Maternal Cognitive Impairment Associated with Gestational Diabetes Mellitus—A Review of Potential Contributing Mechanisms

 ,
,
Abstract
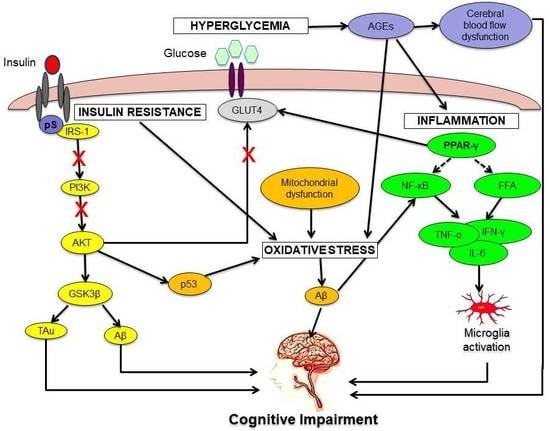
:
1. Introduction
2. Pathophysiology of GDM That Can Lead to Cognitive Impairment
2.1. Hyperglycemia
2.2. Insulin Resistance
2.3. Oxidative Stress
2.4. Neuroinflammation
3. Future Directions and Conclusions
- (a)
- Potential biomarkers and molecules for cognitive decline includes AGEs, serine-phosphorylated IRS-1, and cytokines such as TNF-α, hs-CRP, leptin, IL-1β, and IL-6. The level of these molecules should be evaluated in GDM cases in the future and in relation to cognitive decline.
- (b)
- The PI3K and AKT signalling cascade is important in the downstream insulin pathway in GDM. Moreover, there is also crosstalk with other pathways that are important in maintaining cognitive function through increasing or decreasing key regulators of cognitive function such as GSK3β.
Funding
Conflicts of Interest
References
- American Diabetes Association. (2) Classification and Diagnosis of Diabetes. Diabetes Care 2015, 38, S8–S16. [Google Scholar] [CrossRef] [PubMed]
- Ogurtsova, K.; da Rocha Fernandes, J.D.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.H.; Cavan, D.; Shaw, J.E.; Makaroff, L.E. Idf Diabetes Atlas: Global Estimates for the Prevalence of Diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 2017, 128, 40–50. [Google Scholar] [CrossRef] [PubMed]
- World Health Organisation. Global Report on Diabetes; WHO Press: Geneva, Switzerland, 2016. [Google Scholar]
- Buchanan, A.T.; Xiang, A.H. Gestational Diabetes Mellitus. J. Clin. Investig. 2005, 115, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Weijers, N.R.; Bekedam, D.J. Relationship between Gestational Diabetes Mellitus and Type 2 Diabetes: Evidence of Mitochondrial Dysfunction. Clin. Chem. 2007, 53, 377–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herath, H.R.; Wickremasinghe, R. Gestational Diabetes Mellitus and Risk of Type 2 Diabetes 10 Years after the Index Pregnancy in Sri Lankan Women-a Community Based Retrospective Cohort Study. PLoS ONE 2017, 12, e0179647. [Google Scholar] [CrossRef] [PubMed]
- Karamanos, B.A.; Thanopoulou, E.; Anastasiou, S.; Assaad-Khalil, N.; Albache, M.; Bachaoui, C.B.; Slama, H.; El Ghomari, A.; Jotic, N.; Lalic, A.; et al. Relation of the Mediterranean Diet with the Incidence of Gestational Diabetes. Eur. J. Clin. Nutr. 2014, 68, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Salib, M.M.; Hickman, P.E.; Oakman, C.; Potter, J.M. Retrospective Reassessment of Gestational Diabetes Mellitus Diagnosis by Using the New Classification. Pathology 2015, 47, 391–392. [Google Scholar] [CrossRef] [PubMed]
- Alewijn, O.; Stolk, R.P.; Van Harskamp, F.; Pols, H.A.P.; Hofman, A.; Breteler, M.M.B. Diabetes Mellitus and the Risk of Dementia the Rotterdam Study. Neurology 1999, 53, 1937. [Google Scholar]
- Matioli, M.N.P.D.S.; Suemoto, C.K.; Rodriguez, R.D.; Farias, D.S.; da Silva, M.M.; Paraizo Leite, R.E.; Ferretti-Rebustini, R.E.L.; Pasqualucci, C.A.; Jacob, W.F.; Grinberg, L.T. Association between Diabetes and Causes of Dementia: Evidence from a Clinicopathological Study. Dement. Neuropsychol. 2017, 11, 406–412. [Google Scholar] [CrossRef]
- Ricci, G.; Pirillo, I.; Tomassoni, D.; Sirignano, A.; Grappasonni, I. Metabolic Syndrome, Hypertension, and Nervous System Injury: Epidemiological Correlates. Clin. Exp. Hypertens. 2017, 39, 8–16. [Google Scholar] [CrossRef]
- Kodl, C.T.; Seaquist, E.R. Cognitive Dysfunction and Diabetes Mellitus. Endocr. Rev. 2008, 29, 494–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutski, M.; Weinstein, G.; Goldbourt, U.; Tanne, D. Insulin Resistance and Future Cognitive Performance and Cognitive Decline in Elderly Patients with Cardiovascular Disease. J. Alzheimer’s Dis. 2017, 57, 633–643. [Google Scholar] [CrossRef]
