novel diagnostic and prognostic marker of prostate cancer. Int. J. Mol. Sci. 2017, 18, 261. [Google Scholar] [CrossRef] [PubMed]
- Hessels, D.; Gunnewiek, J.M.K.; van Oort, I.; Karthaus, H.F.; van Leenders, G.J.; van Balken, B.; Kiemeney, L.A.; Witjes, J.A.; Schalken, J.A. DD3PCA3-based molecular urine analysis for the diagnosis of prostate cancer. Eur. Urol. 2003, 44, 8–16. [Google Scholar] [CrossRef]
- Kulkarni, P.; Uversky, V.N. Cancer/Testis Antigens:“Smart” biomarkers for diagnosis and prognosis of prostate and other cancers. Int. J. Mol. Sci. 2017, 18, 740. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H.; Groskopf, J.; Fritsche, H.A.; Bhadkamkar, V.; Blase, A.; Kumar, S.V.; Davis, J.W.; Troncoso, P.; Rittenhouse, H.; Babaian, R.J. PCA3 molecular urine assay correlates with prostate cancer tumor volume: Implication in selecting candidates for active surveillance. J. Urol. 2008, 179, 1804–1810. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Laxman, B.; Varambally, S.; Cao, X.; Yu, J.; Helgeson, B.E.; Cao, Q.; Prensner, J.R.; Rubin, M.A.; Shah, R.B. Role of the TMPRSS2-ERG gene fusion in prostate cancer. Neoplasia 2008, 10, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Tseng, T.-L.; Shih, Y.-P.; Huang, Y.-C.; Wang, C.-K.; Chen, P.-H.; Chang, J.-G.; Yeh, K.-T.; Chen, Y.-M.A.; Buetow, K.H. Genotypic and phenotypic characterization of a putative tumor susceptibility gene, GNMT, in liver cancer. Cancer Res. 2003, 63, 647–654. [Google Scholar] [PubMed]
- Huang, Y.-C.; Lee, C.-M.; Chen, M.; Chung, M.-Y.; Chang, Y.-H.; Huang, W.J.-S.; Ho, D.M.-T.; Pan, C.-C.; Wu, T.T.; Yang, S. Haplotypes, loss of heterozygosity, and expression levels of glycine N-methyltransferase in prostate cancer. Clin. Cancer Res. 2007, 13, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Hatziapostolou, M.; Iliopoulos, D. Epigenetic aberrations during oncogenesis. Cell. Mol. Life Sci. 2011, 68, 1681–1702. [Google Scholar] [CrossRef] [PubMed]
- Kelavkar, U.; Harya, N.; Hutzley, J.; Bacich, D.; Monzon, F.; Chandran, U.; Dhir, R.; O’Keefe, D. DNA methylation paradigm shift: 15-lipoxygenase-1 upregulation in prostatic intraepithelial neoplasia and prostate cancer by atypical promoter hypermethylation. Prostagl. Lipid Med. 2007, 82, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Pierconti, F.; Martini, M.; Pinto, F.; Cenci, T.; Capodimonti, S.; Calarco, A.; Bassi, P.F.; Larocca, L.M. Epigenetic silencing of SOCS3 identifies a subset of prostate cancer with an aggressive behavior. Prostate 2011, 71, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Ianni, M.; Porcellini, E.; Carbone, I.; Potenzoni, M.; Pieri, A.; Pastizzaro, C.; Benecchi, L.; Licastro, F. Genetic factors regulating inflammation and DNA methylation associated with prostate cancer. Prost. Cancer Prost. Dis. 2013, 16, 56. [Google Scholar] [CrossRef] [PubMed]
- Luka, Z.; Mudd, S.H.; Wagner, C. Glycine N-methyltransferase and regulation of S-adenosylmethionine levels. J. Biol. Chem. 2009, 284, 22507–22511. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.P.; Rajendiran, T.M.; Bushra, A.; Asangani, I.A.; Athanikar, J.N.; Yocum, A.K.; Mehra, R.; Siddiqui, J.; Palapattu, G.; Wei, J.T. The role of sarcosine metabolism in prostate cancer progression. Neoplasia 2013, 15, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Baronia, R.; Singh, M.; Gupta, R.B.; Karuppiah, S.; Kumar, R.; Belz, J.; Shanker, R.; Sridhar, S.; Singh, S.P. Synthesis and characterization of multifunctional gold nanoclusters for application in radiation therapy. Int. J. Nanomed. 2018, 13, 113. [Google Scholar] [CrossRef] [PubMed]
