Distinct Patterns of PPARγ Promoter Usage, Lipid Degradation Activity, and Gene Expression in Subcutaneous Adipose Tissue of Lean and Obese Swine

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Genome-Wide Identification of the Expressed Genes in Subcutaneous Adipose Tissue
2.2. Spatiotemporal Expression of Subcutaneous Adipose-Associated Genes
2.3. Functional Enrichment Analysis
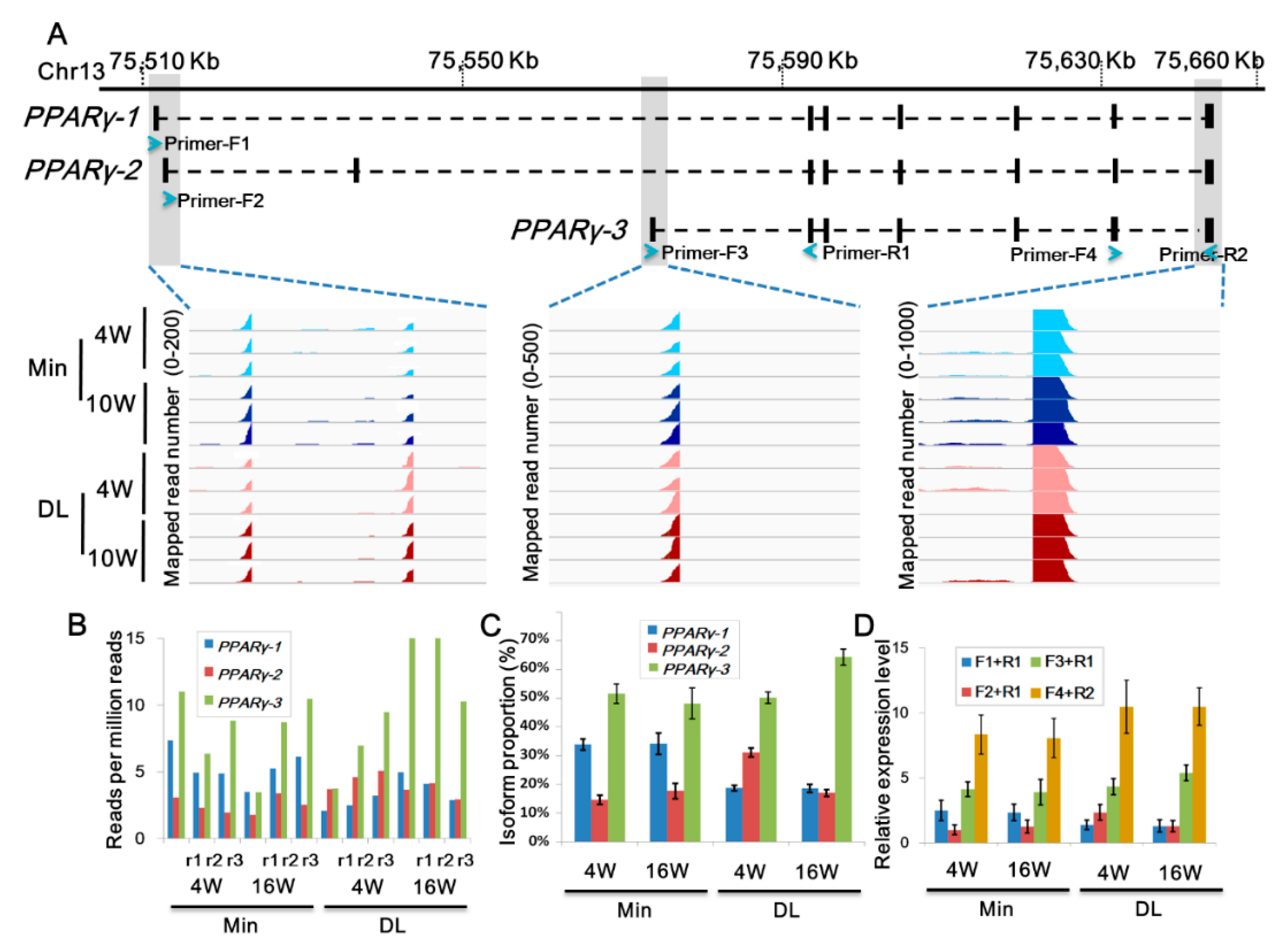
2.4. Distinct PPARγ Promoter Usage Patterns Between Breeds
3. Discussion
4. Materials and Methods
4.1. Animals and Ethics Statement
4.2. Biological Materials and RNA-Seq Experiments
4.3. GO and KEGG Pathway and Functional Enrichment Analysis
4.4. qRT-PCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Groenen, M.A.; Archibald, A.L.; Uenishi, H.; Tuggle, C.K.; Takeuchi, Y.; Rothschild, M.F.; Schook, L.B. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 2012, 491, 393–398. [Google Scholar] [CrossRef] [Green Version]
- Frantz, L.A.; Schraiber, J.G.; Madsen, O.; Megens, H.J.; Bosse, M.; Paudel, Y.; Groenen, M.A. Genome sequencing reveals fine scale diversification and reticulation history during speciation in Sus. Genome Biol. 2013, 14, R107. [Google Scholar] [CrossRef]
- Frantz, L.A.; Madsen, O.; Megens, H.J.; Schraiber, J.G.; Paudel, Y.; Bosse, M.; Groenen, M.A. Evolution of Tibetan wild boars. Nat. Genet. 2015, 47, 188–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groenen, M.A. A decade of pig genome sequencing: A window on pig domestication and evolution. Genet. Sel. Evol. 2016, 48, 23. [Google Scholar] [CrossRef] [PubMed]
- Megens, H.J.; Crooijmans, R.P.; San Cristobal, M.; Hui, X.; Li, N.; Groenen, M.A. Biodiversity of pig breeds from China and Europe estimated from pooled DNA samples: Differences in microsatellite variation between two areas of domestication. Genet. Sel. Evol. 2008, 40, 103–128. [Google Scholar] [PubMed]
- Davoli, R.; Braglia, S. Molecular approaches in pig breeding to improve meat quality. Brief Funct. Genomic Proteomic 2007, 6, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Lopez, B.I.M.; Song, C.; Seo, K. Genetic parameters and trends for production traits and their relationship with litter traits in Landrace and Yorkshire pigs. Anim. Sci. J. 2018, 89, 1381–1388. [Google Scholar] [CrossRef]
- Huang, J.; Liu, R.; Su, L.; Xiao, Q.; Yu, M. Transcriptome Analysis Revealed the Embryo-Induced Gene Expression Patterns in the Endometrium from Meishan and Yorkshire Pigs. Int. J. Mol. Sci. 2015, 16, 22692–22710. [Google Scholar] [CrossRef] [Green Version]