- Hamed, S.A. Brain Injury with Diabetes Mellitus: Evidence, Mechanisms and Treatment Implications. Expert Rev. Clin. Pharmacol. 2017, 10, 409–428. [Google Scholar] [CrossRef] [PubMed]
- Mittal, K.; Katare, D.P. Shared Links between Type 2 Diabetes Mellitus and Alzheimer’s Disease: A Review. Diabetes Metab. Syndr. 2016, 10, S144–S149. [Google Scholar] [CrossRef]
- Okereke, O.I.; Kang, J.H.; Cook, N.R.; Gaziano, J.M.; Manson, J.E.; Buring, J.E.; Grodstein, F. Type 2 Diabetes Mellitus and Cognitive Decline in Two Large Cohorts of Community-Dwelling Older Adults. J. Am. Geriat. Soc. 2008, 56, 1028–1036. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Lourenco, M.V.; Ferreira, S.T. How Does Brain Insulin Resistance Develop in Alzheimer’s Disease? Alzheimer’s Dement. 2014, 10, S26–S32. [Google Scholar] [CrossRef] [PubMed]
- Keskin, F.E.; Ozyazar, M.; Pala, A.S.; Elmali, A.D.; Yilmaz, B.; Uygunoglu, U.; Bozluolcay, M.; Tuten, A.; Bingöl, A.; Hatipoglu, E. Evaluation of Cognitive Functions in Gestational Diabetes Mellitus. Exp. Clin. Endocrinol. Diabetes 2015, 123, 246–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camprubi, R.M.; Campoy, C.; Fernandez, L.G.; Lopez-Pedrosa, J.M.; Rueda, R.; Martin, M.J. Maternal Diabetes and Cognitive Performance in the Offspring: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0142583. [Google Scholar] [CrossRef] [PubMed]
- Linder, K.; Schleger, F.; Kiefer-Schmidt, I.; Fritsche, L.; Kummel, S.; Heni, M.; Weiss, M.; Haring, H.U.; Preissl, H.; Fritsche, A. Gestational Diabetes Impairs Human Fetal Postprandial Brain Activity. J. Clin. Endocrinol. Metab. 2015, 100, 4029–4036. [Google Scholar] [CrossRef] [Green Version]
- Carson, M.B.; Liu, C.; Lu, Y.; Jia, C.; Lu, H. A Disease Similarity Matrix Based on the Uniqueness of Shared Genes. BMC Med. Genom. 2017, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Goh, K.I.; Cusick, M.E.; Valle, D.; Childs, B.; Vidal, M.; Barabási, A.L. The Human Disease Network. Proc. Natl. Acad. Sci. USA 2007, 104, 8685–8690. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhu, C.; Jacom, A.; Lu, L.J.; Jegg, A.G. The Orphan Disease Networks. Am. J. Hum. Genet. 2011, 88, 755–766. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.J.; Hiscock, R.J.; Wein, P.; Walker, S.P.; Permezel, M. Gestational Diabetes Mellitus: Clinical Predictors and Long-Term Risk of Developing Type 2 Diabetes: A Retrospective Cohort Study Using Survival Analysis. Diabetes Care 2007, 30, 878–883. [Google Scholar] [CrossRef]
- Hopkins, R.; Shaver, K.; Weinstock, R.S. Management of Adults with Diabetes and Cognitive Problems. Diabetes Spectr. 2016, 29, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Spauwen, P.J.J.; van Eupen, M.G.A.; Köhler, S.; Stehouwer, C.D.A.; Verhey, F.R.J.; van der Kallen, C.J.H.; Sep, S.J.S.; Koster, A.; Schaper, N.C.; Dagnelie, P.C.; et al. Associations of Advanced Glycation End-Products with Cognitive Functions in Individuals with and without Type 2 Diabetes: The Maastricht Study. J. Clin. Endocrinol. Metab. 2015, 100, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Gorelick, P.B.; Scuteri, A.; Black, S.E.; Decarli, C.; Greenberg, S.M.; Iadecola, C.; Launer, L.J.; Laurent, S.; Lopez, O.L.; Nyenhuis, D.; et al. Vascular Contributions to Cognitive Impairment and Dementia: A Statement for Healthcare Professionals from the American Heart Association/American Stroke Association. Stroke 2011, 42, 2672–2713. [Google Scholar] [CrossRef] [PubMed]
- Dorsemans, A.C.; Couret, D.; Hoarau, A.; Meilhac, O.; d’Hellencourt, C.L.; Diotel, N. Diabetes, Adult Neurogenesis and Brain Remodeling: New Insights from Rodent and Zebrafish Models. Neurogenesis (Austin) 2017, 4, e1281862. [Google Scholar] [CrossRef]