- De Boer, B.; Kahlman, J.; Jansen, T.; Duric, H.; Veen, J. An integrated and sensitive detection platform for magneto-resistive biosensors. Biosens. Bioelectron. 2007, 22, 2366–2370. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Qi, C.; Li, Y.; Han, Q.; Yang, R.; Wang, C. PtCo bimetallic nanoparticles with high oxidase-like catalytic activity and their applications for magnetic-enhanced colorimetric biosensing. J. Mater. Chem. B 2016, 4, 1869–1877. [Google Scholar] [CrossRef]
- Patel, P.; Kansara, K.; Singh, R.; Shukla, R.K.; Singh, S.; Dhawan, A.; Kumar, A. Cellular internalization and antioxidant activity of cerium oxide nanoparticles in human monocytic leukemia cells. Int. J. Nanomed. 2018, 13, 39. [Google Scholar] [CrossRef] [PubMed]
- Purohit, R.; Singh, S. Fluorescent gold nanoclusters for efficient cancer cell targeting. Int. J. Nanomed. 2018, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Siow, W.X.; Chang, Y.-T.; Babič, M.; Lu, Y.-C.; Horák, D.; Ma, Y.-H. Interaction of poly-L-lysine coating and heparan sulfate proteoglycan on magnetic nanoparticle uptake by tumor cells. Int. J. Nanomed. 2018, 13, 1693. [Google Scholar] [CrossRef] [PubMed]
- Unterweger, H.; Dézsi, L.; Matuszak, J.; Janko, C.; Poettler, M.; Jordan, J.; Bäuerle, T.; Szebeni, J.; Fey, T.; Boccaccini, A.R. Dextran-coated superparamagnetic iron oxide nanoparticles for magnetic resonance imaging: Evaluation of size-dependent imaging properties, storage stability and safety. Int. J. Nanomed. 2018, 13, 1899–1915. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Duan, J.; Wang, J.; Liu, Q.; Shang, R.; Yang, X.; Lu, P.; Xia, C.; Wang, L.; Dou, K. superparamagnetic iron oxide nanoparticles modified with polyethylenimine and galactose for siRNA targeted delivery in hepatocellular carcinoma therapy. Int. J. Nanomed. 2018, 13, 1851. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, L.; Qiao, Z.; Lei, C.; Fu, Y.; Xie, Q.; Yao, S.; Li, Y.; Ying, Y. Electrochemical Conversion of Fe3O4 Magnetic Nanoparticles to Electroactive Prussian Blue Analogues for Self-Sacrificial Label Biosensing of Avian Influenza Virus H5N1. Anal. Chem. 2017, 89, 12145–12151. [Google Scholar] [CrossRef] [PubMed]
- Haun, J.B.; Yoon, T.J.; Lee, H.; Weissleder, R. Magnetic nanoparticle biosensors. Wiley Interdiscip. Rev. 2010, 2, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Peng, Y.; Bai, J.; Zhang, X.; Cui, Y.; Ning, B.; Cui, J.; Gao, Z. Magnetic nanoparticle enhanced surface plasmon resonance sensor for estradiol analysis. Sens. Actuators B Chem. 2018, 254, 629–635. [Google Scholar] [CrossRef]
- Pakapongpan, S.; Poo-Arporn, R.P. Self-assembly of glucose oxidase on reduced graphene oxide-magnetic nanoparticles nanocomposite-based direct electrochemistry for reagentless glucose biosensor. Mater. Sci. Eng. 2017, 76, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Ding, Y.; Sun, Y.; Tian, F.; Yang, M. A Nanoporous Alumina Membrane Based Impedance Biosensor for Histamine Detection with Magnetic Nanoparticles Separation and Amplification. Proc. Technol. 2017, 27, 116–117. [Google Scholar] [CrossRef]
- Zhao, J.; Dong, W.; Zhang, X.; Chai, H.; Huang, Y. FeNPs@ Co3O4 hollow nanocages hybrids as effective peroxidase mimics for glucose biosensing. Sens. Actuators B Chem. 2018, 263, 575–584. [Google Scholar] [CrossRef]
- Yuan, L.-F.; He, Y.-J.; Zhao, H.; Zhou, Y.; Gu, P. Colorimetric detection of D-amino acids based on anti-aggregation of gold nanoparticles. Chin. Chem. Lett. 2014, 25, 995–1000. [Google Scholar] [CrossRef]
- Zhang, F.X.; Han, L.; Israel, L.B.; Daras, J.G.; Maye, M.M.; Ly, N.K.; Zhong, C.-J. Colorimetric detection of thiol-containing amino acids using gold nanoparticles. Analyst 2002, 127, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, D.; Lu, Y.; Xu, G.; Yao, Y.; Li, S.; Liu, Q. Label-free amino acid detection based on nanocomposites of graphene oxide hybridized with gold nanoparticles. Biosens. Bioelectron. 2016, 77, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Nikitin, M.; Orlov, A.; Znoyko, S.; Bragina, V.; Gorshkov, B.; Ksenevich, T.; Cherkasov, V.; Nikitin, P. Multiplex biosensing with highly sensitive magnetic nanoparticle quantification method. J. Magn. Magn. Mater. 2017. [Google Scholar] [CrossRef]
