iTRAQ-Based Quantitative Proteomic Analysis of Embryogenic and Non-embryogenic Calli Derived from a Maize (Zea mays L.) Inbred Line Y423

Abstract
:1. Introduction
2. Results
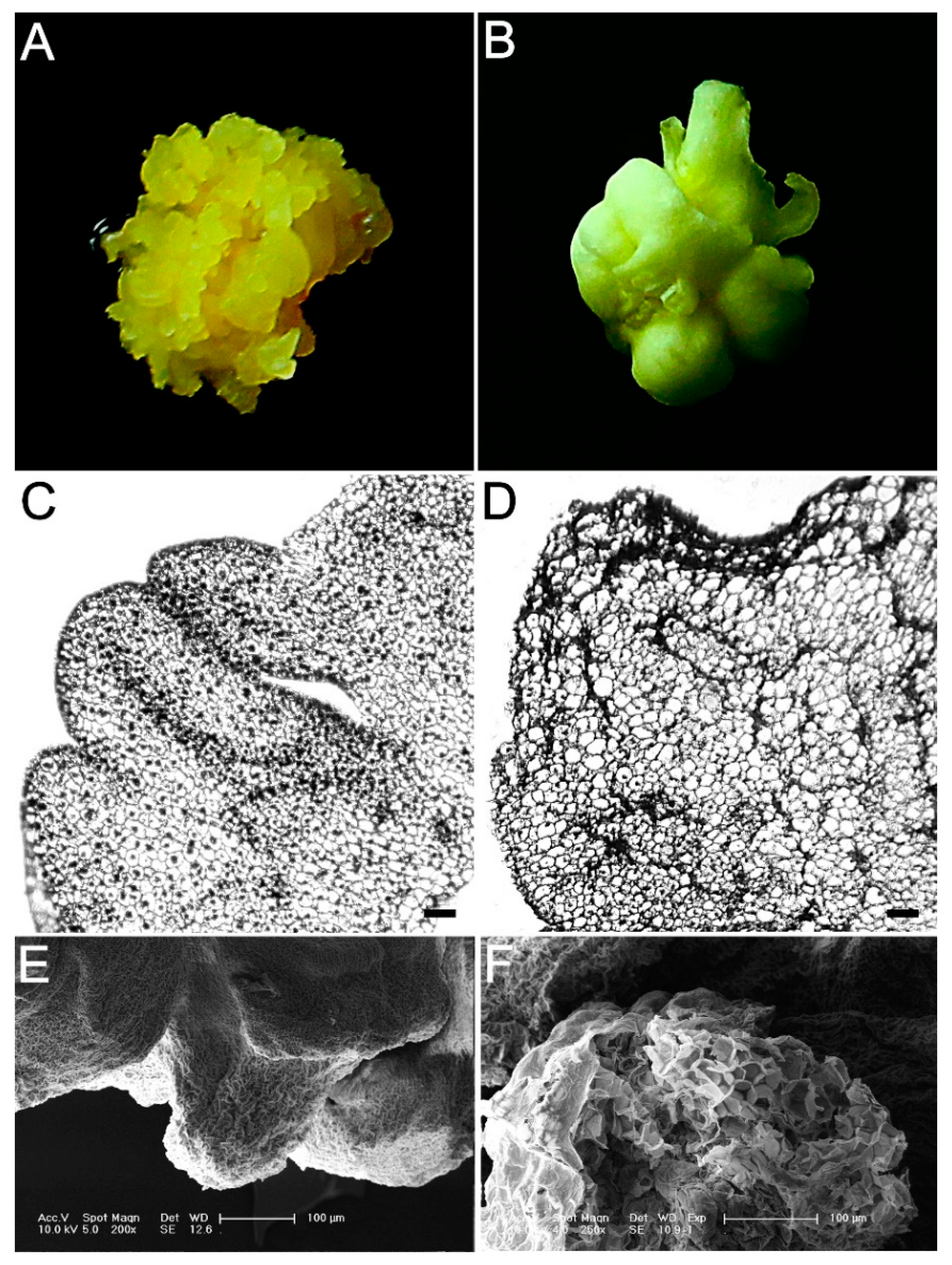
2.1. Induction of Embryogenic and Nonembryogenic Calli
2.2. Primary Data Analysis and Protein Identification
2.3. Identification of Differentially Accumulated Protein Species (DAPS) by iTRAQ
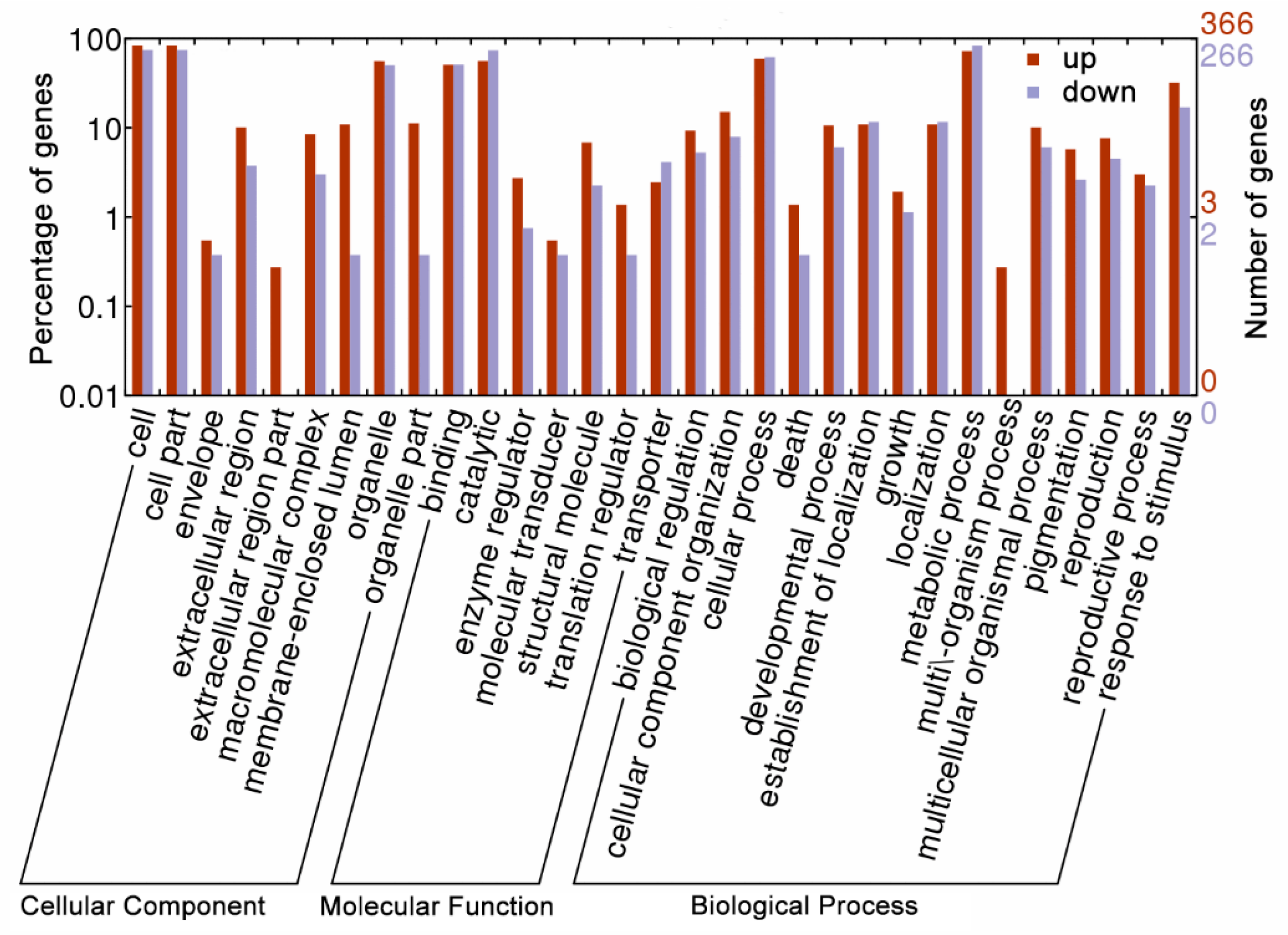
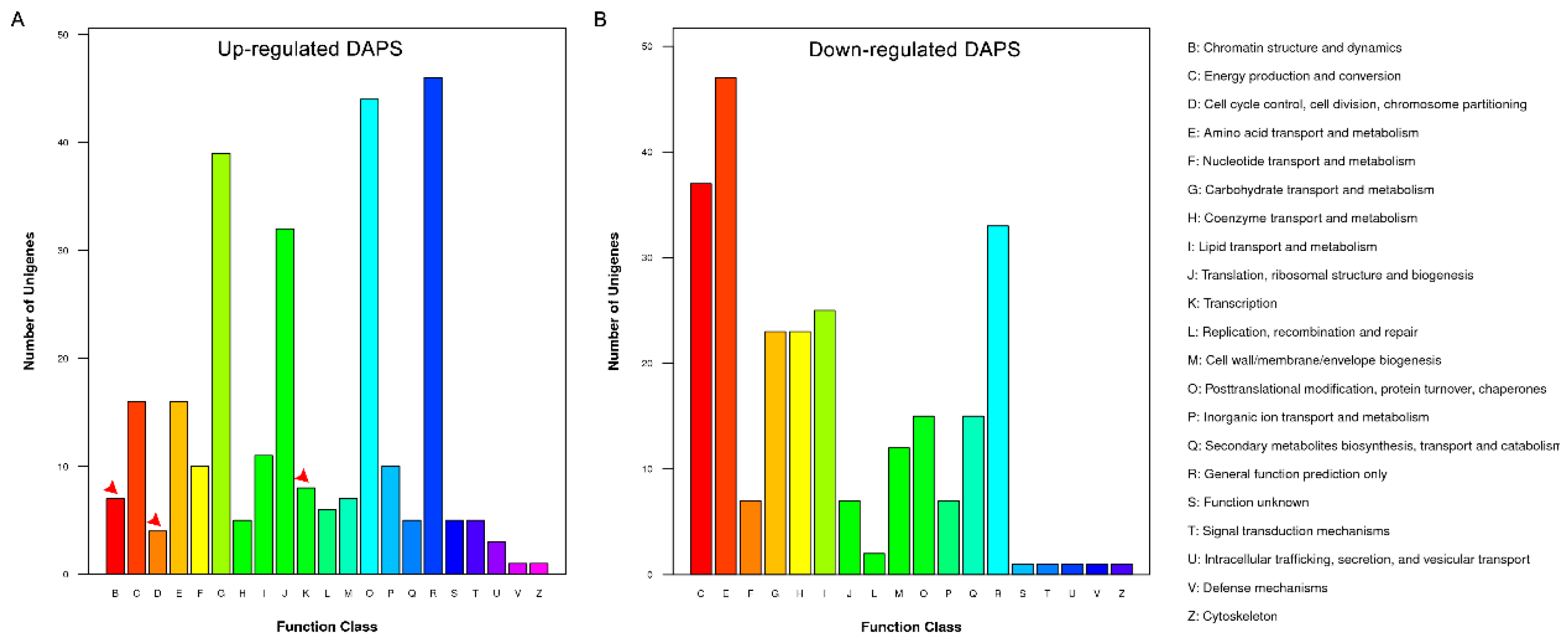
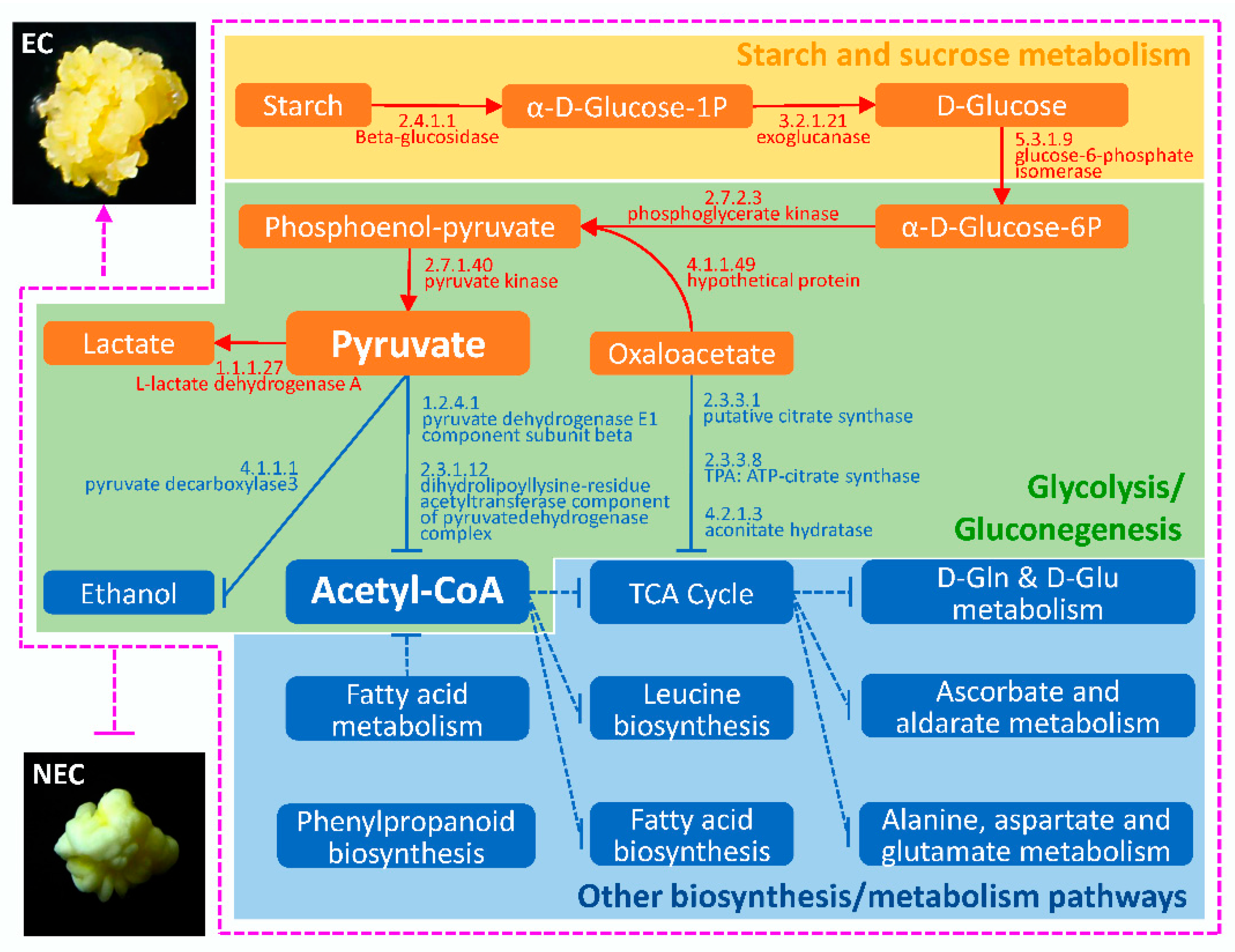
2.4. Bioinformatics Analysis of DAPS Identified by iTRAQ
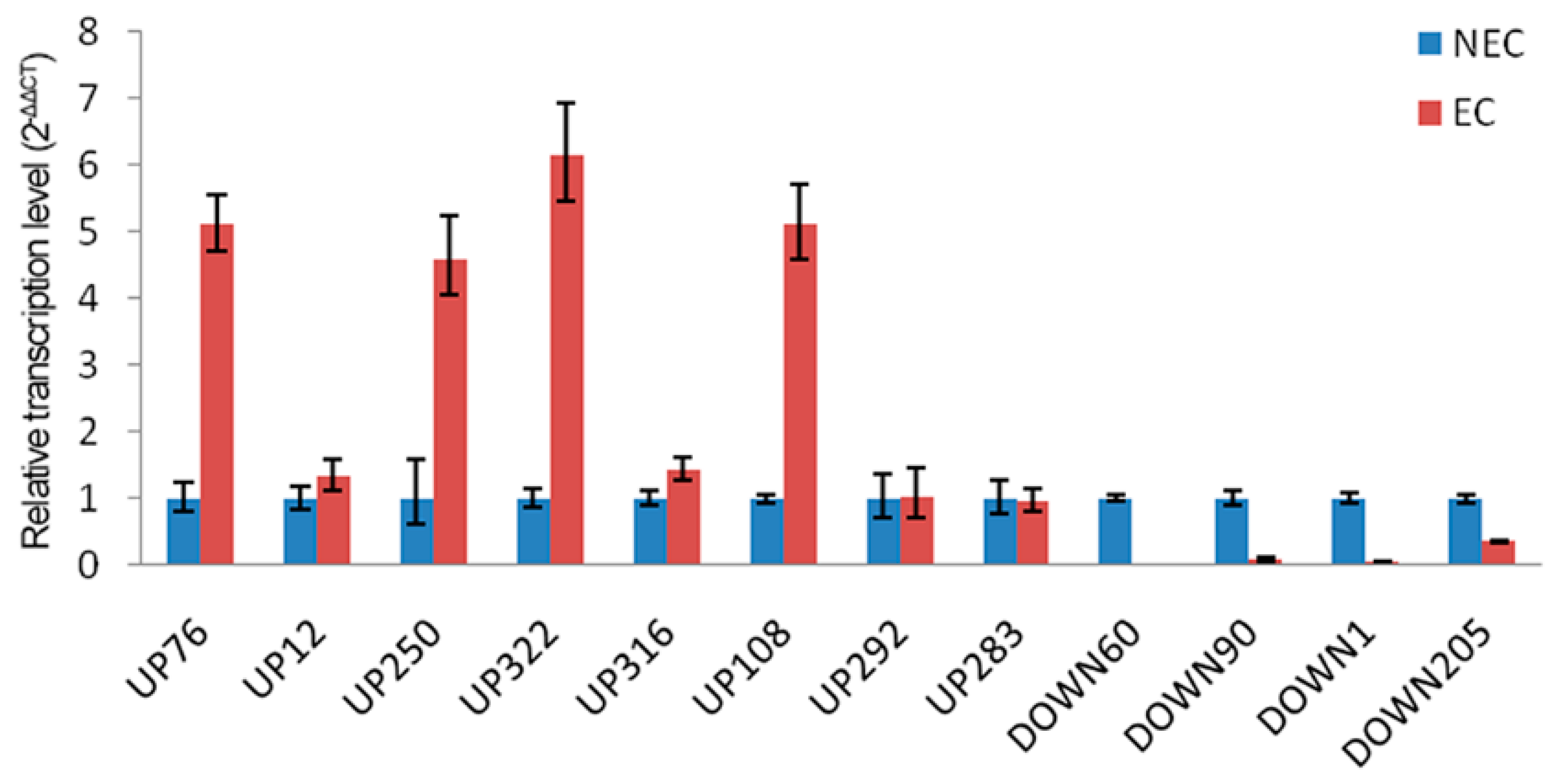
2.5. Transcriptional Analysis of Selected Genes for the Differentially Expressed Proteins
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Tissue Culture Conditions
4.2. Protein Preparation
4.3. iTRAQ Labeling and SCX Fractionation
4.4. LC-ESI-MS/MS Analysis
4.5. Protein Identification and Data Analysis
4.6. Bioinformatics Analysis
4.7. Quantitative Real-Time PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| COG | clusters of orthologous groups of proteins |
| DAPS | differential abundance protein species |
| EC | embryogenic calli |
| FDR | false discovery rate |
| GM | genetically modified |
| GO | gene ontology |
| iTRAQ | isobaric tags for relative and absolute quantification |
| NEC | nonembryogenic calli |
| SE | Somatic embryo |
| TCA | tricarboxylic acid |
| UDP | uracil-diphosphate |
References
- Ash, C.; Jasny, B.R.; Malakoff, D.A.; Sugden, A.M. Food security. Feeding the future. Introduction. Science 2010, 327, 797. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.L.; Romero-Severson, J.; Hodges, T.K. Improved tissue culture response of an elite maize inbred through backcross breeding, and identification of chromosomal regions important for regeneration by RFLP analysis. Theor. Appl. Genet. 1992, 84, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Landi, P.; Chiappetta, L.; Salvi, S.; Frascaroli, E.; Lucchese, C.; Tuberosa, R. Responses and allelic frequency changes associated with recurrent selection for plant regeneration from callus cultures in maize. Maydica 2002, 47, 21–32. [Google Scholar]
- Hodges, T.K.; Kamo, K.K.; Imbrie, C.W.; Becwar, M.R. Genotype Specificity of Somatic Embryogenesis and Regeneration in Maize. Nat. Biotechnol. 1986, 4, 219–223. [Google Scholar] [CrossRef]
- Garrocho-Villegas, V.; De Jesus-Olivera, M.T.; Quintanar, E.S. Maize somatic embryogenesis: Recent features to improve plant regeneration. Methods Mol. Biol. 2012, 877, 173–182. [Google Scholar] [PubMed]
- Dennis, J.G.P.D.; Amul, P.P.D.; Triglano, R.N. Somatic embryogenesis and development of synthetic seed technology. Crit. Rev. Plant Sci. 1991, 10, 33–61. [Google Scholar]
- Murray, S.C.; Eckhoff, P.; Wood, L.; Paterson, A.H. A proposal to use gamete cycling in vitro to improve crops and livestock. Nat. Biotechnol. 2013, 31, 877–880. [Google Scholar] [CrossRef]