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory Response in the Cns: Friend or Foe? Mol. Neurobiol. 2017, 54, 8071–8089. [Google Scholar] [CrossRef]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The Cholinergic Hypothesis of Alzheimer’s Disease: A. Review of Progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef]
- Saltiel, A.R.; Kahn, C.R. Insulin Signalling and the Regulation of Glucose and Lipid Metabolism. Nature 2001, 414, 799. [Google Scholar] [CrossRef]
- Grillo, C.A.; Piroli, G.G.; Lawrence, R.C.; Wrighten, S.A.; Green, A.J.; Wilson, S.P.; Sakai, R.R.; Kelly, S.J.; Wilson, M.A.; Mott, D.D.; et al. Hippocampal Insulin Resistance Impairs Spatial Learning and Synaptic Plasticity. Diabetes 2015, 64, 3927–3936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S. Demonstrated Brain Insulin Resistance in Alzheimer’s Disease Patients Is Associated with Igf-1 Resistance, Irs-1 Dysregulation, and Cognitive Decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed]
- Tello, P.S.; Matamoros, A.O.; Arias, C. Gsk3 Function in the Brain during Development, Neuronal Plasticity, and Neurodegeneration. Int. J. Alzheimer’s Dis. 2011, 2011. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Chen, H.; Xu, H.; Moore, E.; Meiri, N.; Quon, M.J.; Alkon, D.L. Brain Insulin Receptors and Spatial Memory. Correlated Changes in Gene Expression, Tyrosine Phosphorylation, and Signaling Molecules in the Hippocampus of Water Maze Trained Rats. J. Biol. Chem. 1999, 274, 34893–34902. [Google Scholar] [CrossRef] [PubMed]
- Droge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [Green Version]
- De la Monte, S.M.; Wands, J.R. Molecular Indices of Oxidative Stress and Mitochondrial Dysfunction Occur Early and Often Progress with Severity of Alzheimer’s Disease. J. Alzheimer’s Dis. 2006, 9, 167–181. [Google Scholar] [CrossRef]
- Karacay, O.; Sepici-Dincel, A.; Karcaaltincaba, D.; Sahin, D.; Yalvaç, S.; Akyol, M.; Kandemir, O.; Altan, N. A Quantitative Evaluation of Total Antioxidant Status and Oxidative Stress Markers in Preeclampsia and Gestational Diabetic Patients in 24–36 Weeks of Gestation. Diabetes Res. Clin. Pract. 2010, 89, 231–238. [Google Scholar] [CrossRef]
- Liu, B.; Chen, Y.; St Clair, D.K. Ros and P53: A Versatile Partnership. Free Radic. Biol. Med. 2008, 44, 1529–1535. [Google Scholar] [CrossRef]
- Muriach, M.; Flores-Bellver, M.; Francisco, J.R.; Jorge, M.B. Diabetes and the Brain: Oxidative Stress, Inflammation, and Autophagy. Oxid. Med. Cell. Longev. 2014, 2014. [Google Scholar] [CrossRef] [Green Version]
- Pinheiro, L.; de Melo, A.D.; Andreazzi, A.E.; de Caires Júnior, L.C.; Costa, M.; Gonzalez Garcia, M.R. Protocol of Insulin Therapy for Streptozotocin-Diabetic Rats Based on a Study of Food Ingestion and Glycemic Variation. Scand. J. Lab. Anim. Sci. 2011, 38, 117–127. [Google Scholar]
- Yan, H.; Harding, J.J. Glycation-Induced Inactivation and Loss of Antigenicity of Catalase and Superoxide Dismutase. Biochem. J. 1997, 328, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Sözmen, E.Y.; Sözmen, B.; Delen, Y.; Onat, T. Catalase/Superoxide Dismutase (Sod) and Catalase/Paraoxonase (Pon) Ratios May Implicate Poor Glycemic Control. Arch. Med. Res. 2001, 32, 283–287. [Google Scholar] [CrossRef]
- Aziz, S.H.A.; John, C.M.; Yusof, N.I.S.M.; Nordin, M.; Ramasamy, R.; Adam, A.; Fauzi, F.M. Animal Model of Gestational Diabetes Mellitus with Pathophysiological Resemblance to the Human Condition Induced by Multiple Factors (Nutritional, Pharmacological, and Stress) in Rats. BioMed Res. Int. 2016, 2016. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butterfield, D.A.; Di Domenico, F.; Barone, E. Elevated Risk of Type 2 Diabetes for Development of Alzheimer Disease: A Key Role for Oxidative Stress in Brain. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 1693–1706. [Google Scholar] [CrossRef] [PubMed]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free Radicals, Antioxidants and Functional Foods: Impact on Human Health. Pharm. Rev. 2010, 4, 118. [Google Scholar] [CrossRef] [PubMed]