- Tamanaha, C.; Mulvaney, S.; Rife, J.; Whitman, L. Magnetic labeling, detection, and system integration. Biosens. Bioelectron. 2008, 24, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Chi, B.; Wu, F.; Ma, S.; Zhan, S.; Yi, M.; Xu, H.; Mao, C. A sensitive label-free immunosensor for detection α-Fetoprotein in whole blood based on anticoagulating magnetic nanoparticles. Biosens. Bioelectron. 2017, 95, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Heger, Z.; Cernei, N.; Krizkova, S.; Masarik, M.; Kopel, P.; Hodek, P.; Zitka, O.; Adam, V.; Kizek, R. Paramagnetic nanoparticles as a platform for FRET-based sarcosine picomolar detection. Sci. Rep. 2015, 5, 8868. [Google Scholar] [CrossRef] [PubMed]
- Pamme, N. On-chip bioanalysis with magnetic particles. Curr. Opin. Chem. Biol. 2012, 16, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Adam, V.; Huska, D.; Hubalek, J.; Kizek, R. Easy to use and rapid isolation and detection of a viral nucleic acid by using paramagnetic microparticles and carbon nanotubes-based screen-printed electrodes. Microfluid. Nanofluid. 2010, 8, 329–339. [Google Scholar] [CrossRef]
- Ng, A.H.; Choi, K.; Luoma, R.P.; Robinson, J.M.; Wheeler, A.R. Digital microfluidic magnetic separation for particle-based immunoassays. Anal. Chem. 2012, 84, 8805–8812. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, D.; Kim, S. A rapid real-time quantification in hybrid paper-polymer centrifugal optical devices. Biosens. Bioelectron. 2018. [Google Scholar] [CrossRef] [PubMed]
- Trinder, P. Determination of glucose in blood using glucose oxidase with an alternative oxygen acceptor. Ann. Clin. Biochem. 1969, 6, 24–27. [Google Scholar] [CrossRef]
- Wiewiorka, O.; Dastych, M.; Cermakova, Z. Trinder Reaction in Clinical Biochemistry-Benefits and Limits. Chem. Listy 2017, 111, 186–191. [Google Scholar]
- Yamkamon, V.; Phakdee, B.; Yainoy, S.; Suksrichawalit, T.; Tatanandana, T.; Sangkum, P.; Eiamphungporn, W. Development of sarcosine quantification in urine based on enzyme-coupled colorimetric method for prostate cancer diagnosis. EXCLI J. 2018, 17, 467. [Google Scholar] [PubMed]
- Cernei, N.; Zitka, O.; Skalickova, S.; Gumulec, J.; Masarik, M.; Hrabec, R.; Adam, V.; Kizek, R. Sarcosine in urine of patients with prostate carcinome. Prakt. Lek. 2012, 92, 444–448. [Google Scholar]
- El-Boubbou, K.; Gruden, C.; Huang, X. Magnetic glyco-nanoparticles: A unique tool for rapid pathogen detection, decontamination, and strain differentiation. J. Am. Chem. Soc. 2007, 129, 13392–13393. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Teng, X.; Rahman, S.; Yang, H. Patterned Langmuir—Blodgett films of monodisperse nanoparticles of iron oxide using soft lithography. J. Am. Chem. Soc. 2003, 125, 630–631. [Google Scholar] [CrossRef] [PubMed]
- Haviv, A.H.; Grenèche, J.-M.; Lellouche, J.-P. Aggregation control of hydrophilic maghemite (γ-Fe2O3) nanoparticles by surface doping using cerium atoms. J. Am. Chem. Soc. 2010, 132, 12519–12521. [Google Scholar] [CrossRef] [PubMed]
- Scialabba, C.; Licciardi, M.; Mauro, N.; Rocco, F.; Ceruti, M.; Giammona, G. Inulin-based polymer coated SPIONs as potential drug delivery systems for targeted cancer therapy. Eur. J. Pharm. Biopharm. 2014, 88, 695–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inamdar, N.N.; Mourya, V. Chitosan and Low Molecular Weight Chitosan: Biological and Biomedical Applications. Adv. Biomater. Biodev. 2014, 183–242. [Google Scholar]