- Liu, B.; Su, S.; Wu, Y.; Li, Y.; Shan, X.; Li, S.; Liu, H.; Dong, H.; Ding, M.; Han, J.; et al. Histological and transcript analyses of intact somatic embryos in an elite maize (Zea mays L.) inbred line Y423. Plant Physiol. Biochem. 2015, 92, 81–91. [Google Scholar] [CrossRef]
- Sun, L.; Wu, Y.; Su, S.; Liu, H.; Yang, G.; Li, S.; Shan, X.; Yuan, Y. Differential gene expression during somatic embryogenesis in the maize (Zea mays L.) inbred line H99. Plant Cell Tissue Organ Cult. 2012, 109, 271–286. [Google Scholar] [CrossRef]
- Šamaj, J.; Baluška, F.; Bobák, M.; Volkmann, D. Extracellular matrix surface network of embryogenic units of friable maize callus contains arabinogalactan-proteins recognized by monoclonal antibody JIM4. Plant Cell Rep. 1999, 18, 369–374. [Google Scholar] [CrossRef]
- Letarte, J.; Simion, E.; Miner, M.; Kasha, K.J. Arabinogalactans and arabinogalactan-proteins induce embryogenesis in wheat (Triticum aestivum L.) microspore culture. Plant Cell Rep. 2006, 24, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Stacey, N.J.; Roberts, K.; Knox, J.P. Patterns of expression of the JIM4 arabinogalactan-protein epitope in cell cultures and during somatic embryogenesis in Daucus carota L. Planta 1990, 180, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Neutelings, G.; Domon, J.M.; Membre, N.; Bernier, F.; Meyer, Y.; David, A.; David, H. Characterization of a germin-like protein gene expressed in somatic and zygotic embryos of pine (Pinus caribaea Morelet). Plant Mol. Biol. 1998, 38, 1179–1190. [Google Scholar] [CrossRef]
- Almeida, A.M.; Parreira, J.R.; Santos, R.; Duque, A.S.; Francisco, R.; Tome, D.F.; Ricardo, C.P.; Coelho, A.V.; Fevereiro, P. A proteomics study of the induction of somatic embryogenesis in Medicago truncatula using 2DE and MALDI-TOF/TOF. Physiol. Plant. 2012, 146, 236–249. [Google Scholar] [CrossRef] [PubMed]
- Imin, N.; De Jong, F.; Mathesius, U.; Van Noorden, G.; Saeed, N.A.; Wang, X.D.; Rose, R.J.; Rolfe, B.G. Proteome reference maps of Medicago truncatula embryogenic cell cultures generated from single protoplasts. Proteomics 2004, 4, 1883–1896. [Google Scholar] [CrossRef] [PubMed]
- Winkelmann, T.; Heintz, D.; Van Dorsselaer, A.; Serek, M.; Braun, H.P. Proteomic analyses of somatic and zygotic embryos of Cyclamen persicum Mill. reveal new insights into seed and germination physiology. Planta 2006, 224, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Rode, C.; Lindhorst, K.; Braun, H.P.; Winkelmann, T. From callus to embryo: A proteomic view on the development and maturation of somatic embryos in Cyclamen persicum. Planta 2012, 235, 995–1011. [Google Scholar] [CrossRef]
- Sun, L.; Wu, Y.; Zou, H.; Su, S.; Li, S.; Shan, X.; Xi, J.; Yuan, Y. Comparative proteomic analysis of the H99 inbred maize (Zea mays L.) line in embryogenic and non-embryogenic callus during somatic embryogenesis. Plant Cell Tissue Organ Cult. 2013, 113, 103–119. [Google Scholar] [CrossRef]