- Eckert, A.; Schmitt, K.; Götz, J. Mitochondrial Dysfunction-the Beginning of the End in Alzheimer’s Disease? Separate and Synergistic Modes of Tau and Amyloid-Β Toxicity. Alzheimer’s Res. Ther. 2011, 3, 15. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Carvalho, C.; Zhu, X.; Smith, M.A.; Perry, G. Mitochondrial Dysfunction Is a Trigger of Alzheimer’s Disease Pathophysiology. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2010, 1802, 2–10. [Google Scholar] [CrossRef]
- Valla, J.; Schneider, L.; Niedzielko, T.; Coon, K.D.; Caselli, R.; Sabbagh, M.N.; Ahern, G.L.; Baxter, L.; Alexander, G.; Walker, D.G. Impaired Platelet Mitochondrial Activity in Alzheimer’s Disease and Mild Cognitive Impairment. Mitochondrion 2006, 6, 323–330. [Google Scholar] [CrossRef]
- Readnower, R.D.; Sauerbeck, A.D.; Sullivan, P.G. Mitochondria, Amyloid Β, and Alzheimer’s Disease. Int. J. Alzheimer’s Dis. 2011, 2011, 104545. [Google Scholar] [CrossRef] [PubMed]
- Abell, S.K.; De Courten, B.; Boyle, J.A.; Teede, H.J. Inflammatory and Other Biomarkers: Role in Pathophysiology and Prediction of Gestational Diabetes Mellitus. Int. J. Mol. Sci. 2015, 16, 13442–13473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, C.C.; Pimentel, D.; Jor’dan, A.J.; Hao, Y.; Milberg, W.; Novak, V. Inflammation-Associated Declines in Cerebral Vasoreactivity and Cognition in Type 2 Diabetes. Neurology 2015, 85, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Grigsby, J.G.; Cardona, S.M.; Pouw, C.E.; Muniz, A.; Mendiola, A.S.; Tsin, A.T.; Allen, D.M.; Cardona, A.E. The Role of Microglia in Diabetic Retinopathy. J. Ophthalmol. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Kuhad, A.; Bishnoi, M.; Tiwari, V.; Chopra, K. Suppression of Nf-Κβ Signaling Pathway by Tocotrienol Can Prevent Diabetes Associated Cognitive Deficits. Pharmacol. Biochem. Behav. 2009, 92, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, J.P.; Hauguel-De Mouzon, S.; Lepercq, J.; Challier, J.C.; Huston-Presley, L.; Friedman, J.E.; Kalhan, S.C.; Catalano, P.M. Tnf-A Is a Predictor of Insulin Resistance in Human Pregnancy. Diabetes 2002, 51, 2207–2213. [Google Scholar] [CrossRef] [PubMed]
- Catalano, P.M.; Nizielski, S.E.; Shao, J.; Preston, L.; Qiao, L.; Friedman, J.E. Downregulated Irs-1 and Ppargamma in Obese Women with Gestational Diabetes: Relationship to Ffa during Pregnancy. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E522–E533. [Google Scholar] [CrossRef]
- Sawada, M.; Sawada, H.; Nagatsu, T. Effects of Aging on Neuroprotective and Neurotoxic Properties of Microglia in Neurodegenerative Diseases. Neurodegener. Dis. 2008, 5, 254–256. [Google Scholar] [CrossRef]
- Wojtera, M.; Sikorska, B.; Sobow, T. Microglial Cells in Neurodegenerative Disorders. Folia Neuropathol. 2005, 43, 311–321. [Google Scholar]
- Kreutzberg, G.W. Microglia: A Sensor for Pathological Events in the Cns. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef]
- Song, I.-U.; Chung, S.-W.; Kim, Y.-D.; Maeng, L.-S. Relationship between the Hs-Crp as Non-Specific Biomarker and Alzheimer’s Disease According to Aging Process. Int. J. Med. Sci. 2015, 12, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Denner, L.A.; Rodriguez-Rivera, J.; Haidacher, S.J.; Jahrling, J.B.; Carmical, J.R.; Hernandez, C.M.; Zhao, Y.; Sadygov, R.G.; Starkey, J.M.; Spratt, H. Cognitive Enhancement with Rosiglitazone Links the Hippocampal Pparγ and Erk Mapk Signaling Pathways. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 16725–16735. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Rivera, J.; Denner, L.; Dineley, K.T. Rosiglitazone Reversal of Tg2576 Cognitive Deficits Is Independent of Peripheral Gluco-Regulatory Status. Behav. Brain Res. 2011, 216, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Yan, L.; Yang, Z.; Zhong, B.; Xie, W. Hba1c, Diabetes and Cognitive Decline: The English Longitudinal Study of Ageing. Diabetologia 2018, 61, 839–848. [Google Scholar] [CrossRef]