- Kumar, P.; Jaiwal, R.; Pundir, C. An improved amperometric creatinine biosensor based on nanoparticles of creatininase, creatinase and sarcosine oxidase. Anal. Biochem. 2017, 537, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Narwal, V.; Kumar, P.; Joon, P.; Pundir, C. Fabrication of an amperometric sarcosine biosensor based on sarcosine oxidase/chitosan/CuNPs/c-MWCNT/Au electrode for detection of prostate cancer. Enzyme Microbiol. Technol. 2018, 113, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Lad, U.; Kale, G.M.; Bryaskova, R. Sarcosine oxidase encapsulated polyvinyl alcohol-silica-AuNP hybrid films for sarcosine sensing electrochemical bioelectrode. J. Electrochem. Soc. 2014, 161, B98–B101. [Google Scholar] [CrossRef]
- Nguy, T.P.; van Phi, T.; Tram, D.T.; Eersels, K.; Wagner, P.; Lien, T.T. Development of an impedimetric sensor for the label-free detection of the amino acid sarcosine with molecularly imprinted polymer receptors. Sens. Actuators B Chem. 2017, 246, 461–470. [Google Scholar] [CrossRef]
- Hirsch, V.; Kinnear, C.; Moniatte, M.; Rothen-Rutishauser, B.; Clift, M.J.; Fink, A. Surface charge of polymer coated SPIONs influences the serum protein adsorption, colloidal stability and subsequent cell interaction in vitro. Nanoscale 2013, 5, 3723–3732. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Yu, M.K.; Jeong, Y.Y.; Kim, J.W.; Lee, K.; Phan, V.N.; Jon, S. Antibiofouling amphiphilic polymer-coated superparamagnetic iron oxide nanoparticles: Synthesis, characterization, and use in cancer imaging in vivo. J. Mater. Chem. 2009, 19, 6412–6417. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Cregg, P. Modelling the effect of SPION size in a stent assisted magnetic drug targeting system with interparticle interactions. Sci. World J. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Kapri, A.; Zaidi, M.; Satlewal, A.; Goel, R. SPION-accelerated biodegradation of low-density polyethylene by indigenous microbial consortium. Int. Biodeter. Biodegrad. 2010, 64, 238–244. [Google Scholar] [CrossRef]
- Herve, K.; Douziech-Eyrolles, L.; Munnier, E.; Cohen-Jonathan, S.; Souce, M.; Marchais, H.; Limelette, P.; Warmont, F.; Saboungi, M.; Dubois, P. The development of stable aqueous suspensions of PEGylated SPIONs for biomedical applications. Nanotechnology 2008, 19, 465608. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Xu, C.; Kohler, N.; Hou, Y.; Sun, S. Controlled PEGylation of monodisperse Fe3O4 nanoparticles for reduced non-specific uptake by macrophage cells. Adv. Mater. 2007, 19, 3163–3166. [Google Scholar] [CrossRef]
- HUANG, Z.r. Development and evaluation of lipid nanoparticles for camptothecin delivery: A comparison of solid lipid nanoparticles, nanostructured lipid carriers, and lipid emulsion. Acta Pharmacolog. Sinica 2008, 29, 1094–1102. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.H.; MaÈder, K.; Gohla, S. Solid lipid nanoparticles (SLN) for controlled drug delivery—A review of the state of the art. Eur. J. Pharm. Biopharm. 2000, 50, 161–177. [Google Scholar] [CrossRef]
- Mahmoudi, M.; Sant, S.; Wang, B.; Laurent, S.; Sen, T. Superparamagnetic iron oxide nanoparticles (SPIONs): Development, surface modification and applications in chemotherapy. Adv. Drug Dev. Rev. 2011, 63, 24–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubey, S.P.; Lahtinen, M.; Sillanpää, M. Green synthesis and characterizations of silver and gold nanoparticles using leaf extract of Rosa rugosa. Colloids Surf. A 2010, 364, 34–41. [Google Scholar] [CrossRef]
- Dubey, S.P.; Lahtinen, M.; Sillanpää, M. Tansy fruit mediated greener synthesis of silver and gold nanoparticles. Process Biochem. 2010, 45, 1065–1071. [Google Scholar] [CrossRef]
- Sonavane, G.; Tomoda, K.; Makino, K. Biodistribution of colloidal gold nanoparticles after intravenous administration: Effect of particle size. Colloids Surf. B 2008, 66, 274–280. [Google Scholar] [CrossRef] [PubMed]