- Marsoni, M.; Bracale, M.; Espen, L.; Prinsi, B.; Negri, A.S.; Vannini, C. Proteomic analysis of somatic embryogenesis in Vitis vinifera. Plant Cell Rep. 2008, 27, 347–356. [Google Scholar] [CrossRef]
- Pan, Z.; Guan, R.; Zhu, S.; Deng, X. Proteomic analysis of somatic embryogenesis in Valencia sweet orange (Citrus sinensis Osbeck). Plant Cell Rep. 2009, 28, 281–289. [Google Scholar] [CrossRef]
- Silva Rde, C.; Carmo, L.S.; Luis, Z.G.; Silva, L.P.; Scherwinski-Pereira, J.E.; Mehta, A. Proteomic identification of differentially expressed proteins during the acquisition of somatic embryogenesis in oil palm (Elaeis guineensis Jacq.). J. Proteom. 2014, 104, 112–127. [Google Scholar] [CrossRef] [PubMed]
- Gilany, K. Proteomics a Key Tool for a Better Understanding of Endometriosis: A Mini-Review. J. Paramed. Sci. 2011, 2, 51–58. [Google Scholar]
- Zieske, L.R. A perspective on the use of iTRAQ reagent technology for protein complex and profiling studies. J. Exp. Bot. 2006, 57, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.G.; Cheng, W.H.; Tian, W.G.; Li, Y.J.; Liu, F.; Xue, F.; Zhu, Q.H.; Sun, Y.Q.; Sun, J. iTRAQ-based comparative proteomic analysis provides insights into somatic embryogenesis in Gossypium hirsutum L. Plant Mol. Biol. 2017, 96, 89–102. [Google Scholar] [CrossRef]
- Zhao, J.; Li, H.; Fu, S.; Chen, B.; Sun, W.; Zhang, J.; Zhang, J. An iTRAQ-based proteomics approach to clarify the molecular physiology of somatic embryo development in Prince Rupprecht’s larch (Larix principis-rupprechtii Mayr). PLoS ONE 2015, 10, e0119987. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Vasil, V.; Vasil, I.K. Improved efficiency of somatic embryogenesis and plant regeneration in tissue cultures of maize (Zea mays L.). Theor. Appl. Genet. 1983, 66, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Saito, H.; Ohta, S.; Hiei, Y.; Komari, T.; Kumashiro, T. High efficiency transformation of maize (Zea mays L.) mediated by Agrobacterium tumefaciens. Nat. Biotechnol. 1996, 14, 745–750. [Google Scholar] [CrossRef]
- Zhao, Z.Y.; Gu, W.N.; Cai, T.S.; Tagliani, L.; Hondred, D.; Bond, D.; Schroeder, S.; Rudert, M.; Pierce, D. High throughput genetic transformation mediated by Agrobacterium tumefaciens in maize. Mol. Breed. 2002, 8, 323–333. [Google Scholar] [CrossRef]
- Yokoya, N.S.; West, J.A.; Luchi, A.E. Effects of plant growth regulators on callus formation, growth and regeneration in axenic tissue cultures of Gracilaria tenuistipitata and Gracilaria perplexa (Gracilariales, Rhodophyta). Phycol. Res. 2004, 52, 244–254. [Google Scholar] [CrossRef]
- Cheng, W.H.; Wang, F.L.; Cheng, X.Q.; Zhu, Q.H.; Sun, Y.Q.; Zhu, H.G.; Sun, J. Polyamine and Its Metabolite H2O2 Play a Key Role in the Conversion of Embryogenic Callus into Somatic Embryos in Upland Cotton (Gossypium hirsutum L.). Front. Plant Sci. 2015, 6, 1063. [Google Scholar] [CrossRef]