- Gupta, A.; Singh, A.; Deka, R.C.; Gupta, R.; Jha, R. To Investigate Role of Glycosylated Hemoglobin (Hba1c) as a Biomarker for Prediction of Dementia and Cognitive Dysfunction in Type 2 Diabetic Patients. J. Alzheimers Dis. Parkinsonism 2018, 8, 437. [Google Scholar] [CrossRef]
- Wang, P.; Huang, R.; Lu, S.; Xia, W.; Cai, R.; Sun, H.; Wang, S. Rage and Ages in Mild Cognitive Impairment of Diabetic Patients: A Cross-Sectional Study. PLoS ONE 2016, 11, e0145521. [Google Scholar] [CrossRef] [PubMed]
- Mayeda, E.R.; Haan, M.N.; Neuhaus, J.; Yaffe, K.; Knopman, D.S.; Sharrett, A.R.; Griswold, M.E.; Mosley, T.H. Type 2 Diabetes and Cognitive Decline over 14 Years in Middle-Aged African Americans and Whites: The Aric Brain Mri Study. Neuroepidemiology 2014, 43, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A. Type 2 Diabetes and Cognitive Impairment: Linking Mechanisms. J. Alzheimers Dis. 2012, 30, S185–S198. [Google Scholar] [CrossRef]
- Craft, S. Insulin Resistance and Alzheimer’s Disease Pathogenesis: Potential Mechanisms and Implications for Treatment. Curr. Alzheimer Res. 2007, 4, 147–152. [Google Scholar] [CrossRef]
- Dineley, K.T.; Jahrling, J.B.; Denner, L. Insulin Resistance in Alzheimer’s Disease. Neurobiol. Dis. 2014, 72, 92–103. [Google Scholar] [CrossRef]
- Diehl, T.; Mullins, R.; Kapogiannis, D. Insulin Resistance in Alzheimer’s Disease. Transl. Res. 2017, 183, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.; Qin, W.; Pompl, P.N.; Xiang, Z.; Wang, J.; Zhao, Z.; Peng, Y.; Cambareri, G.; Rocher, A.; Mobbs, C.V.; et al. Diet-Induced Insulin Resistance Promotes Amyloidosis in a Transgenic Mouse Model of Alzheimer’s Disease. FASEB J. 2004, 18, 902–904. [Google Scholar] [CrossRef] [PubMed]
- Al-Noaemi, M.C.; Shalayel, M.H.F. Pathophysiology of Gestational Diabetes Mellitus: The Past, the Present and the Future. In Gestational Diabetes; InTech: Rijeka, Croatia, 2011. [Google Scholar] [Green Version]
- de la Monte, M.S. Brain Insulin Resistance and Deficiency as Therapeutic Targets in Alzheimer’s Disease. Curr. Alzheimer Res. 2012, 9, 35–66. [Google Scholar] [CrossRef] [PubMed]
- Johnston, A.M.; Pirola, L.; Van Obberghen, E. Molecular Mechanisms of Insulin Receptor Substrate Protein-Mediated Modulation of Insulin Signalling. FEBS Lett. 2003, 546, 32–36. [Google Scholar] [CrossRef]
- Šalković-Petrišić, M.; Zdravko, L. Insulin Resistant Brain State and Its Link to Diabetes Mellitus. Period. Biol. 2005, 107, 137–146. [Google Scholar]
- Nees, S.; Weiss, D.R.; Senftl, A.; Knott, M.; Förch, S.; Schnurr, M.; Weyrich, P.; Juchem, G. Isolation, Bulk Cultivation, and Characterization of Coronary Microvascular Pericytes: The Second Most Frequent Myocardial Cell Type in Vitro. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H69–H84. [Google Scholar] [CrossRef] [PubMed]
- Ertl, L.; Sanftenberg, L.; Schelling, J. Medical Certificates and Examinations in Family Doctor’s Office. Indications, Barriers and Relevance of Standardization. MMW Fortschr. Med. 2016, 158, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Bosco, D.; Fava, A.; Plastino, M.; Montalcini, T.; Pujia, A. Possible Implications of Insulin Resistance and Glucose Metabolism in Alzheimer’s Disease Pathogenesis. J. Cell. Mol. Med. 2011, 15, 1807–1821. [Google Scholar] [CrossRef] [PubMed]