- Schwab, C.R.; Baas, T.J.; Stalder, K.J. Results from six generations of selection for intramuscular fat in Duroc swine using real-time ultrasound. II. Genetic parameters and trends. J. Anim. Sci. 2010, 88, 69–79. [Google Scholar] [CrossRef]
- Miar, Y.; Plastow, G.; Bruce, H.; Moore, S.; Manafiazar, G.; Kemp, R.; Wang, Z. Genetic and phenotypic correlations between performance traits with meat quality and carcass characteristics in commercial crossbred pigs. PLoS ONE 2014, 9, e110105. [Google Scholar] [CrossRef]
- Suzuki, K.; Irie, M.; Kadowaki, H.; Shibata, T.; Kumagai, M.; Nishida, A. Genetic parameter estimates of meat quality traits in Duroc pigs selected for average daily gain, longissimus muscle area, backfat thickness, and intramuscular fat content. J. Anim. Sci. 2005, 83, 2058–2065. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Jin, C.; Xuan, Y.; Zhou, P.; Fang, Z.; Che, L.; Wu, D. Effect of maternal or post-weaning methyl donor supplementation on growth performance, carcass traits and meat quality of pig offspring. J. Sci. Food Agricult. 2018. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, X.; Jing, X.; He, X.; Wang, L.; Liu, Y.; Liu, D. Transcriptomics Analysis on Excellent Meat Quality Traits of Skeletal Muscles of the Chinese Indigenous Min Pig Compared with the Large White Breed. Int. J. Mol. Sci. 2017, 19, 21. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.S.; Huh, J.Y.; Hwang, I.J.; Kim, J.I.; Kim, J.B. Adipose Tissue Remodeling: Its Role in Energy Metabolism and Metabolic Disorders. Front. Endocrinol. 2016, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.C.; Stenesen, D.; Zeve, D.; Graff, J.M. The developmental origins of adipose tissue. Development 2013, 140, 3939–3949. [Google Scholar] [CrossRef] [Green Version]
- Corrales, P.; Vidal-Puig, A.; Medina-Gomez, G. PPARs and Metabolic Disorders Associated with Challenged Adipose Tissue Plasticity. Int. J. Mol. Sci. 2018, 19, 2124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, X.; Xie, X.B.; Cheng, X.C.; Wang, R.L. Multitargeted bioactive ligands for PPARs discovered in the last decade. Chem. Biol. Drug Des. 2016, 88, 635–663. [Google Scholar] [CrossRef] [PubMed]
- Grygiel-Gorniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications--a review. Nutrit. J. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Ge, K. Transcriptional and epigenetic regulation of PPARgamma expression during adipogenesis. Cell Biosci. 2014, 4, 29. [Google Scholar] [CrossRef]
- Kim, D.M.; Choi, H.R.; Park, A.; Shin, S.M.; Bae, K.H.; Lee, S.C.; Kim, W.K. Retinoic acid inhibits adipogenesis via activation of Wnt signaling pathway in 3T3-L1 preadipocytes. Biochem. Biophys. Res. Commun. 2013, 434, 455–459. [Google Scholar] [CrossRef]