- Shang, H.H.; Liu, C.L.; Zhang, C.J.; Li, F.L.; Hong, W.D.; Li, F.G. Histological and ultrastructural observation reveals significant cellular differences between Agrobacterium transformed embryogenic and non-embryogenic calli of cotton. J. Integr. Plant Biol. 2009, 51, 456–465. [Google Scholar] [CrossRef]
- Rajasekaran, K. Regeneration of plants from cryopreserved embryogenic cell suspension and callus cultures of cotton (Gossypium hirsutum L.). Plant Cell Rep. 1996, 15, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Overvoorde, P.J.; Grimes, H.D. The Role of Calcium and Calmodulin in Carrot Somatic Embryogenesis. Plant Cell Physiol. 1994, 35, 135–144. [Google Scholar]
- Suprasanna, P.; Desai, N.S.; Nishanth, G.; Ghosh, S.B.; Laxmi, N.; Bapat, V.A. Differential gene expression in embryogenic, non-embryogenic and desiccation induced cultures of sugarcane. Sugar Tech 2004, 6, 305–309. [Google Scholar] [CrossRef]
- Kawade, K.; Ishizaki, T.; Masuda, K. Differential expression of ribosome-inactivating protein genes during somatic embryogenesis in spinach (Spinacia oleracea). Physiol. Plant. 2008, 134, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Varhanikova, M.; Uvackova, L.; Skultety, L.; Pretova, A.; Obert, B.; Hajduch, M. Comparative quantitative proteomic analysis of embryogenic and non-embryogenic calli in maize suggests the role of oxylipins in plant totipotency. J. Proteom. 2014, 104, 57–65. [Google Scholar] [CrossRef]
- Andriotis, V.M.; Kruger, N.J.; Pike, M.J.; Smith, A.M. Plastidial glycolysis in developing arabidopsis embryos. New Phytol. 2010, 185, 649–662. [Google Scholar] [CrossRef]
- Zhang, J.; Khvorostov, I.; Hong, J.S.; Oktay, Y.; Vergnes, L.; Nuebel, E.; Wahjudi, P.N.; Setoguchi, K.; Wang, G.; Do, A.; et al. UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells. EMBO J. 2011, 30, 4860–4873. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.M.; Liu, X.; Shen, J.; Jovanovic, O.; Pohl, E.E.; Gerson, S.L.; Finkel, T.; Broxmeyer, H.E.; Qu, C.K. Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell 2013, 12, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Corbet, C. Stem Cell Metabolism in Cancer and Healthy Tissues. Pyruvate in the Limelight. Front. Pharmacol. 2017, 8, 958. [Google Scholar] [CrossRef]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simsek, T.; Kocabas, F.; Zheng, J.; Deberardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, V.M.; Bangerth, F. Hormonal status of maize initial explants and of the embryogenic and non-embryogenic callus cultures derived from them as related to morphogenesis in vitro. Plant Sci. 2001, 160, 247–257. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Accession | Fold Change | Description | COG Code |
|---|---|---|---|
| gi|195605264|gb|ACG24462.1| | 1.311 | Histone H2A [Zea mays] | B |
| gi|195635409|gb|ACG37173.1| | 1.314 | Histone H4 [Zea mays] | B |
| gi|162458988|ref|NP_001105357.1| | 1.41 | Histone H2A [Zea mays] | B |
| gi|195618876|gb|ACG31268.1| | 1.467 | Histone H2A [Zea mays] | B |
| gi|195616386|gb|ACG30023.1| | 1.541 | Histone H2A [Zea mays] | B |