- Solano, D.C.; Sironi, M.; Bonfini, C.; Solerte, S.B.; Govoni, S.; Racchi, M. Insulin Regulates Soluble Amyloid Precursor Protein Release Via Phosphatidyl Inositol 3 Kinase-Dependent Pathway. FASEB J. 2000, 14, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, L.; Gouras, G.K.; Wang, R.; Gross, R.S.; Beal, M.F.; Greengard, P.; Xu, H. Stimulation of Β-Amyloid Precursor Protein Trafficking by Insulin Reduces Intraneuronal Β-Amyloid and Requires Mitogen-Activated Protein Kinase Signaling. J. Neurosci. 2001, 21, 2561–2570. [Google Scholar] [CrossRef]
- Mattson, M.P. Cellular Actions of Beta-Amyloid Precursor Protein and Its Soluble and Fibrillogenic Derivatives. Physiol. Rev. 1997, 77, 1081–1132. [Google Scholar] [CrossRef] [PubMed]
- Vincent, B.; Govitrapong, P. Activation of the A-Secretase Processing of Aβpp as a Therapeutic Approach in Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 24, 75–94. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, S.T.; Klein, W.L. The Aβ Oligomer Hypothesis for Synapse Failure and Memory Loss in Alzheimer’s Disease. Neurobiol. Learn. Mem. 2011, 96, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Shu, H.; Ye, Q.; Wang, Z.; Xie, C.; Yuan, B.; Zhang, Z.; Bai, F. Brain Insulin Resistance Deteriorates Cognition by Altering the Topological Features of Brain Networks. NeuroImage Clin. 2017, 13, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Pivarnik, J.M.; Stein, A.D.; Rivera, J.M. Effect of Pregnancy on Heart Rate/Oxygen Consumption Calibration Curves. Med. Sci. Sports Exerc. 2002, 34, 750–755. [Google Scholar] [CrossRef]
- Li, H.; Yin, Q.; Li, N.; Ouyang, Z.; Zhong, M. Plasma Markers of Oxidative Stress in Patients with Gestational Diabetes Mellitus in the Second and Third Trimester. Obs. Gynecol. Int. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Yang, H.; Geng, Q.; Ma, Q.; Long, Y.; Zhou, C.; Chen, M. Association of Oxidative Stress Biomarkers with Gestational Diabetes Mellitus in Pregnant Women: A Case-Control Study. PLoS ONE 2015, 10, e0126490. [Google Scholar] [CrossRef] [Green Version]
- Genc, S.; Kusku-Kiraz, Z.; Dervisoglu, E.; Oztop, N.; Dinccag, N.; Gurdol, F. The Relation of Oxidative Stress Biomarkers with Proinflammatory Cytokines in Gestational Diabetes. Clin. Investig. 2017, 7, 44–48. [Google Scholar] [CrossRef]
- Pan, H.Z.; Zhang, L.; Guo, M.Y.; Sui, H.; Li, H.; Wu, W.H.; Qu, N.Q.; Liang, M.H.; Chang, D. The Oxidative Stress Status in Diabetes Mellitus and Diabetic Nephropathy. Acta Diabetol. 2010, 47, 71–76. [Google Scholar] [CrossRef]
- Peerapatdit, T.; Sriratanasathavorn, C. Lipid Peroxidation and Antioxidant Enzyme Activities in Erythrocytes of Type 2 Diabetic Patients. J. Med. Assoc. Thai 2010, 93, 682–693. [Google Scholar]
- Chang, C.; Hsieh, C.-J.; Huang, J.-C.; Huang, I.-C. Acute and Chronic Fluctuations in Blood Glucose Levels Can Increase Oxidative Stress in Type 2 Diabetes Mellitus. Acta Diabetol. 2012, 49, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.M.; Scapozza, L.; Ruegg, U.T.; Dorchies, O.M. Diapocynin, a Dimer of the Nadph Oxidase Inhibitor Apocynin, Reduces Ros Production and Prevents Force Loss in Eccentrically Contracting Dystrophic Muscle. PLoS ONE 2014, 9, e110708. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, M.T.; Vervaart, P.P.; Permezel, M.; Georgiou, H.M.; Rice, G.E. Altered Placental Oxidative Stress Status in Gestational Diabetes Mellitus. Placenta 2004, 25, 78–84. [Google Scholar] [CrossRef]