- De Palma, R.; Peeters, S.; van Bael, M.J.; van den Rul, H.; Bonroy, K.; Laureyn, W.; Mullens, J.; Borghs, G.; Maes, G. Silane ligand exchange to make hydrophobic superparamagnetic nanoparticles water-dispersible. Chem. Mater. 2007, 19, 1821–1831. [Google Scholar] [CrossRef]
- Shukla, A.; Mishra, V.; Bhoop, B.S.; Katare, O.P. Alginate coated chitosan microparticles mediated oral delivery of diphtheria toxoid.(Part A). Systematic optimization, development and characterization. Int. J. Pharm. 2015, 495, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Burda, C.; Chen, X.; Narayanan, R.; El-Sayed, M.A. Chemistry and properties of nanocrystals of different shapes. Chemical Rev. 2005, 105, 1025–1102. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Wamer, W.; Xia, Q.; Yin, J.-j.; Fu, P.P. Enzyme-like activity of nanomaterials. J. Environ. Sci. Health 2014, 32, 186–211. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Liu, S.; Bao, J.; Ju, H. Nanostructured FeS as a mimic peroxidase for biocatalysis and biosensing. Chem. A Eur. J. 2009, 15, 4321–4326. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhuang, J.; Nie, L.; Zhang, J.; Zhang, Y.; Gu, N.; Wang, T.; Feng, J.; Yang, D.; Perrett, S. Intrinsic peroxidase-like activity of ferromagnetic nanoparticles. Nat. Nanotechnol. 2007, 2, 577. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Wu, X.; Liu, J.; Hu, X.; Zhang, K.; Hou, S.; Zhou, W.; Xie, S. Design of AgM bimetallic alloy nanostructures (M = Au, Pd, Pt) with tunable morphology and peroxidase-like activity. Chem. Mater. 2010, 22, 2988–2994. [Google Scholar] [CrossRef]
- Jv, Y.; Li, B.; Cao, R. Positively-charged gold nanoparticles as peroxidiase mimic and their application in hydrogen peroxide and glucose detection. Chem. Commun. 2010, 46, 8017–8019. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Li, Y.-S.; Yuan, J.; Zhu, L.; Liu, Z.; Tang, H.; Liu, S. Ultrasensitive fluorometric determination of hydrogen peroxide and glucose by using multiferroic BiFeO3 nanoparticles as a catalyst. Talanta 2010, 81, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wang, X.; Zhao, C.; Qu, K.; Ren, J.; Qu, X. Label-free colorimetric detection of single nucleotide polymorphism by using single-walled carbon nanotube intrinsic peroxidase-like activity. Chem. A Eur. J. 2010, 16, 3617–3621. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Wang, E. Fe3O4 magnetic nanoparticles as peroxidase mimetics and their applications in H2O2 and glucose detection. Anal. Chem. 2008, 80, 2250–2254. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.-L.; Wang, L.-Y.; Xie, Z.-J.; Xue, F.; Peng, C.-F. Colorimetric detection of Hg2+ based on inhibiting the peroxidase-like activity of DNA–Ag/Pt nanoclusters. RSC Adv. 2016, 6, 75384–75389. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Z.; Wang, X.; Yang, X. Magnetic nanoparticle-linked colorimetric aptasensor for the detection of thrombin. Sens. Actuators B Chem. 2010, 147, 428–433. [Google Scholar] [CrossRef]
- Asati, A.; Santra, S.; Kaittanis, C.; Nath, S.; Perez, J.M. Oxidase-like activity of polymer-coated cerium oxide nanoparticles. Angew. Chem. Int. Ed. 2009, 121, 2344–2348. [Google Scholar] [CrossRef]
- Jiang, C.; Zhu, J.; Li, Z.; Luo, J.; Wang, J.; Sun, Y. Chitosan–gold nanoparticles as peroxidase mimic and their application in glucose detection in serum. RSC Advances 2017, 7, 44463–44469. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hu, Y.; Wei, H. Nanozymes in bionanotechnology: From sensing to therapeutics and beyond. Inorg. Chem. Front. 2016, 3, 41–60. [Google Scholar] [CrossRef]
- Shah, J.; Purohit, R.; Singh, R.; Karakoti, A.S.; Singh, S. ATP-enhanced peroxidase-like activity of gold nanoparticles. J. Colloid Interface Sci. 2015, 456, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zeng, F.; Wu, S.; Liu, X.; Hou, C.; Tong, Z. Gold nanoparticles bound on microgel particles and their application as an enzyme support. Nanotechnology 2007, 18, 265704. [Google Scholar] [CrossRef] [PubMed]