- Swarbreck, D.; Wilks, C.; Lamesch, P.; Berardini, T.Z.; Garcia-Hernandez, M.; Foerster, H.; Huala, E. The Arabidopsis Information Resource (TAIR): Gene structure and function annotation. Nucleic Acids Res. 2008, 36, D1009–D1014. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jung, C.; Xu, J.; Wang, H.; Deng, S.; Bernad, L.; Chua, N.H. Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis. Plant Cell 2012, 24, 4333–4345. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chung, P.J.; Liu, J.; Jang, I.C.; Kean, M.J.; Xu, J.; Chua, N.H. Genome-wide identification of long noncoding natural antisense transcripts and their responses to light in Arabidopsis. Genome Res. 2014, 24, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeifer, M.; Kugler, K.G.; Sandve, S.R.; Zhan, B.; Rudi, H.; Hvidsten, T.R.; Olsen, O.A. Genome interplay in the grain transcriptome of hexaploid bread wheat. Science 2014, 345, 1250091. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Fearon, D.T.; Locksley, R.M. The instructive role of innate immunity in the acquired immune response. Science 1996, 272, 50–53. [Google Scholar] [CrossRef]
- Fearon, D.T. The complement system and adaptive immunity. Semin. Immunol. 1998, 10, 355–3561. [Google Scholar] [CrossRef] [PubMed]
- Blom, A.M.; Villoutreix, B.O.; Dahlback, B. Complement inhibitor C4b-binding protein-friend or foe in the innate immune system? Mol. Immunol. 2004, 40, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Artiaga, B.L.; Hackmann, T.J.; Samuel, M.S.; Walters, E.M.; Salek-Ardakani, S.; Driver, J.P. Targeted disruption of CD1d prevents NKT cell development in pigs. Mamm. Genome 2015, 26, 264–270. [Google Scholar] [CrossRef]
- Exley, M.A.; Hand, L.; O′Shea, D.; Lynch, L. Interplay between the immune system and adipose tissue in obesity. J. Endocrinol. 2014, 223, R41–R48. [Google Scholar] [CrossRef] [Green Version]
- Sodhi, S.S.; Park, W.C.; Ghosh, M.; Kim, J.N.; Sharma, N.; Shin, K.Y.; Jeong, D.K. Comparative transcriptomic analysis to identify differentially expressed genes in fat tissue of adult Berkshire and Jeju Native Pig using RNA-seq. Mol. Biol. Rep. 2014, 41, 6305–6315. [Google Scholar] [CrossRef] [PubMed]
- Weber, F.D.; Wiesinger, C.; Forss-Petter, S.; Regelsberger, G.; Einwich, A.; Weber, W.H.; Berger, J. X-linked adrenoleukodystrophy: Very long-chain fatty acid metabolism is severely impaired in monocytes but not in lymphocytes. Human Mol. Genet. 2014, 23, 2542–2550. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Qi, X.; Hu, M.; Lin, R.; Hou, Y.; Wang, Z.; Li, X. Transcriptome Analysis of Adipose Tissue Indicates That the cAMP Signaling Pathway Affects the Feed Efficiency of Pigs. Genes 2018, 9, 336. [Google Scholar] [CrossRef] [PubMed]