- Pistell, P.J.; Morrison, C.D.; Gupta, S.; Knight, A.G.; Keller, J.N.; Ingram, D.K.; Bruce-Keller, A.J. Cognitive Impairment Following High Fat Diet Consumption Is Associated with Brain Inflammation. J. Neuroimmunol. 2010, 219, 25–32. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M.; Luong, T.; Neely, T.R.; Robinson, D.; Wands, J.R. Mitochondrial DNA Damage as a Mechanism of Cell Loss in Alzheimer’s Disease. Lab. Investig. 2000, 80, 1323. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Wei, Y.; James, R.S. Role of Mitochondrial Dysfunction in Insulin Resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Tseng, Y.; White, M.F. Insulin Signaling Meets Mitochondria in Metabolism. Trends Endocrinol. Metab. 2010, 21, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Larsen, R.; Gouveia, Z.; Miguel, P.S.; Gozzelino, R. Heme Cytotoxicity and the Pathogenesis of Immune-Mediated Inflammatory Diseases. Front. Pharm. 2012, 3, 77. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.C.; Santos, R.X.; Santos, M.S.; Casadesus, G.; Lamanna, J.C.; Perry, G.; Smith, M.A.; Moreira, P.I. Mitochondrial Abnormalities in a Streptozotocin-Induced Rat Model of Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2013, 10, 406–419. [Google Scholar] [CrossRef]
- Merad-Boudia, M.; Nicole, A.; Santiard-Baron, D.; Saillé, C.; Ceballos-Picot, I. Mitochondrial Impairment as an Early Event in the Process of Apoptosis Induced by Glutathione Depletion in Neuronal Cells: Relevance to Parkinson’s Disease. Biochem. Pharmacol. 1998, 56, 645–655. [Google Scholar] [CrossRef]
- Alvarez, L.A.; Kovačič, L.; Rodríguez, J.; Gosemann, J.H.; Kubica, M.; Pircalabioru, G.G.; Friedmacher, F.; Cean, A.; Ghişe, A.; Sărăndan, M.B. Nadph Oxidase-Derived H2o2 Subverts Pathogen Signaling by Oxidative Phosphotyrosine Conversion to Pb-Dopa. Proc. Natl. Acad. Sci. USA 2016, 113, 10406–10411. [Google Scholar] [CrossRef] [PubMed]
- Rosales-Corral, S.; Tan, D.-X.; Manchester, L.; Reiter, R.J. Diabetes and Alzheimer Disease, Two Overlapping Pathologies with the Same Background: Oxidative Stress. Oxid. Med. Cell. Longev. 2015, 2015, 14. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Chung, S.S. Contributions of Polyol Pathway to Oxidative Stress in Diabetic Cataract. FASEB J. 1999, 1999, 23–30. [Google Scholar] [CrossRef]
- Pantham, P.; Aye, I.L.; Powell, T.L. Inflammation in Maternal Obesity and Gestational Diabetes Mellitus. Placenta 2015, 36, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Gueuvoghlanian-Silva, B.Y.; Torloni, M.R.; Mattar, R.; de Oliveira, L.S.; Scomparini, F.B.; Nakamura, M.U.; Daher, S. Profile of Inflammatory Mediators in Gestational Diabetes Mellitus: Phenotype and Genotype. Am. J. Reprod. Immunol. 2012, 67, 241–250. [Google Scholar] [CrossRef] [PubMed]
- White, S.L.; Pasupathy, D.; Sattar, N.; Nelson, S.M.; Lawlor, D.A.; Briley, A.L.; Seed, P.T.; Welsh, P.; Poston, L.; UPBEAT Consortium. Metabolic Profiling of Gestational Diabetes in Obese Women during Pregnancy. Diabetologia 2017, 60, 1903–1912. [Google Scholar] [CrossRef]
- Ryan, A.S. Inflammatory Markers in Older Women with a History of Gestational Diabetes and the Effects of Weight Loss. J. Diabetes Res. 2018, 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Korkmazer, E.; Solak, N. Correlation between Inflammatory Markers and Insulin Resistance in Pregnancy. J. Obs. Gynaecol. 2015, 35, 142–145. [Google Scholar] [CrossRef]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory Mechanisms in Obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef]
- Gorska-Ciebiada, M.; Saryusz-Wolska, M.; Borkowska, A.; Ciebiada, M.; Loba, J. Serum Levels of Inflammatory Markers in Depressed Elderly Patients with Diabetes and Mild Cognitive Impairment. PLoS ONE 2015, 10, e0120433. [Google Scholar] [CrossRef]
- Magaki, S.; Mueller, C.; Dickson, C.; Kirsch, W. Increased Production of Inflammatory Cytokines in Mild Cognitive Impairment. Exp. Gerontol. 2007, 42, 233–240. [Google Scholar] [CrossRef] [PubMed]
- McGuire, S.O.; Ling, Z.D.; Lipton, J.W.; Sortwell, C.E.; Collier, T.J.; Carvey, P.M. Tumor Necrosis Factor A Is Toxic to Embryonic Mesencephalic Dopamine Neurons. Exp. Neurol. 2001, 169, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Boje, K.M.; Arora, P.K. Microglial-Produced Nitric Oxide and Reactive Nitrogen Oxides Mediate Neuronal Cell Death. Brain Res. 1992, 587, 250–256. [Google Scholar] [CrossRef]