- Lan, D.; Li, B.; Zhang, Z. Chemiluminescence flow biosensor for glucose based on gold nanoparticle-enhanced activities of glucose oxidase and horseradish peroxidase. Bios. Bioelectron. 2008, 24, 934–938. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Fan, K.; Zhang, D.; Tan, S.; Liang, M.; Liu, Y.; Zhang, J.; Zhang, P.; Liu, W.; Qiu, X. Nanozyme-strip for rapid local diagnosis of Ebola. Biosens. Bioelectron. 2015, 74, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Pan, Y.; Shan, W.; Guo, M.; Nie, Z.; Huang, Y.; Yao, S. Enhanced nonenzymatic sensing of hydrogen peroxide released from living cells based on Fe3 O4/self-reduced graphene nanocomposites. Anal. Methods 2014, 6, 6073–6081. [Google Scholar] [CrossRef]
- Wong, A.C.; Wright, D.W.; Conrad, J.A. Functionalized gold nanoparticles for detection of cancer biomarkers. In General Methods in Biomarker Research and their Applications; Springer: Berlin, Germany, 2015; pp. 1143–1175. [Google Scholar]
- Zurek, M.; Kremplova, M.; Nejdl, L.; Hynek, D.; Kopel, P.; Adam, V.; Kizek, R. Characterization of gold nanoparticles-modified CdTe quantum dots by scanning electrochemical microscopy. NANOCON 2014, 5, 773–778. [Google Scholar]
- Chudobova, D.; Cihalova, K.; Skalickova, S.; Zitka, J.; Rodrigo, M.A.M.; Milosavljevic, V.; Hynek, D.; Kopel, P.; Vesely, R.; Adam, V. 3D-printed chip for detection of methicillin-resistant Staphylococcus aureus labeled with gold nanoparticles. Electrophoresis 2015, 36, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Marquez, L.A.; Dunford, H.B. Mechanism of the oxidation of 3,5,3′,5′-tetramethylbenzidine by myeloperoxidase determined by transient-and steady-state kinetics. Biochemistry 1997, 36, 9349–9355. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Weng, J. Ternary composite of hemin, gold nanoparticles and graphene for highly efficient decomposition of hydrogen peroxide. Sci. Rep. 2013, 3, 3285. [Google Scholar] [CrossRef] [PubMed]
- Skalickova, S.; Loffelmann, M.; Gargulak, M.; Kepinska, M.; Docekalova, M.; Uhlirova, D.; Stankova, M.; Fernandez, C.; Milnerowicz, H.; Ruttkay-Nedecky, B. Zinc-Modified nanotransporter of doxorubicin for targeted prostate cancer delivery. Nanomaterials 2017, 7, 435. [Google Scholar] [CrossRef] [PubMed]
- Prochazkova, S.; Vårum, K.M.; Ostgaard, K. Quantitative determination of chitosans by ninhydrin. Carbohydr. Polym. 1999, 38, 115–122. [Google Scholar] [CrossRef]
- Leane, M.; Nankervis, R.; Smith, A.; Illum, L. Use of the ninhydrin assay to measure the release of chitosan from oral solid dosage forms. Int. J. Pharm. 2004, 271, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Lucarelli, G.; Fanelli, M.; Larocca, A.M.V.; Germinario, C.A.; Rutigliano, M.; Vavallo, A.; Selvaggi, F.P.; Bettocchi, C.; Battaglia, M.; Ditonno, P. Serum sarcosine increases the accuracy of prostate cancer detection in patients with total serum PSA less than 4.0 ng/mL. Prostate 2012, 72, 1611–1621. [Google Scholar] [CrossRef] [PubMed]
- Hurst, R.; Hooper, L.; Norat, T.; Lau, R.; Aune, D.; Greenwood, D.C.; Vieira, R.; Collings, R.; Harvey, L.J.; Sterne, J.A. Selenium and prostate cancer: Systematic review and meta-analysis. Am. J. Clin. Nutr. 2012, 96, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Nishiya, Y.; Toda, A.; Imanaka, T. Gene cluster for creatinine degradation in Arthrobacter sp. TE1826. Mol. Gen. Genet. 1998, 257, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Nishiya, Y.; Yamamoto, M.; Takemoto, J.-i.; Kano, S.; Nakano, S. Monomeric sarcosine oxidase exhibiting high substrate affinity and thermostability. Int. J. Anal. Bio Sci. 2016, 4, 55–62. [Google Scholar]