- Sutovsky, P.; Song, W.H. Post-fertilisation sperm mitophagy: The tale of Mitochondrial Eve and Steve. Reprod. Fertil. Dev. 2017, 30, 56–63. [Google Scholar] [CrossRef]
- Sharpe, A.J.; McKenzie, M. Mitochondrial Fatty Acid Oxidation Disorders Associated with Short-Chain Enoyl-CoA Hydratase (ECHS1) Deficiency. Cells 2018, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Bovo, S.; Di Luca, A.; Galimberti, G.; Dall′Olio, S.; Fontanesi, L. A comparative analysis of label-free liquid chromatography-mass spectrometry liver proteomic profiles highlights metabolic differences between pig breeds. PLoS ONE 2018, 13, e0199649. [Google Scholar] [CrossRef]
- Jo, J.L.; Hwang, J.H.; Kwon, S.G.; Park, D.H.; Kim, T.W.; Kang, D.G.; Kim, C.W. Association between a non-synonymous HSD17B4 single nucleotide polymorphism and meat-quality traits in Berkshire pigs. Genet. Mol. Res. 2016, 15. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Wei, P.; Khan, M.A.; Zhang, J.; Guo, L.; Liu, D.; Wang, S. Transcriptome analysis reveals differential gene expression in intramuscular adipose tissues of Jinhua and Landrace pigs. J. Vet. Med. Sci. 2018, 80, 953–959. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Cui, L.; Ma, J.; Chen, C.; Yang, B.; Huang, L. Transcriptome analyses reveal genes and pathways associated with fatty acid composition traits in pigs. Anim. Genet. 2017, 48, 645–652. [Google Scholar] [CrossRef]
- Tao, X.; Liang, Y.; Yang, X.; Pang, J.; Zhong, Z.; Chen, X.; Lv, X. Transcriptomic profiling in muscle and adipose tissue identifies genes related to growth and lipid deposition. PLoS ONE 2017, 12, e0184120. [Google Scholar] [CrossRef]
- Mo, D.; Yu, K.; Chen, H.; Chen, L.; Liu, X.; He, Z.; Chen, Y. Transcriptome Landscape of Porcine Intramuscular Adipocytes during Differentiation. J. Agricult. Food Chem. 2017, 65, 6317–6328. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wang, J.; Song, X.; Zhang, X.; Ge, C.; Gao, S. Impact of dietary protein on lipid metabolism-related gene expression in porcine adipose tissue. Nutrit. Metabol. 2010, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Ji, Y.; Kersten, S.; Qi, L. Mechanisms of inflammatory responses in obese adipose tissue. Annu. Rev. Nutrit. 2012, 32, 261–286. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.K.; Delaney, C.; Shi, H.; Yung, R. Changes in adipose tissue macrophages and T cells during aging. Crit. Rev. Immunol. 2014, 34, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Georgiadi, A.; Kersten, S. Mechanisms of gene regulation by fatty acids. Adv. Nutrit. 2012, 3, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Hausman, G.J. The origin and purpose of layers of subcutaneous adipose tissue in pigs and man. Horm. Mol. Biol. Clin. Investig. 2018, 33. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Zheng, Y.; Zhang, M.; Zheng, H.; Yan, P. Grape seed procyanidin extract inhibits adipogenesis and stimulates lipolysis of porcine adipocytes in vitro. J. Anim. Sci. 2018, 96, 2753–2762. [Google Scholar] [CrossRef]