| gi|195617710|gb|ACG30685.1| | 1.906 | Histone H2A [Zea mays] | B |
| gi|194693822|gb|ACF80995.1| | 2.084 | Unknown [Zea mays] | B |
| gi|226496461|ref|NP_001140401.1| | 1.234 | Calmodulin [Zea mays] | DTZR |
| gi|226501230|ref|NP_001149695.1| | 1.29 | Cell division protein ftsz [Zea mays] | D |
| gi|226494524|ref|NP_001150546.1| | 1.321 | Serine/threonine-protein phosphatase 2A activator 2 [Zea mays] | DT |
| gi|293331121|ref|NP_001167965.1| | 1.334 | Uncharacterized protein LOC100381681 [Zea mays] | D |
| gi|212276110|ref|NP_001130839.1| | 1.315 | Uncharacterized protein LOC100191943 [Zea mays] | K |
| gi|226506726|ref|NP_001141599.1| | 1.372 | Uncharacterized protein LOC100273717 [Zea mays] | KLJ |
| gi|226490952|ref|NP_001152536.1| | 1.412 | ATP-dependent RNA helicase DDX23 [Zea mays] | KLJ |
| gi|226496545|ref|NP_001148353.1| | 1.53 | Small nuclear ribonucleoprotein Sm D1 [Zea mays] | K |
| gi|195656155|gb|ACG47545.1| | 1.565 | Small nuclear ribonucleoprotein Sm D3 [Zea mays] | K |
| gi|219363641|ref|NP_001136911.1| | 1.586 | Uncharacterized protein LOC100217069 [Zea mays] | K |
| gi|226503431|ref|NP_001140836.1| | 1.647 | Uncharacterized protein LOC100272912 [Zea mays] | K |
| gi|194699902|gb|ACF84035.1| | 2.544 | U6 snrna-associated Sm-like protein lsm3 [Zea mays] | K |
| Code | Protein Accession | Fold Change | Description |
|---|---|---|---|
| UP76 | gi|195614828|gb|ACG29244.1| | 4.26 | Nucleotide pyrophosphatase/phosphodiesterase [Zea mays] |
| UP12 | gi|195621264|gb|ACG32462.1| | 4.46 | histone H1 [Zea mays] |
| UP250 | gi|414878739|tpg|DAA55870.1| | 4.272 | TPA: putative O-Glycosyl hydrolase superfamily protein [Zea mays] |
| UP322 | gi|413952293|gb|AFW84942.1| | 4.218 | Putative subtilase family protein [Zea mays] |
| UP316 | gi|414866956|tpg|DAA45513.1| | 4.841 | Peroxidase superfamily protein [Arabidopsis thaliana] |
| UP108 | gi|414586772|tpg|DAA37343.1| | 6.499 | Polygalacturonase inhibiting protein 1 [Arabidopsis thaliana] |
| UP292 | gi|195654029|gb|ACG46482.1| | 6.87 | Rhicadhesin receptor precursor [Zea mays] |
| UP283 | gi|1352469|sp|P49174.1|INVA_MAIZE | 4.771 | Cell-wall invertase 2 [Arabidopsis thaliana] |
| DOWN60 | gi|257626267|emb|CBD24252.1| | 0.152 | Unnamed protein product [Zea mays] |
| DOWN90 | gi|239050557|ref|NP_001132867.2| | 0.233 | S-adenosylmethionine synthetase family protein [Arabidopsis thaliana] |
| DOWN205 | gi|226500748|ref|NP_001140439.1| | 0.199 | NAD(P)-binding Rossmann-fold superfamily protein [Arabidopsis thaliana] |
| DOWN1 | gi|162458192|ref|NP_001105950.1| | 0.156 | Terpenoid cyclases/Protein prenyltransferases superfamily protein [Arabidopsis thaliana] |
| Protein Accession | Enzyme Commission | Fold Change | Regulated | Description |
|---|---|---|---|---|
| gi|212722264|ref|NP_001132360.1| | S12e | 1.436 | UP | 40S ribosomal protein S12 isoform 1 [Zea mays] |
| gi|195605820|gb|ACG24740.1| | S20e | 1.352 | UP | 40S ribosomal protein S20 [Zea mays] |