- Allan, S.M.; Rothwell, N.J. Inflammation in Central Nervous System Injury. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003, 358, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Neuronal Localization of Amyloid Beta Protein Precursor Mrna in Normal Human Brain and in Alzheimer’s Disease. EMBO J. 1987, 6, 3627–3632. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Lieberburg, I. Cellular Mechanisms of Β-Amyloid Production and Secretion. Proc. Natl. Acad. Sci. USA 1999, 96, 11049–11053. [Google Scholar] [CrossRef] [PubMed]
- Gorska-Ciebiada, M.; Saryusz-Wolska, M.; Borkowska, A.; Ciebiada, M.; Loba, J. Adiponectin, Leptin and Il-1 Β in Elderly Diabetic Patients with Mild Cognitive Impairment. Metab. Brain Dis. 2016, 31, 257–266. [Google Scholar] [CrossRef]
- Tsuchida, A.; Yamauchi, T.; Kadowaki, T. Nuclear Receptors as Targets for Drug Development: Molecular Mechanisms for Regulation of Obesity and Insulin Resistance by Peroxisome Proliferator-Activated Receptor &Gamma, Creb-Binding Protein, and Adiponectin. J. Pharmacol. Sci. 2005, 97, 164–170. [Google Scholar]
- Al-Badri, M.R.; Zantout, M.S.; Azar, S.T. The Role of Adipokines in Gestational Diabetes Mellitus. Ther. Adv. Endocrinol. Metab. 2015, 6, 103–108. [Google Scholar] [CrossRef]
- Saini, V.M.; Yadav, K.A.; Jain, A. Role of Leptin and Adiponectin in Gestational Diabetes Mellitus: A Study in a North Indian Tertiary Care Hospital. Int. J. Med. Update 2015, 10, 11–14. [Google Scholar] [CrossRef]
- Yokota, T.; Oritani, K.; Takahashi, I.; Ishikawa, J.; Matsuyama, A.; Ouchi, N.; Kihara, S.; Funahashi, T.; Tenner, A.J.; Tomiyama, Y.; et al. Adiponectin, a New Member of the Family of Soluble Defense Collagens, Negatively Regulates the Growth of Myelomonocytic Progenitors and the Functions of Macrophages. Blood 2000, 96, 1723–1732. [Google Scholar] [PubMed]
- Racke, M.K.; Drew, P.D. Ppars in Neuroinflammation. PPAR Res. 2008, 2008, 638356. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.I.; Ahn, Y.H. Role of Peroxisome Proliferator-Activated Receptor-gamma in the Glucose-Sensing Apparatus of Liver and Β-Cells. Diabetes 2004, 53, S60–S65. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Evidence | Markers | Pathophysiological Changes | Adverse Effects and Outcome |
|---|---|---|---|
| Hyperglycemia | Advanced glycation end products (AGEs) [26]. |
| |
| Insulin resistance | GLUT 4 [31], GluA1, GluN2B [32], IRS-1 [33], GSK3β [34]. |
| |
| Oxidative stress | ROS [37], p53 [38], NOS/NOX, [38,39,40,41,42] SOD, CAT, GPX [43,44,45], GSH [46]. | ||
| Neuro- inflammation | IL-6 and TNF-α [53], IL-2, hs-CRP [54], IL-8,NF-κβ [55] IL-1β, [56], Leptin [57], PPAR-γ [58]. |
|
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
John, C.M.; Mohamed Yusof, N.I.S.; Abdul Aziz, S.H.; Mohd Fauzi, F. Maternal Cognitive Impairment Associated with Gestational Diabetes Mellitus—A Review of Potential Contributing Mechanisms. Int. J. Mol. Sci. 2018, 19, 3894. https://doi.org/10.3390/ijms19123894
John CM, Mohamed Yusof NIS, Abdul Aziz SH, Mohd Fauzi F. Maternal Cognitive Impairment Associated with Gestational Diabetes Mellitus—A Review of Potential Contributing Mechanisms. International Journal of Molecular Sciences. 2018; 19(12):3894. https://doi.org/10.3390/ijms19123894
Chicago/Turabian StyleJohn, Cini Mathew, Nur Intan Saidaah Mohamed Yusof, Siti Hajar Abdul Aziz, and Fazlin Mohd Fauzi. 2018. "Maternal Cognitive Impairment Associated with Gestational Diabetes Mellitus—A Review of Potential Contributing Mechanisms" International Journal of Molecular Sciences 19, no. 12: 3894. https://doi.org/10.3390/ijms19123894