- Medina-Gomez, G.; Gray, S.L.; Yetukuri, L.; Shimomura, K.; Virtue, S.; Campbell, M.; Vidal-Puig, A. PPAR gamma 2 prevents lipotoxicity by controlling adipose tissue expandability and peripheral lipid metabolism. PLoS Genet. 2007, 3, e64. [Google Scholar] [CrossRef]
- Lin, J.C. Impacts of Alternative Splicing Events on the Differentiation of Adipocytes. Int. J. Mol. Sci. 2015, 16, 22169–22189. [Google Scholar] [CrossRef] [Green Version]
- Birsoy, K.; Chen, Z.; Friedman, J. Transcriptional regulation of adipogenesis by KLF4. Cell Metabol. 2008, 7, 339–347. [Google Scholar] [CrossRef]
- Oishi, Y.; Manabe, I.; Tobe, K.; Tsushima, K.; Shindo, T.; Fujiu, K.; Nagai, R. Kruppel-like transcription factor KLF5 is a key regulator of adipocyte differentiation. Cell Metabol. 2005, 1, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Q.; Wang, X.J. GOEAST: A web-based software toolkit for Gene Ontology enrichment analysis. Nucleic Acids Res. 2008, 36, W358–W363. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, J.; Jia, C.; Li, T.; Wu, R.; Wang, J.; Zhu, D. Systematic identification and evolutionary features of rhesus monkey small nucleolar RNAs. BMC Genomics 2010, 11, 61. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, B.; Di, S.; Cui, S.; Chen, N.; Wang, H.; Wang, X.; Gao, Q.; Tong, G.; Wang, H.; Huang, X.; et al. Distinct Patterns of PPARγ Promoter Usage, Lipid Degradation Activity, and Gene Expression in Subcutaneous Adipose Tissue of Lean and Obese Swine. Int. J. Mol. Sci. 2018, 19, 3892. https://doi.org/10.3390/ijms19123892
Song B, Di S, Cui S, Chen N, Wang H, Wang X, Gao Q, Tong G, Wang H, Huang X, et al. Distinct Patterns of PPARγ Promoter Usage, Lipid Degradation Activity, and Gene Expression in Subcutaneous Adipose Tissue of Lean and Obese Swine. International Journal of Molecular Sciences. 2018; 19(12):3892. https://doi.org/10.3390/ijms19123892
Chicago/Turabian StyleSong, Bin, Shengwei Di, Shiquan Cui, Na Chen, Huan Wang, Xuan Wang, Qian Gao, Guizhi Tong, Hongbao Wang, Xuankai Huang, and et al. 2018. "Distinct Patterns of PPARγ Promoter Usage, Lipid Degradation Activity, and Gene Expression in Subcutaneous Adipose Tissue of Lean and Obese Swine" International Journal of Molecular Sciences 19, no. 12: 3892. https://doi.org/10.3390/ijms19123892