| gi|195623210|gb|ACG33435.1| | S20e | 1.247 | UP | 40S ribosomal protein S20 [Zea mays] |
| gi|162463985|ref|NP_001105635.1| | S24e | 1.245 | UP | 40S ribosomal protein S24 [Zea mays] |
| gi|226532924|ref|NP_001146968.1| | S24e | 0.833 | DOWN | 40S ribosomal protein S24 [Zea mays] |
| gi|195634815|gb|ACG36876.1| | S29e | 2.384 | UP | 40S ribosomal protein S29 [Zea mays] |
| gi|226499664|ref|NP_001149323.1| | S30e | 1.394 | UP | 40S ribosomal protein S30 [Zea mays] |
| gi|162464000|ref|NP_001105390.1| | LP1, LP2 | 0.735 | DOWN | 60S acidic ribosomal protein P2B [Zea mays] |
| gi|212275159|ref|NP_001130808.1| | L13Ae | 1.46 | UP | 60S ribosomal protein L13a [Zea mays] |
| gi|195623288|gb|ACG33474.1| | L13Ae | 1.206 | UP | 60S ribosomal protein L13a [Zea mays] |
| gi|195642172|gb|ACG40554.1| | L17e | 1.536 | UP | 60S ribosomal protein L17 [Zea mays] |
| gi|195656195|gb|ACG47565.1| | L17e | 1.336 | UP | 60S ribosomal protein L17 [Zea mays] |
| gi|195621692|gb|ACG32676.1| | L24e | 1.342 | UP | 60S ribosomal protein L24 [Zea mays] |
| gi|195646398|gb|ACG42667.1| | L28e | 1.39 | UP | 60S ribosomal protein L28 [Zea mays] |
| gi|195625790|gb|ACG34725.1| | L35e | 1.535 | UP | 60S ribosomal protein L35 [Zea mays] |
| gi|413922853|gb|AFW62785.1| | L6e | 1.362 | UP | 60S ribosomal protein L6 [Zea mays] |
| gi|195656241|gb|ACG47588.1| | L7Ae | 1.352 | UP | 60S ribosomal protein L7a [Zea mays] |
| gi|212274675|ref|NP_001130101.1| | L10e | 1.791 | UP | Acid phosphatase 1 [Zea mays] |
| gi|195642478|gb|ACG40707.1| | S19 | 1.24 | UP | Glycine-rich RNA-binding protein 2 [Zea mays] |
| gi|212722104|ref|NP_001132260.1| | L11e | 1.24 | UP | Hypothetical protein [Zea mays] |
| gi|226509268|ref|NP_001141489.1| | LP0 | 0.798 | DOWN | Hypothetical protein [Zea mays] |
| gi|162460468|ref|NP_001105280.1| | L31e | 1.643 | UP | Putative 60S ribosomal protein L31 [Zea mays] |
| gi|132668| sp|P08529.2|RK14_MAIZE | L14 | 0.709 | DOWN | Recname: Full=50S ribosomal protein L14, chloroplastic |
| gi|3264605|gb|AAC24573.1| | L23Ae | 1.39 | UP | Ribosomal protein L25 [Zea mays] |
| gi|226497590|ref|NP_001148801.1| | L7Ae | 1.312 | UP | TPA: 60S ribosomal protein L7a [Zea mays] |
| gi|414873848|tpg|DAA52405.1| | S4e | 1.475 | UP | TPA: ribosomal protein S4 [Zea mays] |
| gi|212721318|ref|NP_001131538.1| | L18e | 1.414 | UP | Uncharacterized protein LOC100192878 [Zea mays] |
| gi|224030849|gb|ACN34500.1| | L6e | 1.793 | UP | Unknown [Zea mays] |
| gi|342591|gb|AAA84482.1| | RP-L16 | 0.768 | DOWN | Unknown protein (chloroplast) [Zea mays] |
| gi|257722902|emb|CBD23964.1| | L19e | 1.268 | UP | Unnamed protein product [Zea mays subsp. Mays] |
| Protein Accession | Enzyme Commission | Fold Change | Regulated | Description |
|---|---|---|---|---|
| gi|162462658|ref|NP_001105539.1| | 3.2.1.1 | 2.29 | UP | Alpha-amylase precursor [Zea mays] |
| gi|226530773|ref|NP_001150278.1| | 3.2.1.1 | 1.627 | UP | Alpha-amylase precursor [Zea mays] |