- Gao, C.; Zhu, H.; Chen, J.; Qiu, H. Facile synthesis of enzyme functional metal-organic framework for colorimetric detecting H2O2 and ascorbic acid. Chin. Chem. Lett. 2017, 28, 1006–1012. [Google Scholar] [CrossRef]
- Mateo, D.; Morales, P.; Ávalos, A.; Haza, A.I. Oxidative stress contributes to gold nanoparticle-induced cytotoxicity in human tumor cells. Toxicol. Mech. Methods 2014, 24, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Boenzli, M.G.; Hindagolla, V.; Ding, J.; Miller, J.M.; Hutchison, J.E.; Greenwood, J.A.; Abeliovich, H.; Bakalinsky, A.T. Identification of gold nanoparticle-resistant mutants of Saccharomyces cerevisiae suggests a role for respiratory metabolism in mediating toxicity. Appl. Environ. Microbiol. 2013, 79, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Pokharkar, V.; Dhar, S.; Bhumkar, D.; Mali, V.; Bodhankar, S.; Prasad, B. Acute and subacute toxicity studies of chitosan reduced gold nanoparticles: A novel carrier for therapeutic agents. J. Biomed. Nanotech. 2009, 5, 233–239. [Google Scholar] [CrossRef]
- Thanou, M.; Verhoef, J.; Junginger, H. Oral drug absorption enhancement by chitosan and its derivatives. Adv. Drug Dev. Rev. 2001, 52, 117–126. [Google Scholar] [CrossRef]
- Richardson, S.W.; Kolbe, H.J.; Duncan, R. Potential of low molecular mass chitosan as a DNA delivery system: Biocompatibility, body distribution and ability to complex and protect DNA. Int. J. Pharm. 1999, 178, 231–243. [Google Scholar] [CrossRef]
- Santhosh, S.; Sini, T.K.; Anandan, R.; Mathew, P.T. Hepatoprotective activity of chitosan against isoniazid and rifampicin-induced toxicity in experimental rats. Eur. J. Pharmacol. 2007, 572, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.; Chen, X.G.; Xing, K.; Park, H.J. Antimicrobial properties of chitosan and mode of action: A state of the art review. Int. J. Food Microbiol. 2010, 144, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Tripathi, S.; Mehrotra, G.; Dutta, J. Perspectives for chitosan based antimicrobial films in food applications. Food Chem. 2009, 114, 1173–1182. [Google Scholar] [CrossRef]
- Gupta, D.; Haile, A. Multifunctional properties of cotton fabric treated with chitosan and carboxymethyl chitosan. Carbohydr. Polym. 2007, 69, 164–171. [Google Scholar] [CrossRef]
- Helander, I.; Nurmiaho-Lassila, E.-L.; Ahvenainen, R.; Rhoades, J.; Roller, S. Chitosan disrupts the barrier properties of the outer membrane of Gram-negative bacteria. Int. J. Food. Microbiol. 2001, 71, 235–244. [Google Scholar] [CrossRef]
- Yang, T.-C.; Chou, C.-C.; Li, C.-F. Antibacterial activity of N-alkylated disaccharide chitosan derivatives. Int. J. Food Microbiol. 2005, 97, 237–245. [Google Scholar] [CrossRef]
- Neun, B.W.; Dobrovolskaia, M.A. Method for analysis of nanoparticle hemolytic properties in vitro. In Characterization of Nanoparticles Intended for Drug Delivery; Springer: Berlin, Germany, 2011; pp. 215–224. [Google Scholar]
- Dobrovolskaia, M.A.; Clogston, J.D.; Neun, B.W.; Hall, J.B.; Patri, A.K.; McNeil, S.E. Method for analysis of nanoparticle hemolytic properties in vitro. Nano Lett. 2008, 8, 2180–2187. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Cai, H.; Wang, X.; Cao, X.; Li, K.; Wang, S.H.; Guo, R.; Zheng, L.; Zhang, G.; Shi, X. Facile one-pot preparation, surface functionalization, and toxicity assay of APTS-coated iron oxide nanoparticles. Nanotechnology 2012, 23, 105601. [Google Scholar] [CrossRef] [PubMed]
- Jentzmik, F.; Stephan, C.; Lein, M.; Miller, K.; Kamlage, B.; Bethan, B.; Kristiansen, G.; Jung, K. Sarcosine in prostate cancer tissue is not a differential metabolite for prostate cancer aggressiveness and biochemical progression. J. Urol. 2011, 185, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Cernei, N.; Zitka, O.; Ryvolova, M.; Adam, V.; Masarik, M.; Hubalek, J.; Kizek, R. Spectrometric and electrochemical analysis of sarcosine as a potential prostate carcinoma marker. Int. J. Electrochem. Sci. 2012, 7, 4286–4301. [Google Scholar]