| gi|1352469|sp|P49174.1|INVA_MAIZE | 3.2.1.26 | 4.771 | UP | Beta-fructofuranosidase/cell wall isozyme; |
| gi|226495335|ref|NP_001151458.1| | 3.2.1.4 | 3.941 | UP | Endoglucanase 1 precursor [Zea mays] |
| gi|8809764|gb|AAF79936.1| | 3.2.1.21 | 2.07 | UP | Exoglucanase precursor [Zea mays] |
| gi|195639126|gb|ACG39031.1| | 2.7.1.4 | 1.261 | UP | Fructokinase-2 [Zea mays] |
| gi|162460322|ref|NP_001105368.1| | 5.3.1.9 | 1.324 | UP | Glucose-6-phosphate isomerase, cytosolic [Zea mays] |
| gi|308080756|ref|NP_001183080.1| | 3.2.1.15 | 1.683 | UP | Hypothetical protein precursor [Zea mays] |
| gi|413933069|gb|AFW67620.1| | 3.2.1.21 | 3.115 | UP | Hypothetical protein ZEAMMB73_549956 [Zea mays] |
| gi|413922001|gb|AFW61933.1| | 2.4.1.14 | 1.242 | UP | Putative sucrose-phosphate synthase family protein [Zea mays] |
| gi|413952450|gb|AFW85099.1| | 3.1.3.12/ 2.4.1.15 | 1.325 | UP | Putative trehalose phosphatase/synthase family protein [Zea mays] |
| gi|194740442|gb|ACF94692.1| | 2.4.1.1 | 1.228 | UP | Starch phosphorylase 1 precursor [Zea mays] |
| gi|525344616|ref|NP_001266691.1| | 2.4.1.13 | 1.203 | UP | Sucrose synthase 1 [Zea mays] |
| gi|414867410|tpg|DAA45967.1| | 3.2.1.28 | 2.643 | UP | TPA: hypothetical protein ZEAMMB73_076801 [Zea mays] |
| gi|414879406|tpg|DAA56537.1| | 3.1.1.11 | 2.287 | UP | TPA: pectinesterase [Zea mays] |
| gi|414885330|tpg|DAA61344.1| | 3.1.3.12/ 2.4.1.15 | 1.84 | UP | TPA: putative trehalose phosphatase/synthase family protein [Zea mays] |
| gi|257713456|emb|CBD15747.1| | 3.2.1.37 | 1.917 | UP | Unnamed protein product [Zea mays] |
| gi|257713456|emb|CBD15747.1| | 3.2.1.21 | 1.917 | UP | Unnamed protein product [Zea mays] |
| gi|413946674|gb|AFW79323.1| | 2.7.7.27 | 0.553 | DOWN | ADP-glucose pyrophosphorylase large subunit [Zea mays] |
| gi|413920595|gb|AFW60527.1| | 2.4.1.14 | 0.55 | DOWN | Putative sucrose-phosphate synthase family protein [Zea mays] |
| gi|1351136|sp|P49036.1|SUS2_MAIZE | 2.4.1.13 | 0.808 | DOWN | Sucrose-UDP glucosyltransferase 2 |
| gi|226529913|ref|NP_001146685.1| | 3.1.1.11 | 0.773 | DOWN | TPA: pectinesterase [Zea mays] |
| gi|414865602|tpg|DAA44159.1| | 3.1.3.12/ 2.4.1.15 | 0.728 | DOWN | TPA: putative trehalose phosphatase/synthase family protein [Zea mays] |
| gi|226505764|ref|NP_001149225.1| | 1.1.1.22 | 0.602 | DOWN | UDP-glucose 6-dehydrogenase [Zea mays] |
| Protein Accession | Enzyme Commission | Fold Change | Regulated | Description |
|---|---|---|---|---|
| gi|6225010|sp|P93629.1|ADHX_MAIZE | 1.1.1.1 | 1.386 | UP | Alcohol dehydrogenase class-III; |
| gi|195613242|gb|ACG28451.1| | 5.1.3.3 | 1.7 | UP | Aldose 1-epimerase precursor [Zea mays] |
| gi|162460322|ref|NP_001105368.1| | 5.3.1.9 | 1.324 | UP | Glucose-6-phosphate isomerase, cytosolic [Zea mays] |
| gi|162457962|ref|NP_001105090.1| | 4.2.1.11 | 1.58 | UP | Heat shock factor-binding protein 1 [Zea mays] |
| gi|226528284|ref|NP_001150088.1| | 4.2.1.11 | 1.609 | UP | Heat shock factor-binding protein 1 [Zea mays] |