- Jiang, Y.; Cheng, X.; Wang, C.; Ma, Y. Quantitative determination of sarcosine and related compounds in urinary samples by liquid chromatography with tandem mass spectrometry. Anal. Chem. 2010, 82, 9022–9027. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.E.; Fox, S.D.; Issaq, H.J.; Xu, X.; Chu, L.W.; Veenstra, T.D.; Hsing, A.W. A reproducible and high-throughput HPLC/MS method to separate sarcosine from α-and β-alanine and to quantify sarcosine in human serum and urine. Anal. Chem. 2011, 83, 5735–5740. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Liu, T.; Ma, C.; Xue, R.; Deng, C.; Zeng, H.; Shen, X. GC/MS-based metabolomic approach to validate the role of urinary sarcosine and target biomarkers for human prostate cancer by microwave-assisted derivatization. Anal. Bional. Chem. 2011, 401, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Biavardi, E.; Tudisco, C.; Maffei, F.; Motta, A.; Massera, C.; Condorelli, G.G.; Dalcanale, E. Exclusive recognition of sarcosine in water and urine by a cavitand-functionalized silicon surface. Proc. Nat. Acad. Sci. USA 2012, 109, 2263–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, F.; Dugheri, S.; Musci, M.; Bonacchi, A.; Salvadori, E.; Arcangeli, G.; Cupelli, V.; Lanciotti, M.; Masieri, L.; Serni, S. Fully automated solid-phase microextraction–fast gas chromatography–mass spectrometry method using a new ionic liquid column for high-throughput analysis of sarcosine and N-ethylglycine in human urine and urinary sediments. Anal. Chim. Acta 2011, 707, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Xu, W.; Wan, Q.; Zhang, X.; Lin, J.; Chen, J.; Chen, J. Colorimetric determination of sarcosine in urine samples of prostatic carcinoma by mimic enzyme palladium nanoparticles. Anal. Chim. Acta 2014, 825, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, S.; Block, L.H. Chitosan as an enabling excipient for drug delivery systems: I. Molecular modifications. Int. J. Biol. Macromol. 2000, 27, 181–186. [Google Scholar] [CrossRef]
- Owen, J.; Iggo, B.; Scandrett, F.; Stewart, C. The determination of creatinine in plasma or serum, and in urine; a critical examination. Biochem. J. 1954, 58, 426. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance | Regresion Equation | Linear Dynamic Range (µM) | Correlation Coefficient (r) | LOD (µM) | LOQ (µM) | RSD (%) |
|---|---|---|---|---|---|---|
| Sarcosine # | y = 0.0011x − 0.0001 | 17–100 | 0.9992 | 5 | 17 | 6.35 |
| Sarcosine * | y = 0.0010x + 0.0006 | 59–100 | 0.9912 | 18 | 59 | 21.35 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uhlirova, D.; Stankova, M.; Docekalova, M.; Hosnedlova, B.; Kepinska, M.; Ruttkay-Nedecky, B.; Ruzicka, J.; Fernandez, C.; Milnerowicz, H.; Kizek, R. A Rapid Method for the Detection of Sarcosine Using SPIONs/Au/CS/SOX/NPs for Prostate Cancer Sensing. Int. J. Mol. Sci. 2018, 19, 3722. https://doi.org/10.3390/ijms19123722
Uhlirova D, Stankova M, Docekalova M, Hosnedlova B, Kepinska M, Ruttkay-Nedecky B, Ruzicka J, Fernandez C, Milnerowicz H, Kizek R. A Rapid Method for the Detection of Sarcosine Using SPIONs/Au/CS/SOX/NPs for Prostate Cancer Sensing. International Journal of Molecular Sciences. 2018; 19(12):3722. https://doi.org/10.3390/ijms19123722
Chicago/Turabian StyleUhlirova, Dagmar, Martina Stankova, Michaela Docekalova, Bozena Hosnedlova, Marta Kepinska, Branislav Ruttkay-Nedecky, Josef Ruzicka, Carlos Fernandez, Halina Milnerowicz, and Rene Kizek. 2018. "A Rapid Method for the Detection of Sarcosine Using SPIONs/Au/CS/SOX/NPs for Prostate Cancer Sensing" International Journal of Molecular Sciences 19, no. 12: 3722. https://doi.org/10.3390/ijms19123722