| gi|413956284|gb|AFW88933.1| | 4.1.1.49 | 1.316 | UP | Hypothetical protein ZEAMMB73_030639 [Zea mays] |
| gi|195621388|gb|ACG32524.1| | 1.1.1.27 | 1.774 | UP | L-lactate dehydrogenase A [Zea mays] |
| gi|226509797|ref|NP_001142404.1| | 2.7.2.3 | 1.334 | UP | Phosphoglycerate kinase isoform 3 [Zea mays] |
| gi|195620854|gb|ACG32257.1| | 2.7.1.40 | 1.712 | UP | Pyruvate kinase, cytosolic isozyme [Zea mays] |
| gi|226528689|ref|NP_001141131.1| | 1.1.1.1 | 1.288 | UP | TPA: putative alcohol dehydrogenase superfamily protein [Zea mays] |
| gi|291047868|emb|CBK51447.1| | 1.2.1.3 | 1.272 | UP | Unnamed protein product [Zea mays] |
| gi|195614676|gb|ACG29168.1| | 1.2.1.3 | 0.629 | DOWN | Aldehyde dehydrogenase, dimeric NADP-preferring [Zea mays] |
| gi|162460054|ref|NP_001105576.1| | 1.2.1.3 | 0.576 | DOWN | Aldehyde dehydrogenase2 [Zea mays] |
| gi|363543269|ref|NP_001241850.1| | 1.1.1.2 | 0.3 | DOWN | Aldose reductase [Zea mays] |
| gi|293335591|ref|NP_001169718.1| | 1.8.1.4 | 0.766 | DOWN | Dihydrolipoyl dehydrogenase [Zea mays] |
| gi|226499350|ref|NP_001142314.1| | 2.3.1.12 | 0.8 | DOWN | Dihydrolipoyl lysine-residue acetyltransferase component of pyruvate dehydrogenase complex [Zea mays] |
| gi|120670|sp|P08735.2|G3PC1_MAIZE | 1.2.1.12 | 0.69 | DOWN | Glyceraldehyde-3-phosphate dehydrogenase 1, cytosolic |
| gi|226531804|ref|NP_001148246.1| | 1.1.1.2 | 0.533 | DOWN | NAD(P)H-dependent oxidoreductase [Zea mays] |
| gi|413937078|gb|AFW71629.1| | 6.2.1.1 | 0.674 | DOWN | Putative AMP-dependent synthetase and ligase superfamily protein [Zea mays] |
| gi|162457852|ref|NP_001105052.1| | 4.1.1.1 | 0.777 | DOWN | Pyruvate decarboxylase 3 [Zea mays] |
| gi|226529151|ref|NP_001150473.1| | 1.2.4.1 | 0.588 | DOWN | Pyruvate dehydrogenase E1 component subunit beta [Zea mays] |
| gi|226528639|ref|NP_001140759.1| | 1.2.4.1 | 0.613 | DOWN | Uncharacterized protein LOC100272834 [Zea mays] |
| gi|219887835|gb|ACL54292.1| | 1.2.1.12 | 0.832 | DOWN | Unknown [Zea mays] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; Shan, X.; Wu, Y.; Su, S.; Li, S.; Liu, H.; Han, J.; Yuan, Y. iTRAQ-Based Quantitative Proteomic Analysis of Embryogenic and Non-embryogenic Calli Derived from a Maize (Zea mays L.) Inbred Line Y423. Int. J. Mol. Sci. 2018, 19, 4004. https://doi.org/10.3390/ijms19124004
Liu B, Shan X, Wu Y, Su S, Li S, Liu H, Han J, Yuan Y. iTRAQ-Based Quantitative Proteomic Analysis of Embryogenic and Non-embryogenic Calli Derived from a Maize (Zea mays L.) Inbred Line Y423. International Journal of Molecular Sciences. 2018; 19(12):4004. https://doi.org/10.3390/ijms19124004
Chicago/Turabian StyleLiu, Beibei, Xiaohui Shan, Ying Wu, Shengzhong Su, Shipeng Li, Hongkui Liu, Junyou Han, and Yaping Yuan. 2018. "iTRAQ-Based Quantitative Proteomic Analysis of Embryogenic and Non-embryogenic Calli Derived from a Maize (Zea mays L.) Inbred Line Y423" International Journal of Molecular Sciences 19, no. 12: 4004. https://doi.org/10.3390/ijms19124004